MicroRNAs in Cervical Cancer: Evidences for a miRNA Profile Deregulated by HPV and Its Impact on Radio-Resistance


 ,
, 

Abstract
:1. Introduction
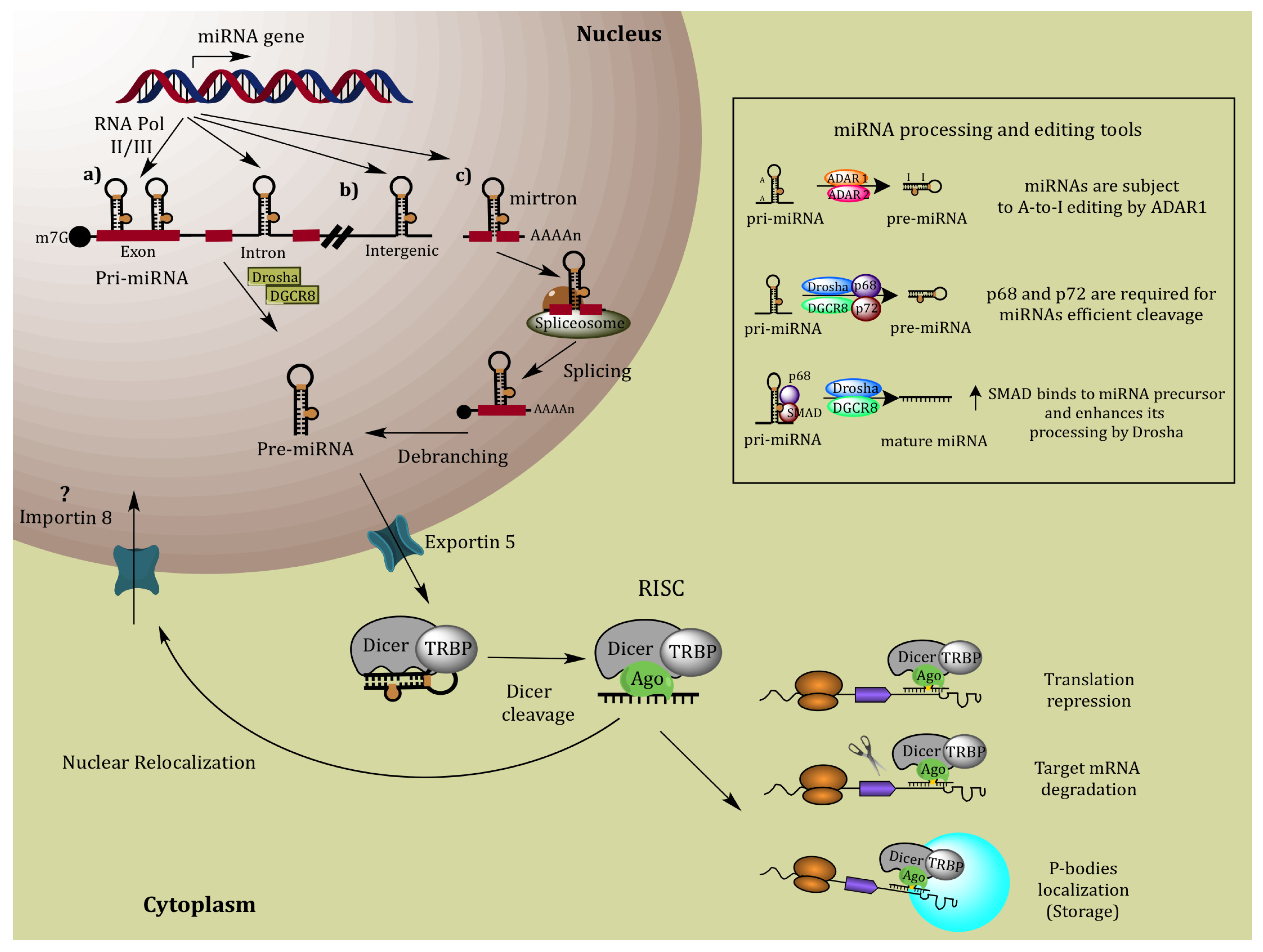
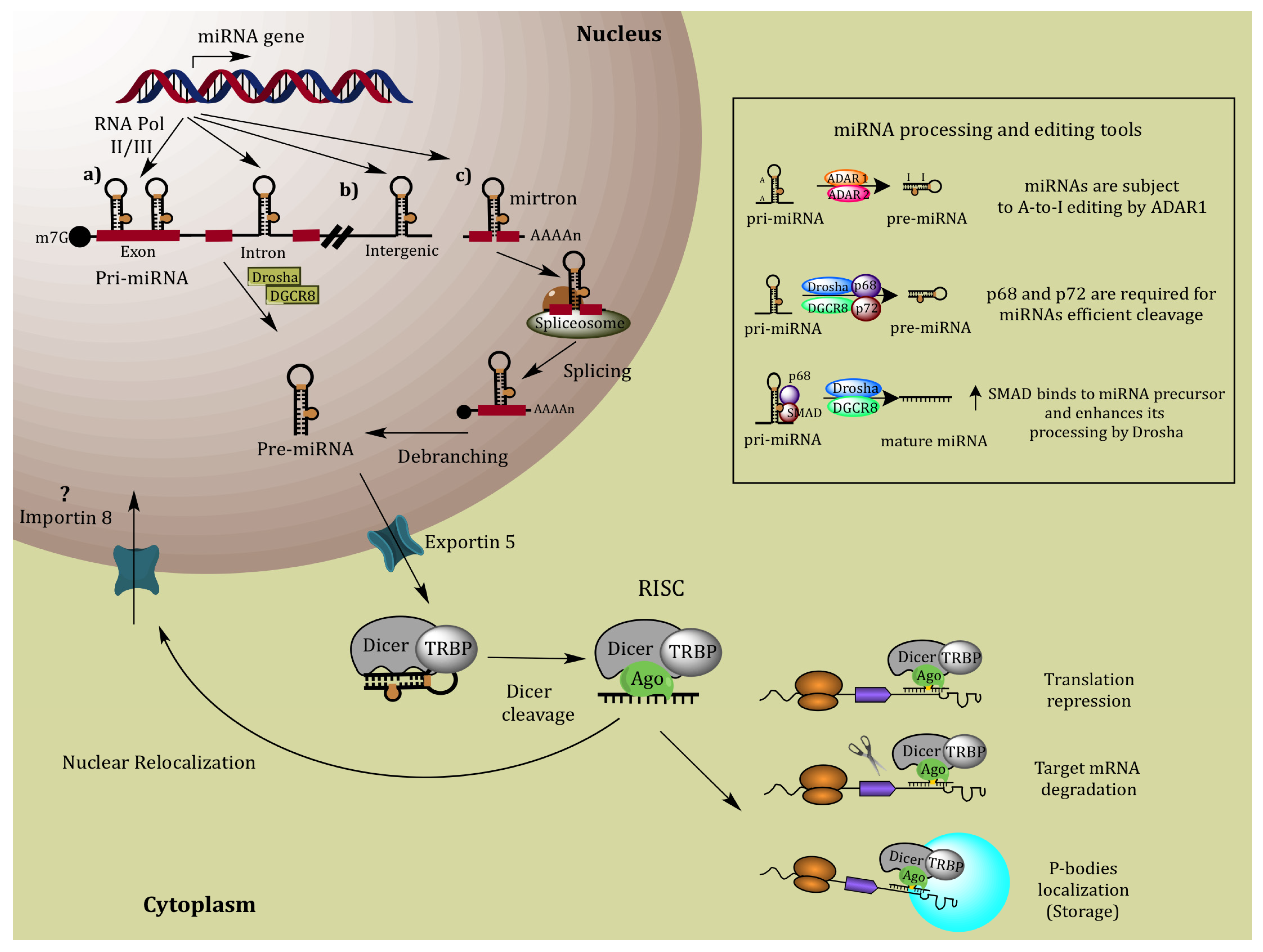
2. MiRNA Biogenesis
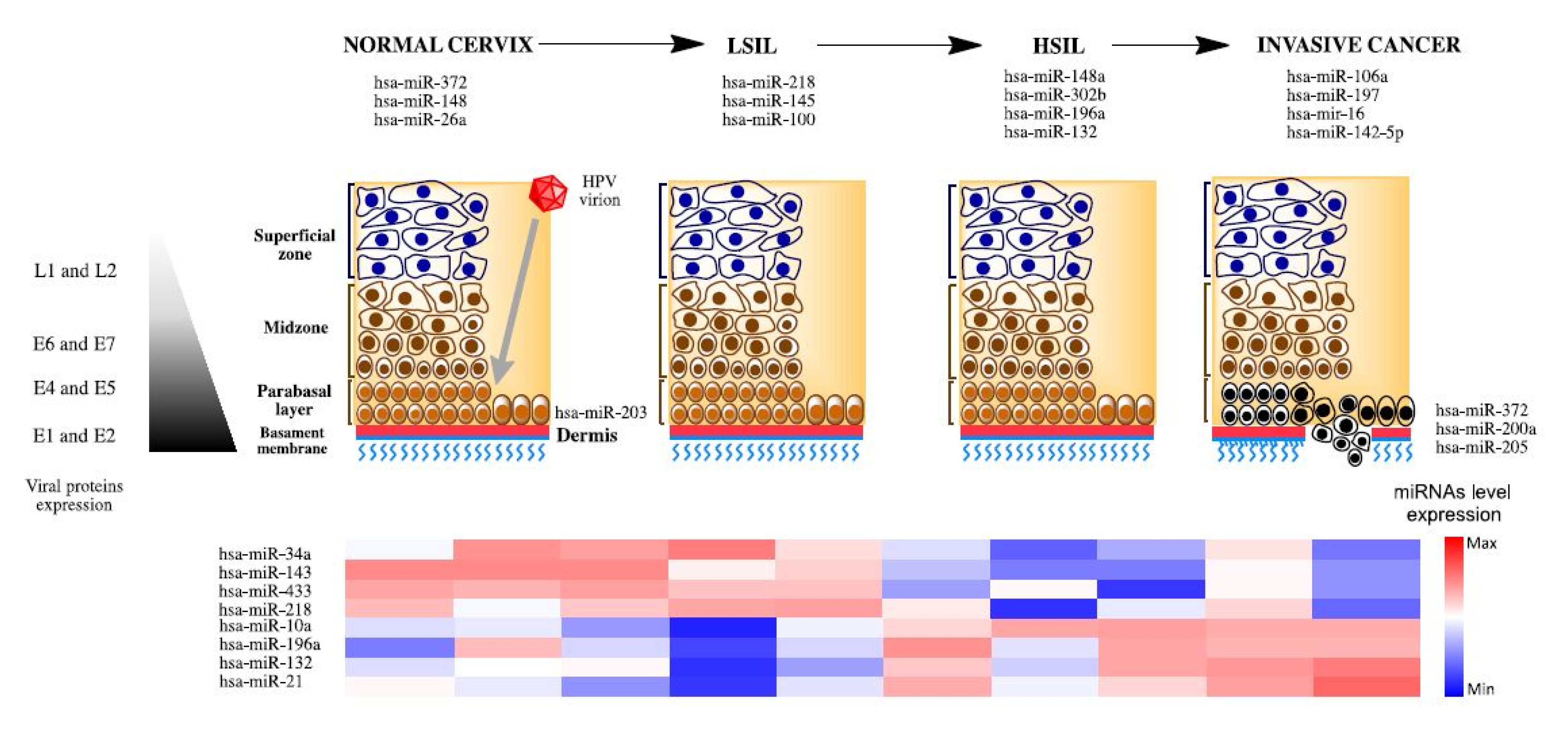
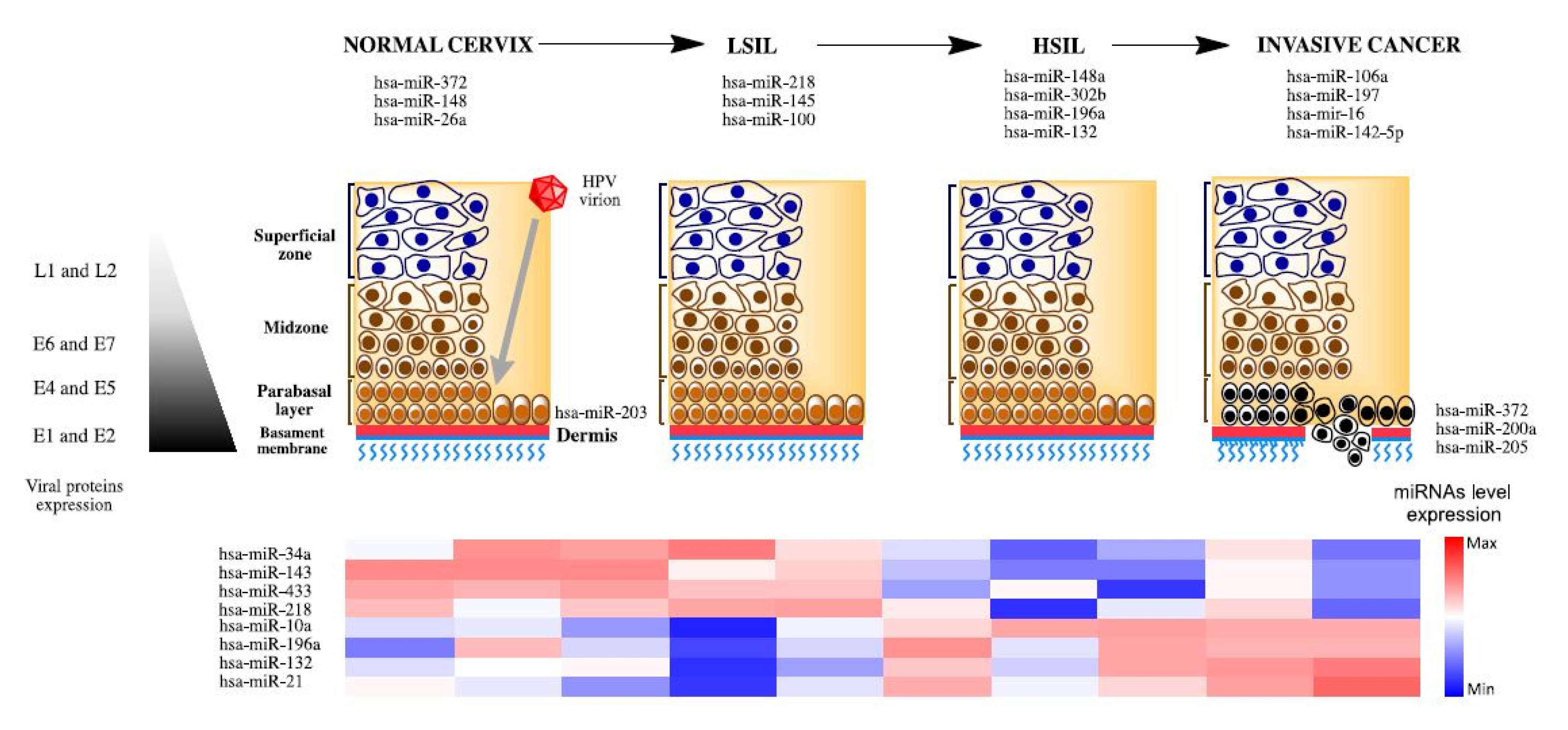
3. MiRNAs Associated to Development and Progression in Cervical Cancer
4. MiRNA Profile Regulated by HR-HPV Oncoproteins

{kind=link}
{kind=link}
{kind=link}
| MiRNA | Up/Down-Regulated | Cellular Process | Target Gene | Clinical Background | Refs. |
|---|---|---|---|---|---|
| miR-34a | Down-regulated | p53-dependent pathway (cell cycle progression, cellular senescence and apoptosis) | p18Ink4c, CDK4, CDK6, Cyclin E2, E2F1, E2F3, E2F5, BCL2, BIRC3, and DcR3 | ↓CIN I, ↓↓CIN II, ↓↓↓CIN III | [40,41] |
| miR-218 | Down-regulated | Focal adhesion | LAMB3 | ↓CIN III, ↓↓↓CaCu | [42] |
| miR-200a, miR-205 | Basal expression | Metastasis (inhibit the epithelial to mesenchymal transition) | ZEB1, ZEB2 and SIP1 | CaCu → CaCu metastasis | [43] |
| miR-9 | Up-regulated | Tumor cell metabolism (ATPase activity, Group transfer coenzyme metabolic process, Glutamine family amino acid metabolic process | No identified | Cervical cancers | [35] |
| miR-127 | Up-regulated | Metastasis | No identified | ↓NSE ‒↑↑ISCCs | [34] |
| miR-199a | Up-regulated | Cell growth | No identified | ↓NSE ‒ ↑↑ISCCs | [34] |
| miR-372 | Down-regulated | Cell growth (induced arrest in the S/G2 phases of cell cycle) | CDK2, Cyclin A1 | Cervical normal tissue → cervical cancer tissues | [44] |
| miR-203 | Up-regulated | Keratinocyte differentiation/maintain HPV episomes | p63-family | Normal epithelia → HPV-infected epithelia | [45] |
| miR-26a | Down-regulated | Cellular Growth and Proliferation | No identified | ↑Normal, ↓CIN, ↓CIN III, ↓Carcinoma | [36] |
| miR-143 | Down-regulated | Cellular Growth and Proliferation | PPAR Signaling | ↑Normal, ↓↓CIN, ↓↓CIN III, ↓↓Carcinoma | [36] |
| miR-145 | Down-regulated | Cellular Movement | IGF-1 | ↑Normal, ↓CIN, ↓CIN III, ↓Carcinoma | [36] |
| miR-99a, miR-203, miR-513, miR-29a | Down-regulated | Cell Death, Tissue Development | IGF-1, BCL2L2, VEGFA and CDK6 | ↑Normal, ↓CIN, ↓CIN III, ↓Carcinoma | [36] |
| miR-522* | Up-regulated | Cell Cycle: G2/M DNA Damage Checkpoint Regulation | No identified | ↑Normal, ↑↑CIN, ↑↑CIN, ↓Carcinoma | [36] |
