The Intramolecular Diels-Alder Reaction of Diarylheptanoids — Quantum Chemical Calculation of Structural Features Favoring the Formation of Phenylphenalenones


Abstract
:
1. Introduction
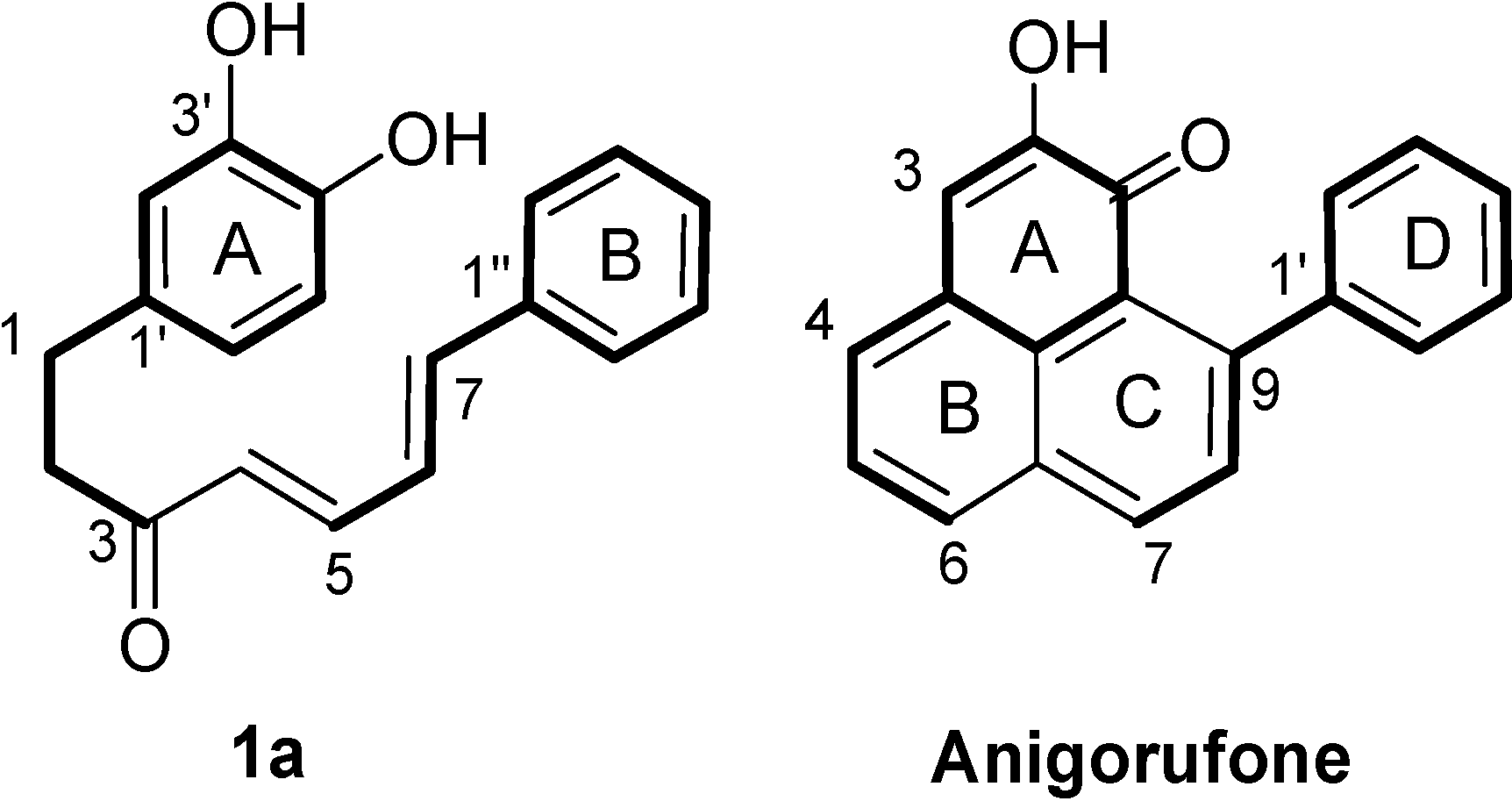
2. Results and Discussion
