Enantioselective Separation of Mirtazapine and Its Metabolites by Capillary Electrophoresis with Acetonitrile Field-Amplified Sample Stacking and Its Application

Abstract
:1. Introduction
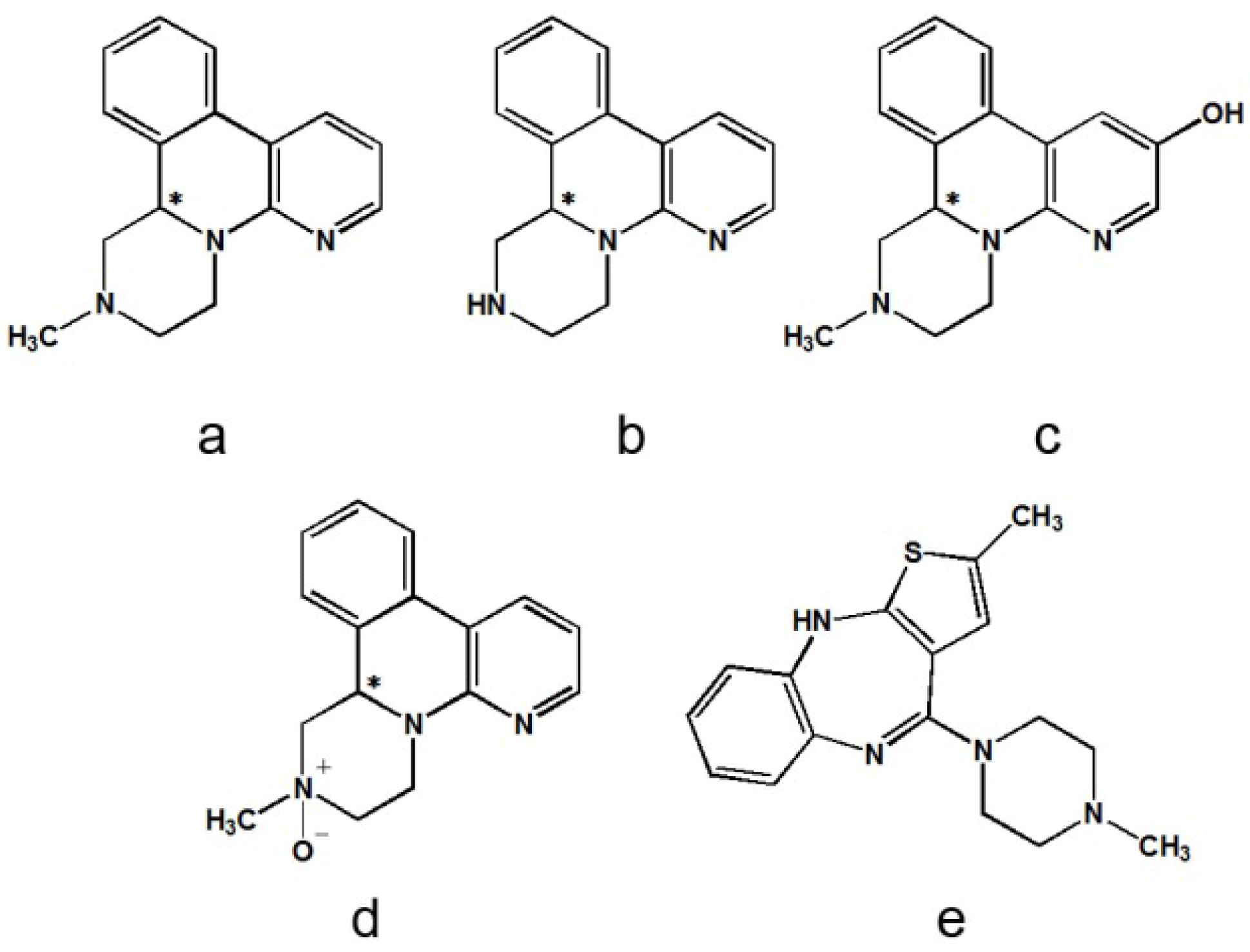
2. Results and Discussion
2.1. Optimization of Enantioseparation Conditions
2.1.1. CD Type and Concentration