| miR-148a | Up-regulated | Tumor supresor genes | PTEN, P53INP1 and TP53INP2 | ↑Normal, ↑↑CIN, ↑↑CIN, ↑↑↑Carcinoma | [36] |
| miR-10a, miR-196a, miR-132 | Up-regulated | Cell transformation and progression | (HOX) genes | ↑Normal, ↑↑↑CIN, ↑↑↑CIN, ↑↑↑Carcinoma | [36] |
| miR-886-5p | Up-regulated | Apoptosis | BAX | ↑ANTT, ↑↑↑CSCC | [46] |
| miR-100 | Down-regulated | Growth, cell cycle, and apoptosis | PLK1 | ↑ Normal, ↓CIN, ↓↓Carcinoma | [37] |
| Protein | MiRNAs | Up-/Down-Regulated | Target Gen | Cellular Process | Refs. |
|---|---|---|---|---|---|
| E5 | mir-146a | Up-regulated | ZNF813 | Cell adhesión and cell cycle | [51] |
| E5 | mir-324-5p | Down-regulated | CDH2, CTNNB1 | Transendothelial migration | [51] |
| E5 | mir-203 | Down-regulated | p63 | Cell juntion, cell migration, and cell motility | [51] |
| E6 | mir-34a | Down-regulated | p18Ink4c, CDK4, CDK6, Cyclin E2 | Cell cycle progression, cellular | [40] [41] |
| E6 | mir-218 | Down-regulated | LAMB3 | No identified | [42] |
| E6 | mir-23b | Down-regulated | uPA | Cell migration | [53] |
| E6/E7 | mir-29 | Down-regulated | YY1 and CDK6 | Restrains cell cycle progression and induces apoptosis | [38] |
| E7 | mir-15b | Down-regulated | CCNA2, CCNB1, CCNB2 MSH6 and MCM7 | Recognition of mismatched nucleotides, prior to their repair, and initiation of eukaryotic genome replication. | [55] |
| E7 | miR-15a/miR-16-1 and miR-203 | Down-regulated | c-Myc, c-Myb, PPAR | Control cell proliferation, survival, and invasion | [52] |
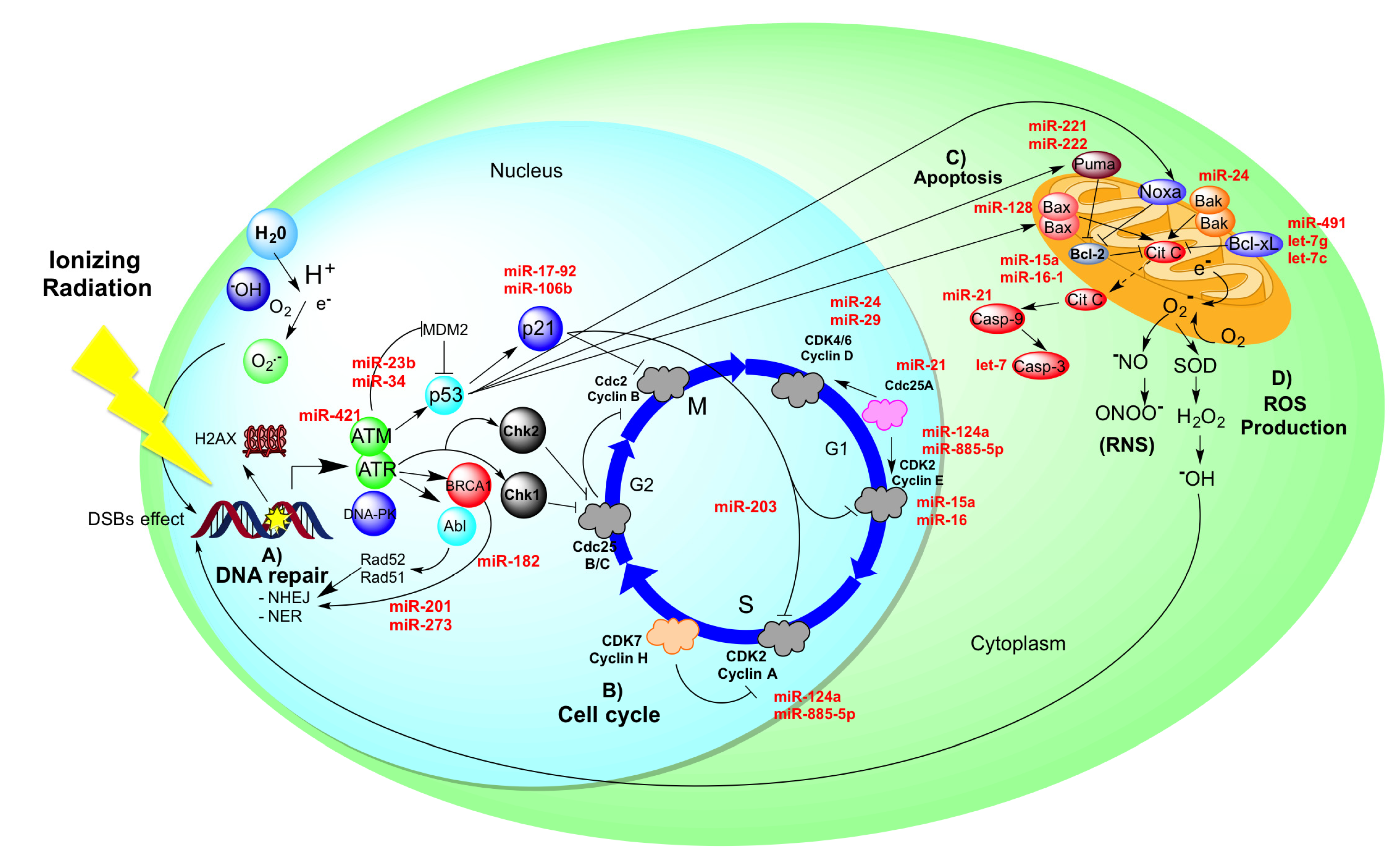
5. Radio-Resistance in Cervical Cancer Could Be Controlled by MiRNAs

6. Summary and Prospects
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Haverkos, H.; Rohrer, M.; Pickworth, W. The cause of invasive cervical cancer could be multifactorial. Biomed. Pharmacother. 2000, 54, 54–59. [Google Scholar]
- Perez-Plasencia, C.; Duenas-Gonzalez, A.; Alatorre-Tavera, B. Second hit in cervical carcinogenesis process: Involvement of wnt/beta catenin pathway. Int. Arch. Med. 2008, 1, 10. [Google Scholar] [CrossRef]
- Landoni, F.; Maneo, A.; Colombo, A.; Placa, F.; Milani, R.; Perego, P.; Favini, G.; Ferri, L.; Mangioni, C. Randomised study of radical surgery versus radiotherapy for stage Ib-IIa cervical cancer. Lancet 1997, 350, 535–540. [Google Scholar] [CrossRef]