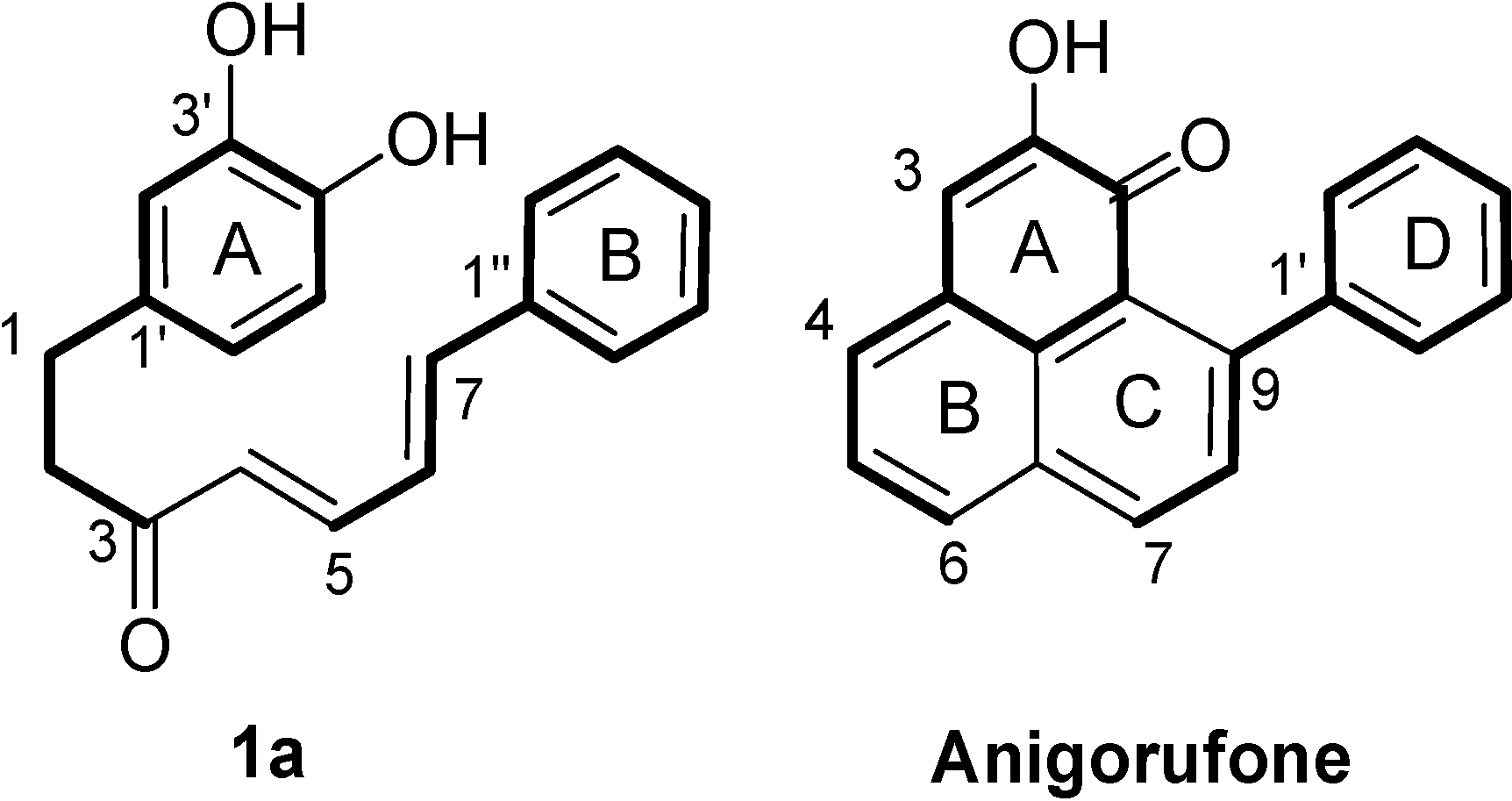
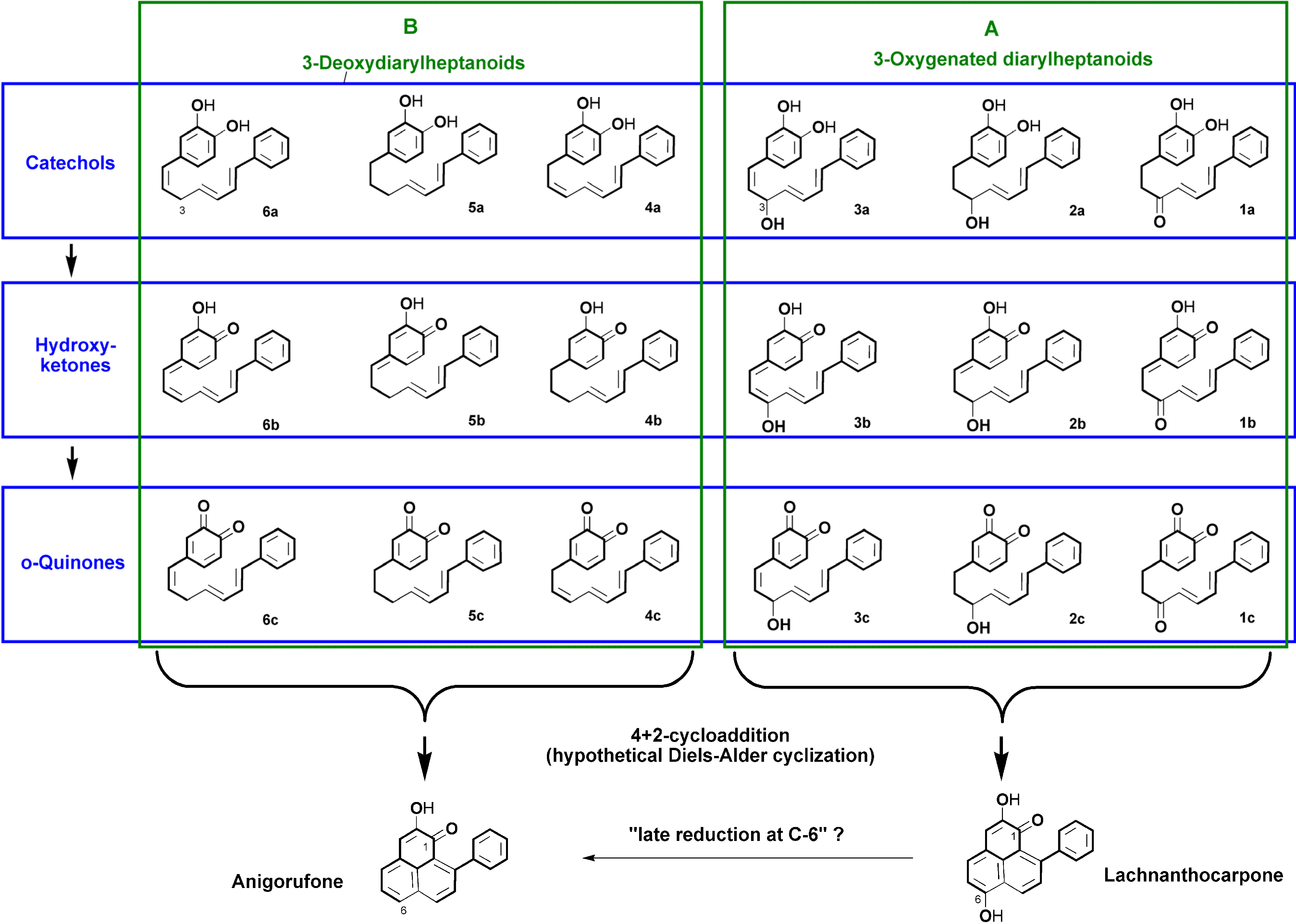
2.1. Diarylheptanoid Candidate Structures for Diels–Alder Cyclization
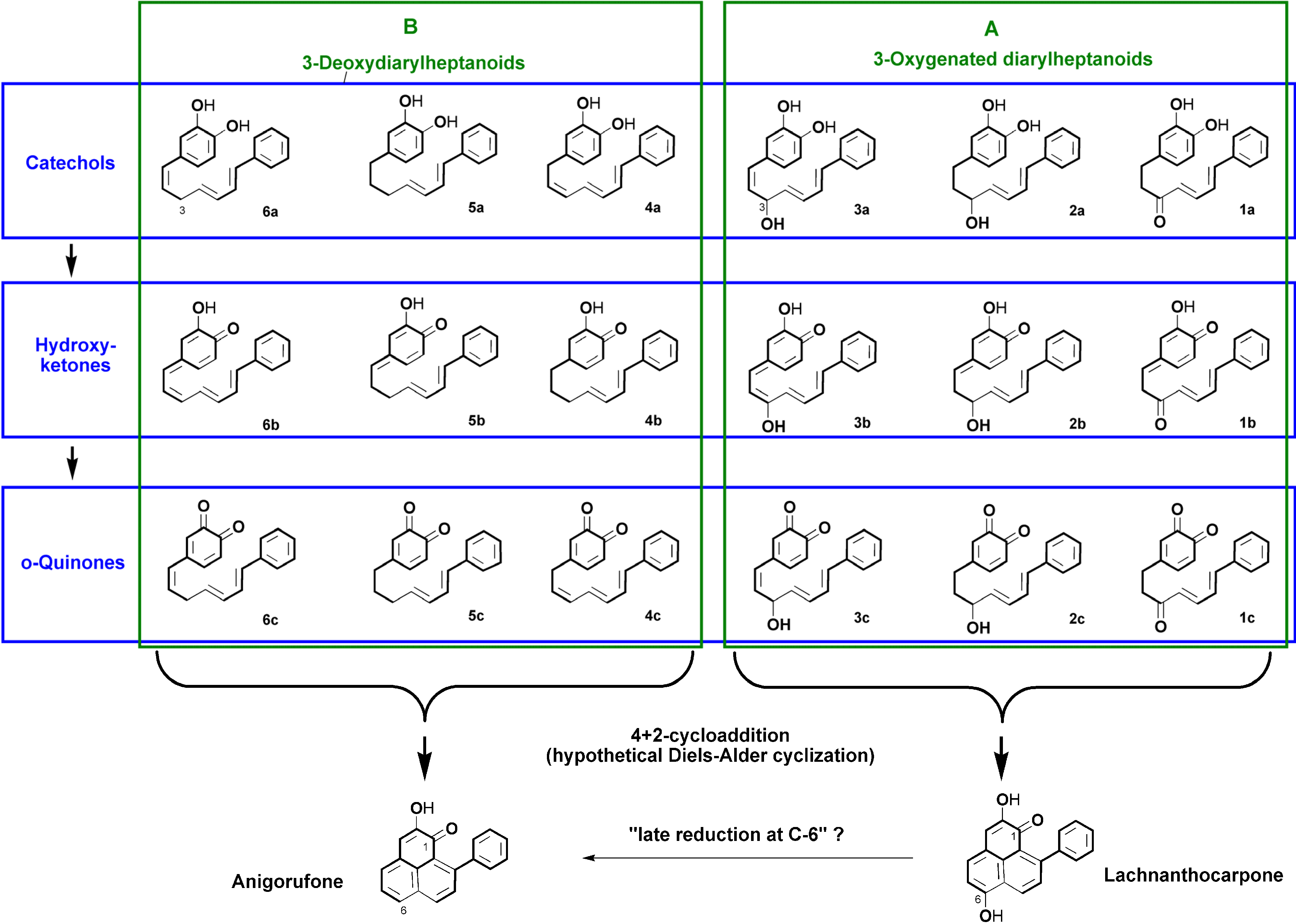
2.2. Molecular Geometry Calculation
- Two oxygen substituents in ortho position (C-3'/C-4') of linear diarylheptanoids (as in all diarylheptanoids shown in Figure 2).
- Conjugated double bonds at C-4 and C-6 in the C7-chain in cisoid conformation (as in all diarylheptanoids shown in Figure 2).