2.1.2. Optimization of Borate-Phosphate Buffer pH and Concentration and Applied Voltage
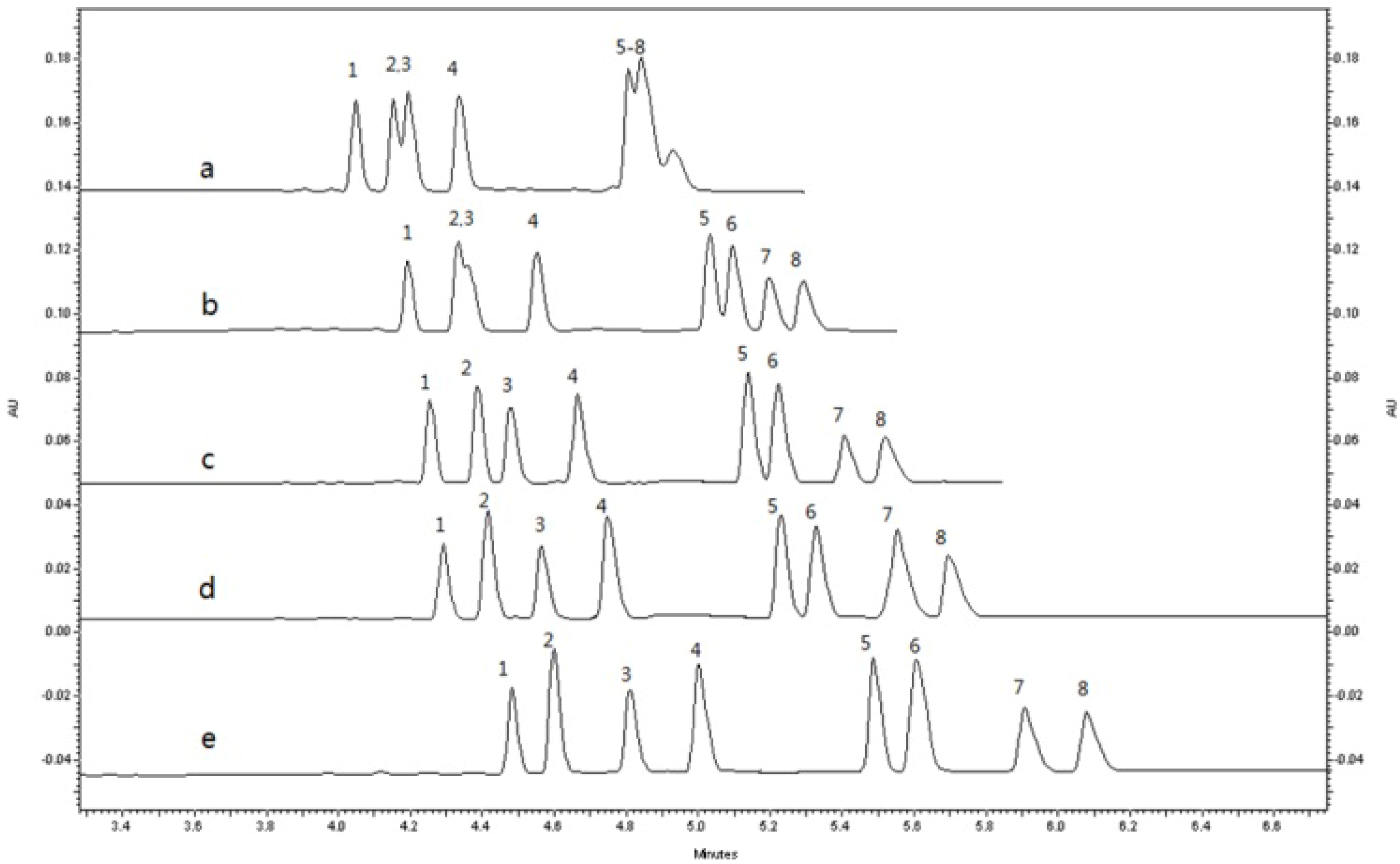

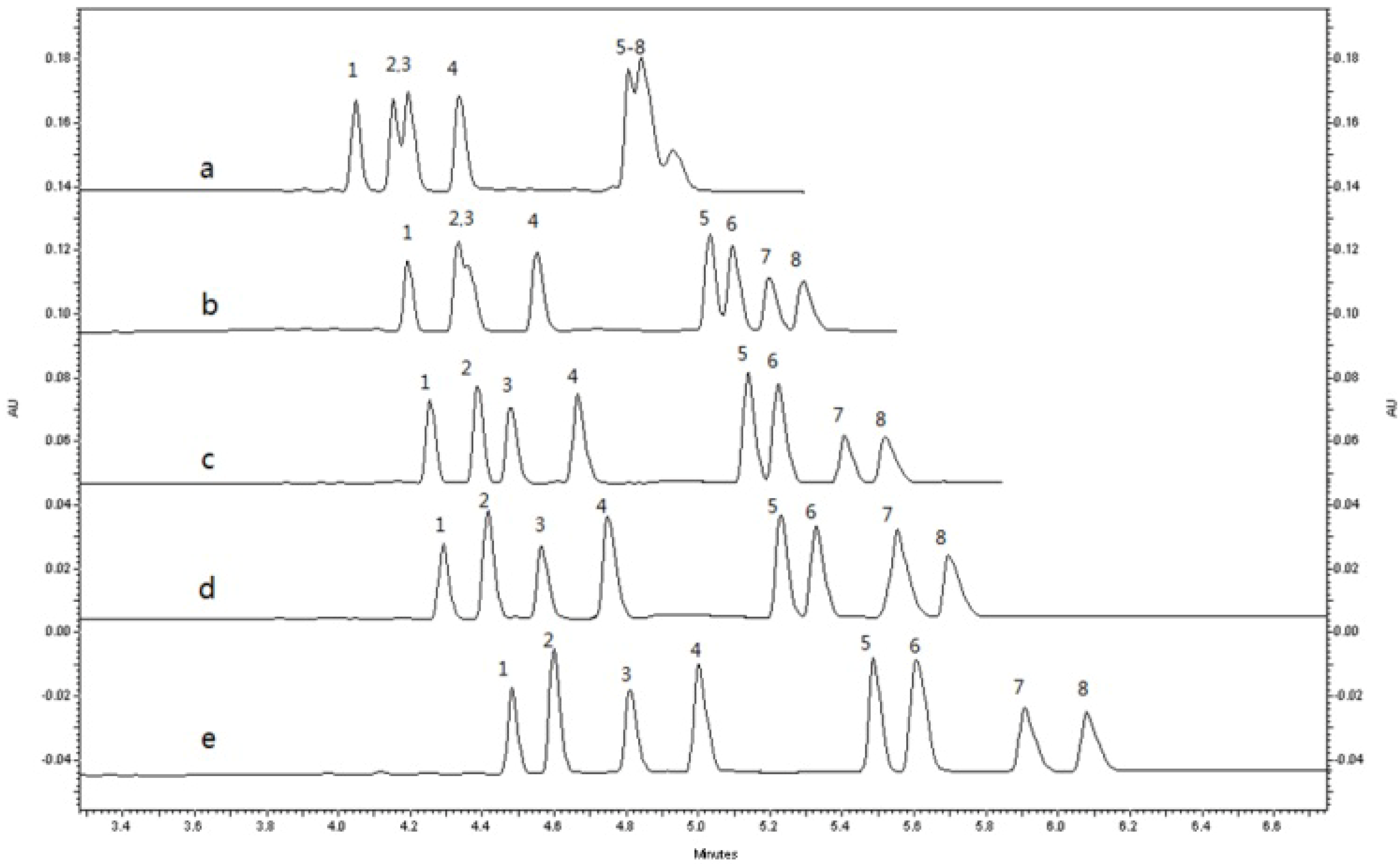