- Quinn, M.A.; Benedet, J.L.; Odicino, F.; Maisonneuve, P.; Beller, U.; Creasman, W.T.; Heintz, A.P.M.; Ngan, H.Y.S.; Pecorelli, S. Carcinoma of the cervix uteri. FIGO 26th annual report on the results of treatment in gynecological cancer. Int. J. Gynaecol. Obstet. 2006, 95 (Suppl. 1), S43–S103. [Google Scholar] [CrossRef]
- Chemoradiotherapy for Cervical Cancer Meta-Analysis Collaboration. Reducing uncertainties about the effects of chemoradiotherapy for cervical cancer: A systematic review and meta-analysis of individual patient data from 18 randomized trials. J. Clin. Oncol. 2008, 26, 5802–5812. [CrossRef]
- Hart, K.; Han, I.; Deppe, G.; Malviya, V.; Malone, J., Jr.; Christensen, C.; Chuba, P.; Porter, A. Postoperative radiation for cervical cancer with pathologic risk factors. Int. J. Radiat. Oncol. Biol. Phys. 1997, 37, 833–838. [Google Scholar] [CrossRef]
- Keys, H.; Gibbons, S.K. Optimal management of locally advanced cervical carcinoma. J. Natl. Cancer Inst. Monogr. 1996, 21, 89–92. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Macfarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, function and role in cancer. Curr. Genomics 2010, 11, 537–561. [Google Scholar] [CrossRef]
- Berezikov, E.; Guryev, V.; van de Belt, J.; Wienholds, E.; Plasterk, R.H.A.; Cuppen, E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell 2005, 120, 21–24. [Google Scholar] [CrossRef]
- Dai, R.; Ahmed, S.A. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl. Res. 2011, 157, 163–179. [Google Scholar]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.-S.; Tam, W.-L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Sedani, A.; Cooper, D.N.; Upadhyaya, M. An emerging role for microRNAs in NF1 tumorigenesis. Hum. Genomics 2012, 6, 23. [Google Scholar] [CrossRef]
- Hwang, H.-W.; Mendell, J.T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 2006, 94, 776–780. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef]
- Liu, J.; Rivas, F.V.; Wohlschlegel, J.; Yates, J.R., III; Parker, R.; Hannon, G.J. A role for the P-body component GW182 in microRNA function. Nat. Cell Biol. 2005, 7, 1261–1266. [Google Scholar] [CrossRef]
- Sen, G.L.; Blau, H.M. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat. Cell Biol. 2005, 7, 633–636. [Google Scholar] [CrossRef]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J. Biol. Chem. 2009, 284, 3728–3738. [Google Scholar]