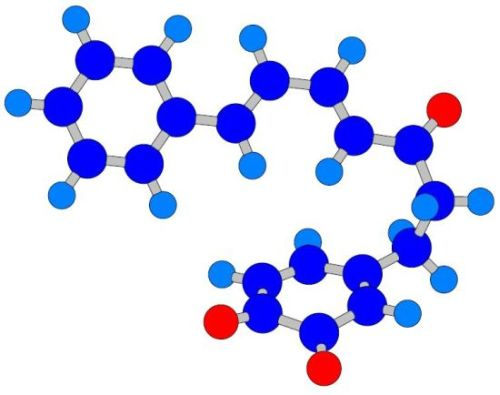
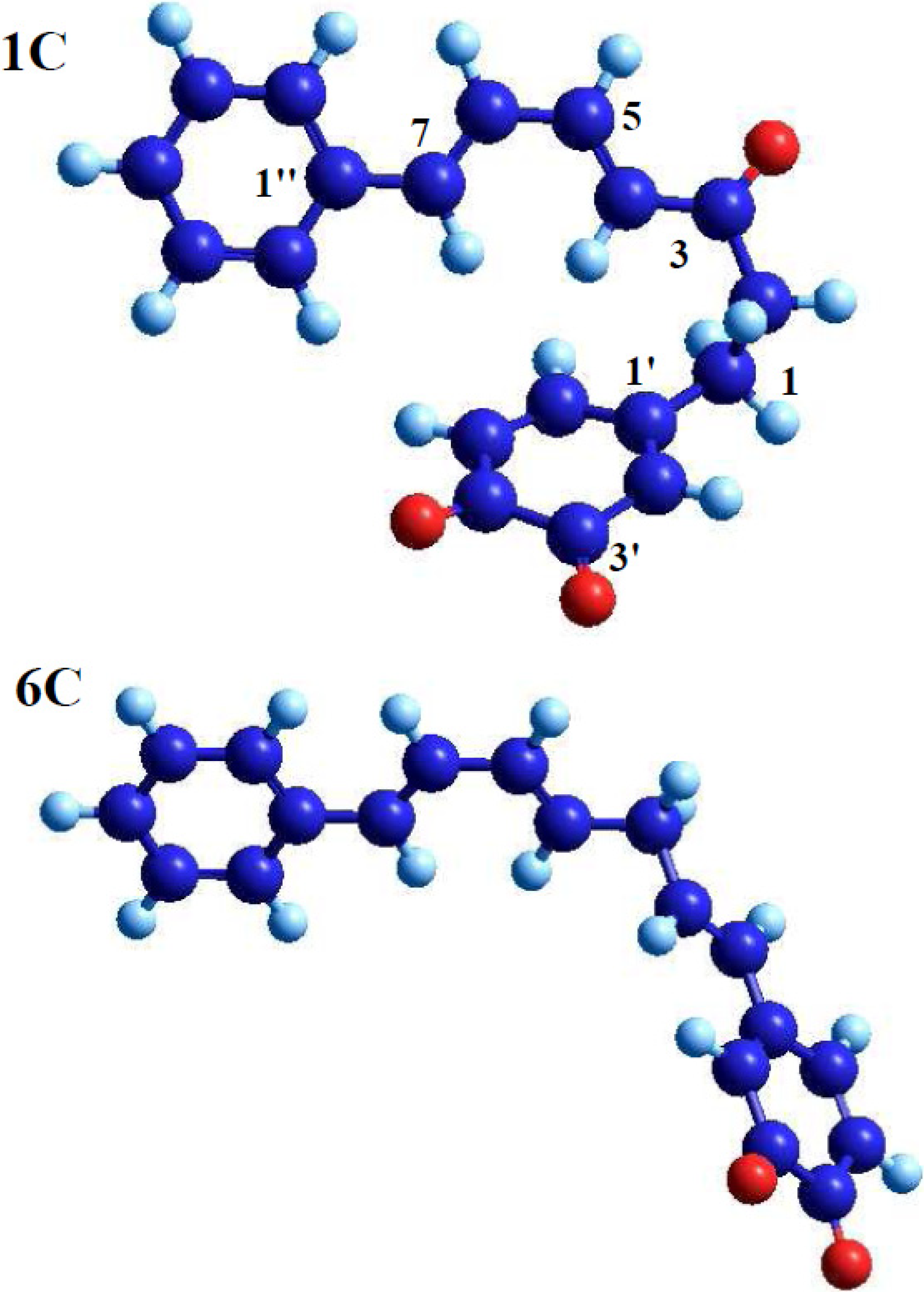
- A saturated segment (sp3-carbon atoms) in the C7-chain next to ring A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
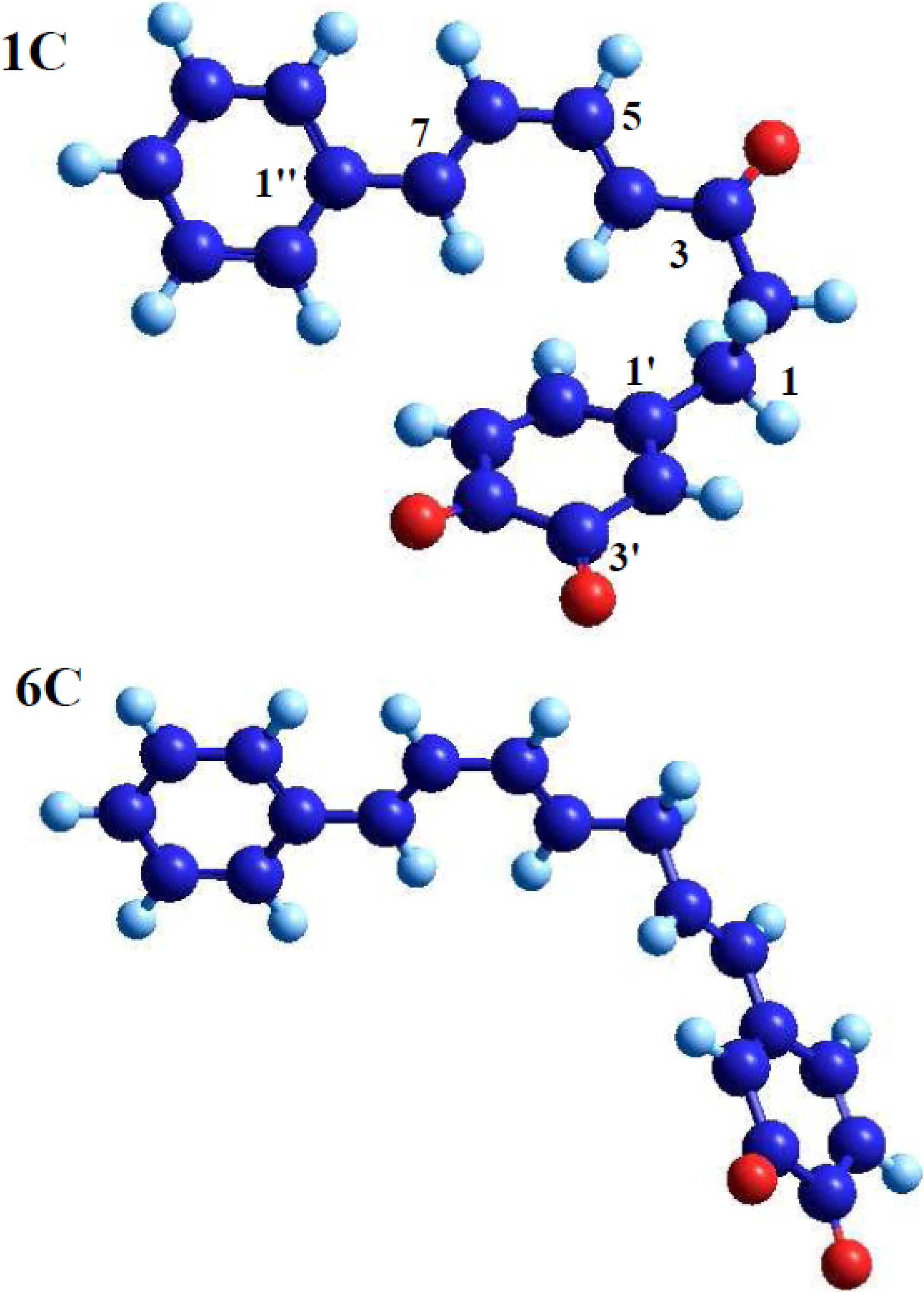
{kind=link}
| Compound | Geometrical Criterion a | Distance Between Atoms [Å] | Mulliken’s Charges | Difference between Atoms’ Charges | |||||
|---|---|---|---|---|---|---|---|---|---|
| (6'-4) | (5'-7) | 6' | 5' | 4 | 7 | (6'-4) | (5'-7) | ||
| 1a | + | 3.39 | 4.01 | −0.11 | −0.12 | −0.29 | −0.07 | 0.16 | 0.06 |
| 2a | + | 3.48 | 4.07 | −0.11 | −0.12 | −0.19 | −0.11 | 0.08 | 0.02 |
| 3a | − | − b | − b | −0.08 | −0.12 | −0.17 | −0.10 | 0.09 | 0.02 |
| 4a | − | − b | − b | −0.09 | −0.12 | −0.11 | −0.01 | 0.02 | 0.02 |
| 5a | + | 3.46 | 4.11 | −0.11 | −0.12 | −0.16 | −0.11 | 0.05 | 0.01 |
| 6a | − | − b | − b | −0.09 | −0.12 | −0.14 | −0.10 | 0.05 | 0.02 |
| 1b | − | − b | − b | −0.03 | −0.26 | −0.21 | −0.05 | 0.18 | 0.21 |
| 2b | − | − b | − b | −0.03 | −0.21 | −0.18 | −0.09 | 0.15 | 0.12 |
| 3b | − | − b | − b | −0.01 | −0.22 | −0.15 | −0.06 | 0.15 | 0.16 |
| 4b | + | 3.47 | 4.10 | −0.09 | −0.19 | −0.17 | −0.10 | 0.08 | 0.09 |
| 5b | − | − b | − b | −0.04 | −0.21 | −0.15 | −0.10 | 0.11 | 0.11 |
| 6b | − | − b | − b | −0.02 | −0.21 | −0.14 | −0.08 | 0.13 | 0.13 |
| 1c | + | 3.38 | 3.93 | −0.05 | −0.20 | −0.27 | −0.06 | 0.22 | 0.14 |
| 2c | + | 3.50 | 4.02 | −0.05 | −0.19 | −0.19 | −0.10 | 0.14 | 0.09 |
| 3c | − | − b | − b | −0.05 | −0.19 | −0.18 | −0.09 | 0.13 | 0.10 |
| 4c | − | − b | − b | −0.05 | −0.19 | −0.12 | −0.09 | 0.08 | 0.10 |
| 5c | + | 3.47 | 4.04 | −0.05 | −0.19 | −0.17 | −0.10 | 0.11 | 0.10 |
| 6c | − | − b | − b | −0.05 | −0.19 | −0.15 | −0.09 | 0.11 | 0.10 |
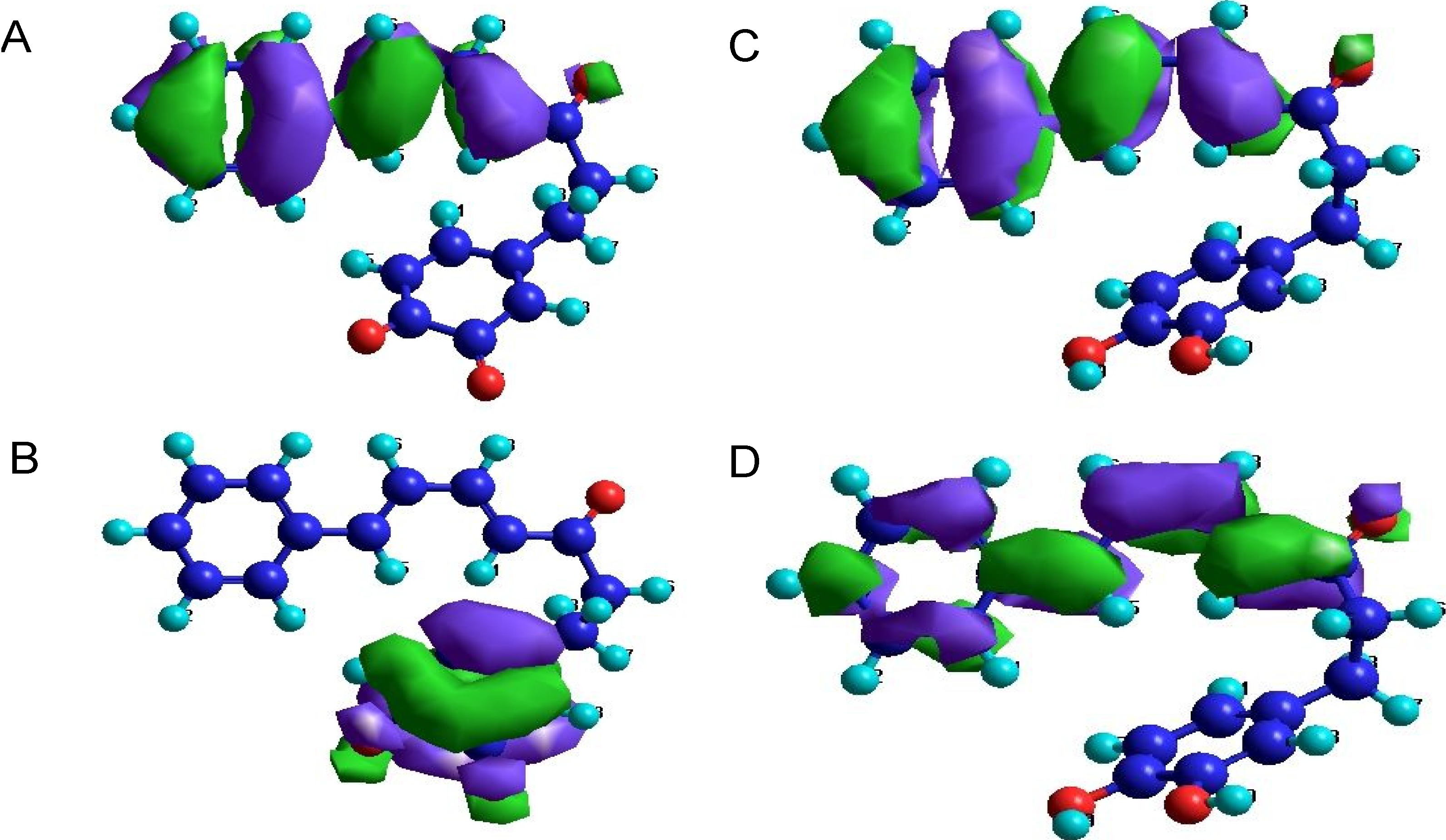
2.3. Orbital Calculation
| Compound | Orbital Criterion a | Energy of Orbitals [eV] | Compound | Orbital Criterion a | Energy of Orbitals [eV] | ||||
|---|---|---|---|---|---|---|---|---|---|