- Takyar, S.; Vasavada, H.; Zhang, J.; Ahangari, F.; Niu, N.; Liu, Q.; Lee, C.G.; Cohn, L.; Elias, J.A. VEGF controls lung Th2 inflammation via the miR-1-Mpl (myeloproliferative leukemia virus oncogene)-P-selectin axis. J. Exp. Med. 2013, 210, 1993–2010. [Google Scholar] [CrossRef]
- Tamura, M.; Uyama, M.; Sugiyama, Y.; Sato, M. Canonical Wnt signaling activates miR-34 expression during osteoblastic differentiation. Mol. Med. Rep. 2013, 8, 1807–1811. [Google Scholar]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef]
- Scott, G.K.; Mattie, M.D.; Berger, C.E.; Benz, S.C.; Benz, C.C. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006, 66, 1277–1281. [Google Scholar] [CrossRef]
- Bandres, E.; Agirre, X.; Bitarte, N.; Ramirez, N.; Zarate, R.; Roman-Gomez, J.; Prosper, F.; Garcia-Foncillas, J. Epigenetic regulation of microRNA expression in colorectal cancer. Int. J. Cancer 2009, 125, 2737–2743. [Google Scholar] [CrossRef]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007, 39, 673–677. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Lee, J.-W.; Choi, C.H.; Choi, J.-J.; Park, Y.-A.; Kim, S.-J.; Hwang, S.Y.; Kim, W.Y.; Kim, T.-J.; Lee, J.-H.; Kim, B.-G. Altered MicroRNA expression in cervical carcinomas. Clin. Cancer Res. 2008, 14, 2535–2542. [Google Scholar] [CrossRef]
- Hu, X.; Schwarz, J.K.; Lewis, J.S., Jr.; Huettner, P.C.; Rader, J.S.; Deasy, J.O.; Grigsby, P.W.; Wang, X. A microRNA expression signature for cervical cancer prognosis. Cancer Res. 2010, 70, 1441–1448. [Google Scholar] [CrossRef]
- Pereira, P.M.; Marques, J.P.; Soares, A.R.; Carreto, L.; Santos, M.A. MicroRNA expression variability in human cervical tissues. PLoS One 2010, 5, e11780. [Google Scholar]
- Li, B.H.; Zhou, J.S.; Ye, F.; Cheng, X.D.; Zhou, C.Y.; Lu, W.G.; Xie, X. Reduced miR-100 expression in cervical cancer and precursors and its carcinogenic effect through targeting PLK1 protein. Eur. J. Cancer 2011, 47, 2166–2174. [Google Scholar]
- Li, Y.; Wang, F.; Xu, J.; Ye, F.; Shen, Y.; Zhou, J.; Lu, W.; Wan, X.; Ma, D.; Xie, X. Progressive miRNA expression profiles in cervical carcinogenesis and identification of HPV-related target genes for miR-29. J. Pathol. 2011, 224, 484–495. [Google Scholar] [CrossRef]
- Cortese, M.S.; Ashrafi, G.H.; Campo, M.S. All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int. J. Cancer 2010, 126, 1675–1682. [Google Scholar]
- Li, B.; Hu, Y.; Ye, F.; Li, Y.; Lv, W.; Xie, X. Reduced miR-34a expression in normal cervical tissues and cervical lesions with high-risk human papillomavirus infection. Int. J. Gynecol. Cancer 2010, 20, 597–604. [Google Scholar] [CrossRef]
- Wang, X.; Meyers, C.; Guo, M.; Zheng, Z.M. Upregulation of p18Ink4c expression by oncogenic HPV E6 via p53-miR-34a pathway. Int. J. Cancer 2011, 129, 1362–1372. [Google Scholar] [CrossRef]
- Martinez, I.; Gardiner, A.S.; Board, K.F.; Monzon, F.A.; Edwards, R.P.; Khan, S.A. Human papillomavirus type 16 reduces the expression ofmicroRNA-218 in cervical carcinoma cells. Oncogene 2008, 27, 2575–2582. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Tian, R.Q.; Wang, X.H.; Hou, L.J.; Jia, W.H.; Yang, Q.; Li, Y.X.; Liu, M.; Li, X.; Tang, H. MicroRNA-372 is down-regulated and targets cyclin-dependent kinase 2 (CDK2) and cyclin A1 in human cervical cancer, which may contribute to tumorigenesis. J. Biol. Chem. 2011, 286, 25556–25563. [Google Scholar]
- Melar-New, M.; Laimins, L.A. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 2010, 84, 5212–5221. [Google Scholar] [CrossRef]
- Li, J.H.; Xiao, X.; Zhang, Y.-N.; Wang, Y.-M.; Feng, L.-M.; Wu, Y.-M.; Zhang, Y.-X. MicroRNA miR-886–5p inhibits apoptosis by down-regulating Bax expression in human cervical carcinoma cells. Gynecol. Oncol. 2011, 120, 145–151. [Google Scholar] [CrossRef]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar]
- Mittal, K.R.; Chan, W.; Demopoulos, R.I. Sensitivity and specificity of various morphological features of cervical condylomas. An in situ hybridization study. Arch. Pathol. Lab. Med. 1990, 114, 1038–1041. [Google Scholar]
- Prasad, C.J.; Sheets, E.; Selig, A.M.; McArthur, M.C.; Crum, C.P. The binucleate squamous cell: Histologic spectrum and relationship to low-grade squamous intraepithelial lesions. Mod. Pathol. 1993, 6, 313–317. [Google Scholar]
- Kabsch, K.; Alonso, A. The human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J. Virol. 2002, 76, 12162–12172. [Google Scholar] [CrossRef]
- Greco, D.; Kivi, N.; Qian, K.; Leivonen, S.K.; Auvinen, P.; Auvinen, E. Human papillomavirus 16 E5 modulates the expression of host microRNAs. PLoS One 2011, 6, e21646. [Google Scholar]
- Zheng, Z.M.; Wang, X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim. Biophys. Acta 2011, 1809, 668–677. [Google Scholar] [CrossRef]