| LUMO | HOMO | LUMO-HOMO Difference | LUMO | HOMO | LUMO-HOMO Difference | ||||
| 1a | − | −0.87 | −8.94 | 8.07 | 4b | + | −0.42 | −5.17 | 4.75 |
| 2a | − | −0.34 | −8.55 | 8.21 | 5b | − | −1.21 | −8.76 | 7.55 |
| 3a | − | −0.49 | −8.66 | 8.17 | 6b | + | −1.73 | −8.31 | 6.58 |
| 4a | − | −0.70 | −8.45 | 7.75 | 1c | + | −1.80 | −9.15 | 7.35 |
| 5a | − | −0.34 | −8.53 | 8.19 | 2c | + | −1.69 | −8.76 | 7.07 |
| 6a | − | −0.52 | −8.58 | 8.06 | 3c | + | −1.77 | −8.81 | 7.04 |
| 1b | − | −1.37 | −9.10 | 7.73 | 4c | + | −1.59 | −8.66 | 7.07 |
| 2b | + | −1.29 | −8.74 | 7.45 | 5c | + | −1.63 | −8.74 | 7.11 |
| 3b | − | −1.74 | −8.16 | 6.42 | 6c | + | −1.67 | −8.66 | 6.99 |
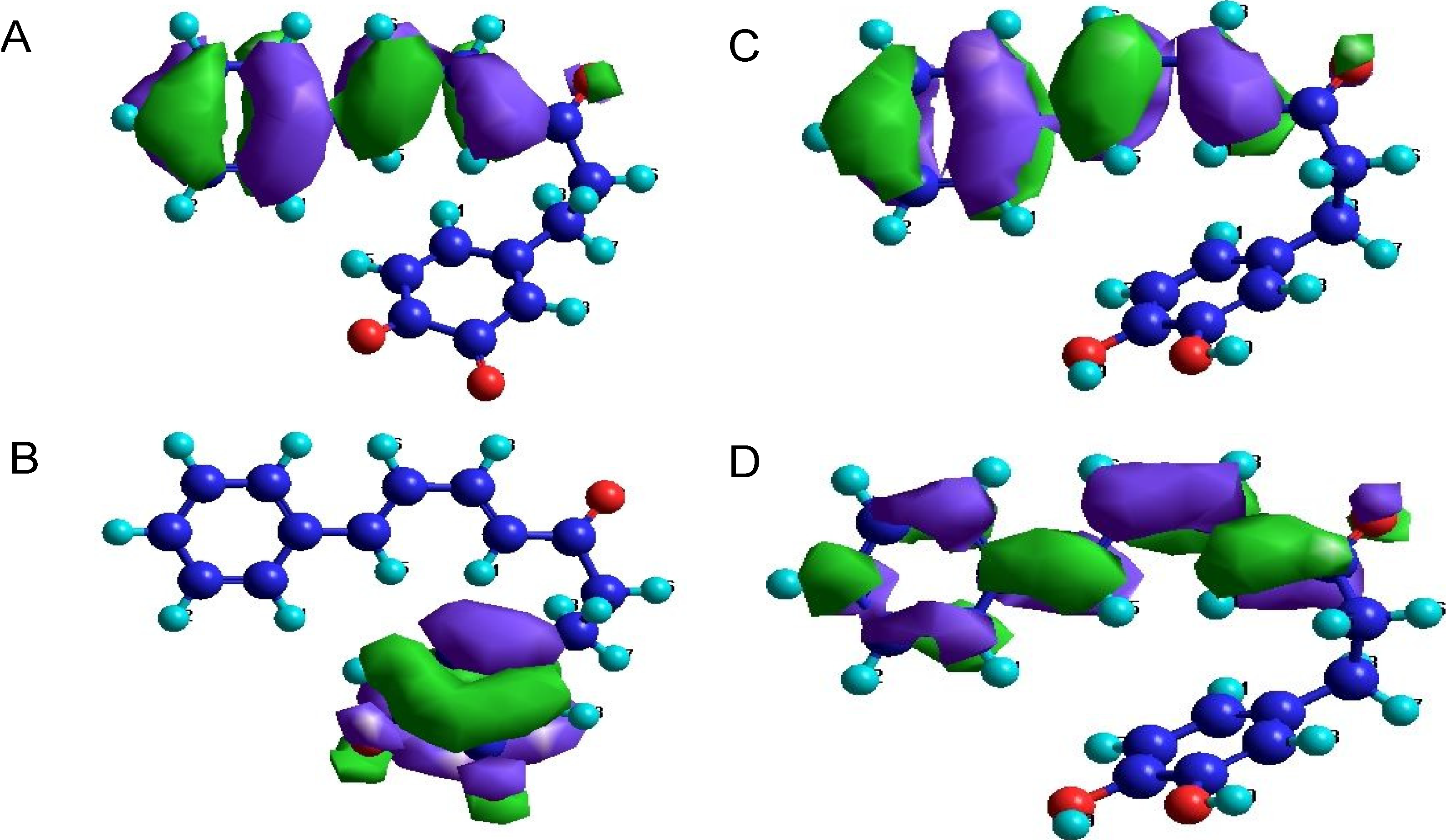
3. Experimental
| Compound | Atom number | Atomic orbital | Type of molecular orbital | Contribution | Compound | Atom number | Atomic orbital | Type of molecular orbital | Contribution |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 7 | Px | LUMO | 0.23 | 4b | 7 | Px | LUMO | 0.24 |
| 4 | Px | LUMO | 0.19 | 4 | Px | LUMO | 0.24 | ||
| 5' | Px | HOMO | 0.00 | 5' | Px | HOMO | 0.03 | ||
| 6' | Py | HOMO | 0.00 | 6' | Pz | HOMO | 0.37 | ||
| 2a | 7 | Py | HOMO | 0.20 | 5b | 7 | Pz | LUMO | 0.03 |
| 4 | Px | HOMO | 0.24 | 4 | Pz | HOMO | 0.32 | ||
| 5' | Pz | LUMO | 0.00 | 5' | Px | LUMO | 0.31 | ||
| 6' | Pz | HOMO | 0.01 | 6' | Px | HOMO | 0.03 | ||
| 3a | 7 | Px | HOMO | 0.21 | 6b | 7 | Pz | HOMO | 0.21 |
| 4 | Py | HOMO | 0.27 | 4 | Pz | LUMO | 0.12 | ||
| 5' | Pz | LUMO | 0.03 | 5' | Pz | LUMO | 0.20 | ||
| 6' | Px | HOMO | 0.07 | 6' | Pz | HOMO | 0.16 | ||
| 4a | 7 | Py | HOMO | 0.33 | 1c | 7 | Px | HOMO | 0.17 |
| 4 | - a | - a | - a | 4 | Py | HOMO | 0.08 | ||
| 5' | Px | HOMO | 0.00 | 5' | Px | LUMO | 0.045 | ||
| 6' | Pz | LUMO | 0.01 | 6' | Py | LUMO | 0.11 | ||
| 5a | 7 | Pz | LUMO | 0.24 | 2c | 7 | Py | HOMO | 0.20 |
| 4 | Px | HOMO | 0.24 | 4 | Px | HOMO | 0.23 | ||
| 5' | S | LUMO | 0.00 | 5' | Px | LUMO | 0.09 | ||
| 6' | Pz | HOMO | 0.00 | 6' | Pz | LUMO | 0.36 | ||