- Au Yeung, C.L.; Tsang, T.Y.; Yau, P.L.; Kwok, T.T. Human papillomavirus type 16 E6 induces cervical cancer cell migration through the p53/microRNA-23b/urokinase-type plasminogen activator pathway. Oncogene 2011, 30, 2401–2410. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, J.; Zhang, L.; Zhu, Z.; Fan, J.; Chen, L.; Zhuang, L.; Luo, J.; Chen, H.; Liu, L.; et al. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology 2013, 145, 1133–1143. [Google Scholar] [CrossRef]
- Myklebust, M.P.; Bruland, O.; Fluge, Ø.; Skarstein, A.; Balteskard, L.; Dahl, O. MicroRNA-15b is induced with E2F-controlled genes in HPV-related cancer. Br. J. Cancer 2011, 105, 1719–1725. [Google Scholar] [CrossRef]
- Chang, T.-C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. P53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef]
- Tazawa, H.; Tsuchiya, N.; Izumiya, M.; Nakagama, H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15472–15477. [Google Scholar]
- Welch, C.; Chen, Y.; Stallings, R.L. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007, 26, 5017–5022. [Google Scholar] [CrossRef]
- Kang, J.; Kim, E.; Kim, W.; Seong, K.M.; Youn, H.; Kim, J.W.; Kim, J.; Youn, B. Rhamnetin and cirsiliol induce radiosensitization and inhibition of epithelial-mesenchymal transition (EMT) by miR-34a-mediated suppression of Notch-1 expression in non-small cell lung cancer cell lines. J. Biol. Chem. 2013, 288, 27343–27357. [Google Scholar]
- Lena, A.M.; Shalom-Feuerstein, R.; di Cervo, R.V.P.; Aberdam, D.; Knight, R.A.; Melino, G.; Candi, E. miR-203 represses “stemness” by repressing DeltaNp63. Cell Death Differ. 2008, 15, 1187–95. [Google Scholar] [CrossRef]
- Yi, R.; Poy, M.N.; Stoffel, M.; Fuchs, E. A skin microRNA promotes differentiation by repressing “stemness”. Nature 2008, 452, 225–229. [Google Scholar] [CrossRef]
- Ju, S.-Y.; Chiou, S.-H.; Su, Y. Maintenance of the stemness in CD44(+) HCT-15 and HCT-116 human colon cancer cells requires miR-203 suppression. Stem Cell Res. 2014, 12, 86–100. [Google Scholar] [CrossRef]
- Gao, X.; McDonald, J.T.; Hlatky, L.; Enderling, H. Acute and fractionated irradiation differentially modulate glioma stem cell division kinetics. Cancer Res. 2013, 73, 1481–1490. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Wang, W.-J.; Wu, S.-P.; Liu, J.-B.; Shi, Y.-S.; Huang, X.; Zhang, Q.-B.; Yao, K.-T. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2013, 73, 1219–1231. [Google Scholar] [CrossRef]
- Marie-Egyptienne, D.T.; Lohse, I.; Hill, R.P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013, 341, 63–72. [Google Scholar]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef]
- Kim, S.J. Multimodal treatment for the locally advanced stage IB, IIA, IIB patients of cervical cancer. Int. J. Gynaecol. Obstet. 1995, 49, S49–S57. [Google Scholar] [CrossRef]
- Satpute, P.S.; Hazarey, V.; Ahmed, R.; Yadav, L. Cancer stem cells in head and neck squamous cell carcinoma: A review. Asian Pac. J. Cancer Prev. 2013, 14, 5579–5587. [Google Scholar]
- Jameel, J.K.A.; Rao, V.S.R.; Cawkwell, L.; Drew, P.J. Radioresistance in carcinoma of the breast. Breast 2004, 13, 452–460. [Google Scholar] [CrossRef]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef]
- Storr, S.J.; Woolston, C.M.; Martin, S.G. Base excision repair, the redox environment and therapeutic implications. Curr. Mol. Pharmacol. 2012, 5, 88–101. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Kitahara, O.; Katagiri, T.; Tsunoda, T.; Harima, Y.; Nakamura, Y. Classification of sensitivity or resistance of cervical cancers to ionizing radiation according to expression profiles of 62 genes selected by cDNA microarray analysis. Neoplasia 2002, 4, 295–303. [Google Scholar] [CrossRef]
- Tewari, D.; Monk, B.J.; Al-Ghazi, M.S.; Parker, R.; Heck, J.D.; Burger, R.A.; Fruehauf, J.P. Gene expression profiling of in vitro radiation resistance in cervical carcinoma: A feasibility study. Gynecol. Oncol. 2005, 99, 84–91. [Google Scholar] [CrossRef]
- Wong, Y.F.; Selvanayagam, Z.E.; Wei, N.; Porter, J.; Vittal, R.; Hu, R.; Lin, Y.; Liao, J.; Shih, J.W.; Cheung, T.H.; et al. Expression genomics of cervical cancer: Molecular classification and prediction of radiotherapy response by DNA microarray. Clin. Cancer Res. 2003, 9, 5486–5492. [Google Scholar]
- Shin, S.; Cha, H.J.; Lee, E.-M.; Lee, S.-J.; Seo, S.-K.; Jin, H.-O.; Park, I.-C.; Jin, Y.-W.; An, S. Alteration of miRNA profiles by ionizing radiation in A549 human non-small cell lung cancer cells. Int. J. Oncol. 2009, 35, 81–86. [Google Scholar]
- Niemoeller, O.M.; Niyazi, M.; Corradini, S.; Zehentmayr, F.; Li, M.; Lauber, K.; Belka, C. MicroRNA expression profiles in human cancer cells after ionizing radiation. Radiat. Oncol. 2011, 6, 29. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, J.; Ren, Z.; Chen, Y.; Li, J.; Miao, X.; Song, Y.; Zhao, T.; Li, Y.; Shi, Y.; et al. A specific miRNA signature promotes radioresistance of human cervical cancer cells. Cancer Cell Int. 2013, 13, 118. [Google Scholar] [CrossRef]