| 6a | 7 | Pz | LUMO | 0.27 | 3c | 7 | Py | HOMO | 0.28 |
| 4 | Py | LUMO | 0.26 | 4 | Px | HOMO | 0.21 | ||
| 5' | Px | LUMO | 0.02 | 5' | Pz | LUMO | 0.13 | ||
| 6' | Px | HOMO | 0.26 | 6' | Px | LUMO | 0.25 | ||
| 1b | 7 | Py | HOMO | 0.08 | 4c | 7 | Pz | HOMO | 0.33 |
| 4 | Pz | HOMO | 0.28 | 4 | Pz | LUMO | 0.02 | ||
| 5' | Px | HOMO | 0.32 | 5' | Py | HOMO | 0.00 | ||
| 6' | Px | LUMO | 0.12 | 6' | Py | LUMO | 0.24 | ||
| 2b | 7 | Px | HOMO | 0.25 | 5c | 7 | Py | HOMO | 0.22 |
| 4 | Py | HOMO | 0.19 | 4 | Px | HOMO | 0.24 | ||
| 5' | Pz | LUMO | 0.11 | 5' | Px | LUMO | 0.11 | ||
| 6' | Px | LUMO | 0.33 | 6' | Pz | LUMO | 0.35 | ||
| 3b | 7 | Px | HOMO | 0.09 | 6c | 7 | Py | HOMO | 0.27 |
| 4 | Py | LUMO | 0.03 | 4 | Px | HOMO | 0.22 | ||
| 5' | Pz | LUMO | 0.08 | 5' | Px | LUMO | 0.27 | ||
| 6' | Pz | HOMO | 0.21 | 6' | Px | LUMO | 0.01 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cooke, R.G.; Thomas, R.L. Colouring matters of Australian plants. XVIII. Constituents of Anigozanthos rufus. Aust. J. Chem. 1975, 28, 1053–1057. [Google Scholar] [CrossRef]
- Hölscher, D.; Schneider, B. HPLC-NMR analysis of phenylphenalenones and a stilbene from Anigozanthos flavidus. Phytochemistry 1999, 50, 155–161. [Google Scholar] [CrossRef]
- Opitz, S.; Hölscher, D.; Oldham, N.J.; Bartram, S.; Schneider, B. Phenylphenalenone-related compounds. Chemotaxonomic markers of the Haemodoraceae from Xiphidium caeruleum. J. Nat. Prod. 2002, 65, 1122–1130. [Google Scholar] [CrossRef]
- Fang, J.; Kai, M.; Schneider, B. Phytochemical profile of aerial parts and roots of Wachendorfia thyrsiflora L. studied by LC-DAD-SPE-NMR. Phytochemistry 2012, 81, 144–152. [Google Scholar] [CrossRef]
- Luis, J.G.; Fletcher, W.Q.; Echeverri, F.; Grillo, T.A. Phenalenone-type phytoalexins from Musa acuminata synthesis of 4-phenyl-phenalenones. Tetrahedron 1994, 50, 10963–10970. [Google Scholar] [CrossRef]
- Kamo, T.; Kato, N.; Hirai, N.; Tsuda, M.; Fujioka, D.; Ohigashi, H. Phenylphenalenone-type phytoalexins from unripe Buñgulan banana fruit. Biosci. Biotechnol. Biochem. 1998, 62, 95–101. [Google Scholar] [CrossRef]