- Xi, Y.; Shalgi, R.; Fodstad, O.; Pilpel, Y.; Ju, J. Differentially regulated micro-RNAs and actively translated messenger RNA transcripts by tumor suppressor p53 in colon cancer. Clin. Cancer Res. 2006, 12, 2014–2024. [Google Scholar] [CrossRef]
- Mansour, W.Y.; Bogdanova, N.V.; Kasten-Pisula, U.; Rieckmann, T.; Köcher, S.; Borgmann, K.; Baumann, M.; Krause, M.; Petersen, C.; Hu, H.; et al. Aberrant overexpression of miR-421 downregulates ATM and leads to a pronounced DSB repair defect and clinical hypersensitivity in SKX squamous cell carcinoma. Radiother. Oncol. 2013, 106, 147–154. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1 and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef]
- Ivanovska, I.; Ball, A.S.; Diaz, R.L.; Magnus, J.F.; Kibukawa, M.; Schelter, J.M.; Kobayashi, S.V.; Lim, L.; Burchard, J.; Jackson, A.L.; et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol. Cell. Biol. 2008, 28, 2167–2174. [Google Scholar] [CrossRef]
- Wu, S.; Huang, S.; Ding, J.; Zhao, Y.; Liang, L.; Liu, T.; Zhan, R.; He, X. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3' untranslated region. Oncogene 2010, 29, 2302–2308. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pedroza-Torres, A.; López-Urrutia, E.; García-Castillo, V.; Jacobo-Herrera, N.; Herrera, L.A.; Peralta-Zaragoza, O.; López-Camarillo, C.; De Leon, D.C.; Fernández-Retana, J.; Cerna-Cortés, J.F.; et al. MicroRNAs in Cervical Cancer: Evidences for a miRNA Profile Deregulated by HPV and Its Impact on Radio-Resistance. Molecules 2014, 19, 6263-6281. https://doi.org/10.3390/molecules19056263
Pedroza-Torres A, López-Urrutia E, García-Castillo V, Jacobo-Herrera N, Herrera LA, Peralta-Zaragoza O, López-Camarillo C, De Leon DC, Fernández-Retana J, Cerna-Cortés JF, et al. MicroRNAs in Cervical Cancer: Evidences for a miRNA Profile Deregulated by HPV and Its Impact on Radio-Resistance. Molecules. 2014; 19(5):6263-6281. https://doi.org/10.3390/molecules19056263
Chicago/Turabian StylePedroza-Torres, Abraham, Eduardo López-Urrutia, Verónica García-Castillo, Nadia Jacobo-Herrera, Luis A. Herrera, Oscar Peralta-Zaragoza, César López-Camarillo, David Cantú De Leon, Jorge Fernández-Retana, Jorge F. Cerna-Cortés, and et al. 2014. "MicroRNAs in Cervical Cancer: Evidences for a miRNA Profile Deregulated by HPV and Its Impact on Radio-Resistance" Molecules 19, no. 5: 6263-6281. https://doi.org/10.3390/molecules19056263