- Otálvaro, F.; Nanclares, J.; Vásquez, L.E.; Quiñónes, W.; Echeverri, F.; Arango, R.; Schneider, B. Phenalenone-type compounds from Musa acuminata var. “Yangambi km 5” (AAA) and their activity against Mycosphaerella fijiensis. J. Nat. Prod. 2007, 70, 887–890. [Google Scholar] [CrossRef]
- Hölscher, D.; Dhakshinamoorthy, S.; Alexandrov, T.; Becker, M.; Bretschneider, T.; Bürkert, A.; Crecelius, A.C.; de Waele, D.; Elsen, A.; Heckel, D.G.; et al. Phenalenone-type phytoalexins mediate resistance of banana plants (Musa spp.) to the burrowing nematode Radopholus similis. Proc. Natl. Acad. Sci. USA 2014, 111, 105–110. [Google Scholar] [CrossRef]
- Thomas, R. Studies in the biosynthesis of fungal metabolites. Biochem. J. 1961, 78, 807–813. [Google Scholar]
- Bazan, A.C.; Edwards, J.M.; Weiss, U. Synthesis of lachnanthocarpone [9-phenyl-2,6-dihydroxyphenalen-1(6)-one] by intramolecular Diels-Alder cyclization of a 1,7-diarylheptanoid orthoquinone; possible biosynthetic significance of Diels-Alder reactions. Tetrahedron 1978, 34, 3005–3015. [Google Scholar] [CrossRef]
- Hölscher, D.; Schneider, B. A diarylheptanoid intermediate in the biosynthesis of phenylphenalenones in Anigozanthos preissii. J. Chem. Soc. Chem. Comm. 1995, 525–526. [Google Scholar]
- Munde, T.; Brand, S.; Hidalgo, W.; Maddula, R.K.; Svatoš, A.; Schneider, B. Biosynthesis of tetraoxygenated phenylphenalenones in Wachendorfia thyrsiflora. Phytochemistry 2013, 91, 165–176. [Google Scholar] [CrossRef]
- Schmitt, B.; Schneider, B. Dihydrocinnamic acids are involved in the biosynthesis of phenylphenalenones in Anigozanthos preissii. Phytochemistry 1999, 52, 45–53. [Google Scholar] [CrossRef]
- Houk, K.N.; González, J.; Li, Y. Pericyclic reaction transition states: Passions and punctilios, 1935–1995. Acc. Chem. Res. 1995, 28, 81–90. [Google Scholar] [CrossRef]
- Brocksom, T.J.; Nakamura, J.; Ferreira, M.L.; Brocksom, U. The Diels-Alder reaction: An update. J. Braz. Chem. Soc. 2001, 12, 597–622. [Google Scholar] [CrossRef]
- Granovsky, A.A. PC GAMESS version 7.1. Available online: http://classic.chem.msu.su/gran/gamess/index.html (accessed on 22 April 2014).
- Nemukhin, A.V.; Grigorenko, B.L.; Granovsky, A.A. Molecular modeling by using the PC GAMESS program: From diatomic molecules to enzymes. Moscow Univ. Chem. Bull. 2004, 45, 75–102. [Google Scholar]
- Guest, M.F.; Bush, I.J.; van Dam, H.J.J.; Sherwood, P.; Thomas, J.M.H.; van Lenthe, J.H.; Havenith, R.W.A.; Kendrick, J. The GAMESS-UK electronic structure package: Algorithms, developments and applications. Mol. Phys. 2005, 103, 719–747. [Google Scholar] [CrossRef]
- Stewart, J.J.P. “PM3” in Encyclopedia of Computational Chemistry; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Pople, J.A.; Nesbet, R.K. Self-consistent orbitals for radicals. J. Chem. Phys. 1954, 22, 571–572. [Google Scholar] [CrossRef]
- Harihara, P.C.; Pople, J.A. Influence of polarization functions on molecular-orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Sample Availability: Sample of the compound 1a is available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Monakhova, Y.; Schneider, B. The Intramolecular Diels-Alder Reaction of Diarylheptanoids — Quantum Chemical Calculation of Structural Features Favoring the Formation of Phenylphenalenones. Molecules 2014, 19, 5231-5242. https://doi.org/10.3390/molecules19045231
Monakhova Y, Schneider B. The Intramolecular Diels-Alder Reaction of Diarylheptanoids — Quantum Chemical Calculation of Structural Features Favoring the Formation of Phenylphenalenones. Molecules. 2014; 19(4):5231-5242. https://doi.org/10.3390/molecules19045231
Chicago/Turabian StyleMonakhova, Yulia, and Bernd Schneider. 2014. "The Intramolecular Diels-Alder Reaction of Diarylheptanoids — Quantum Chemical Calculation of Structural Features Favoring the Formation of Phenylphenalenones" Molecules 19, no. 4: 5231-5242. https://doi.org/10.3390/molecules19045231