2.1.3. Optimization of Sample Stacking Performance
2.1.3.1. Selection of Injection Modes, Time and Voltage
2.1.3.2. Selection of Sample Solvent
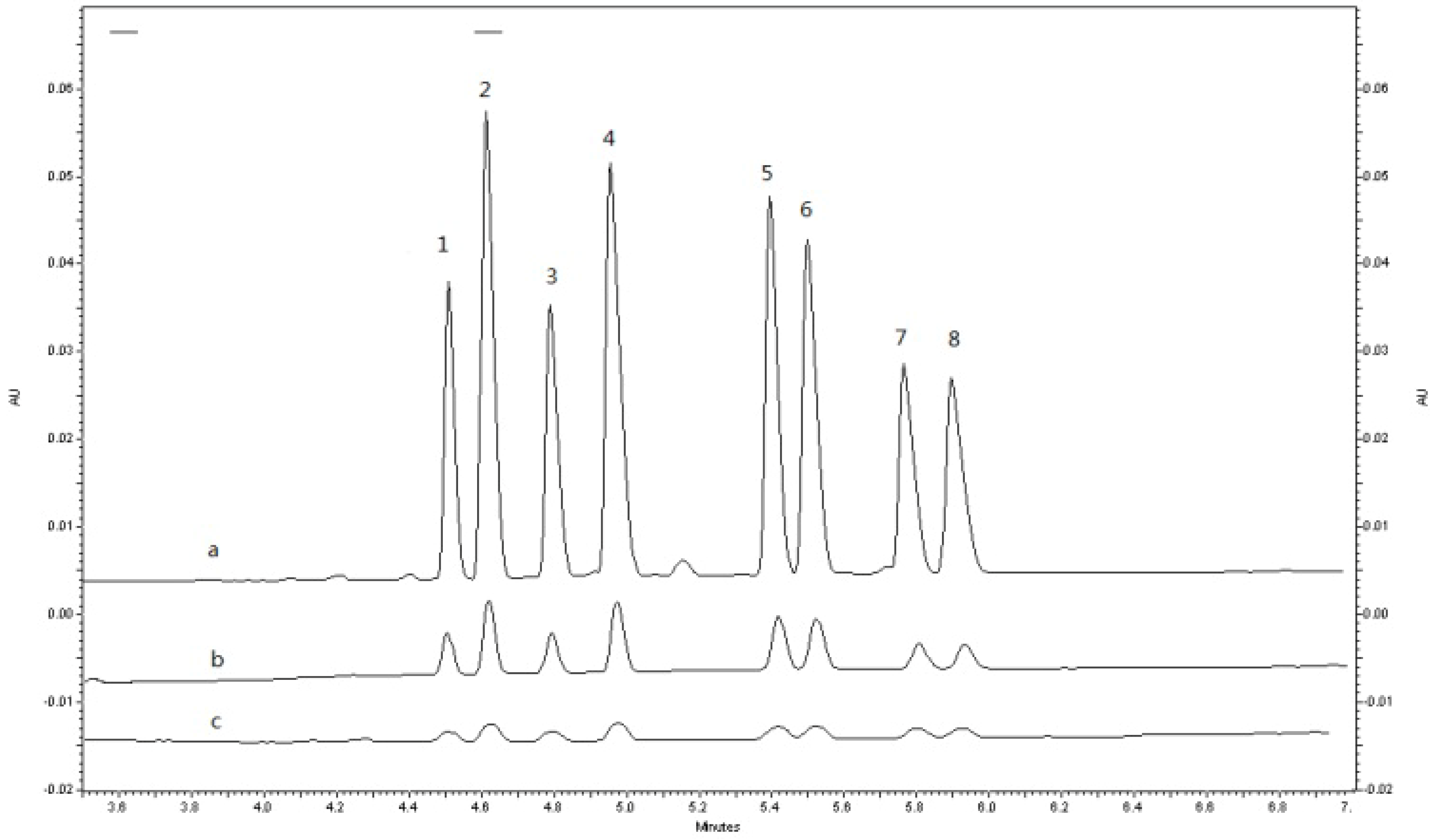
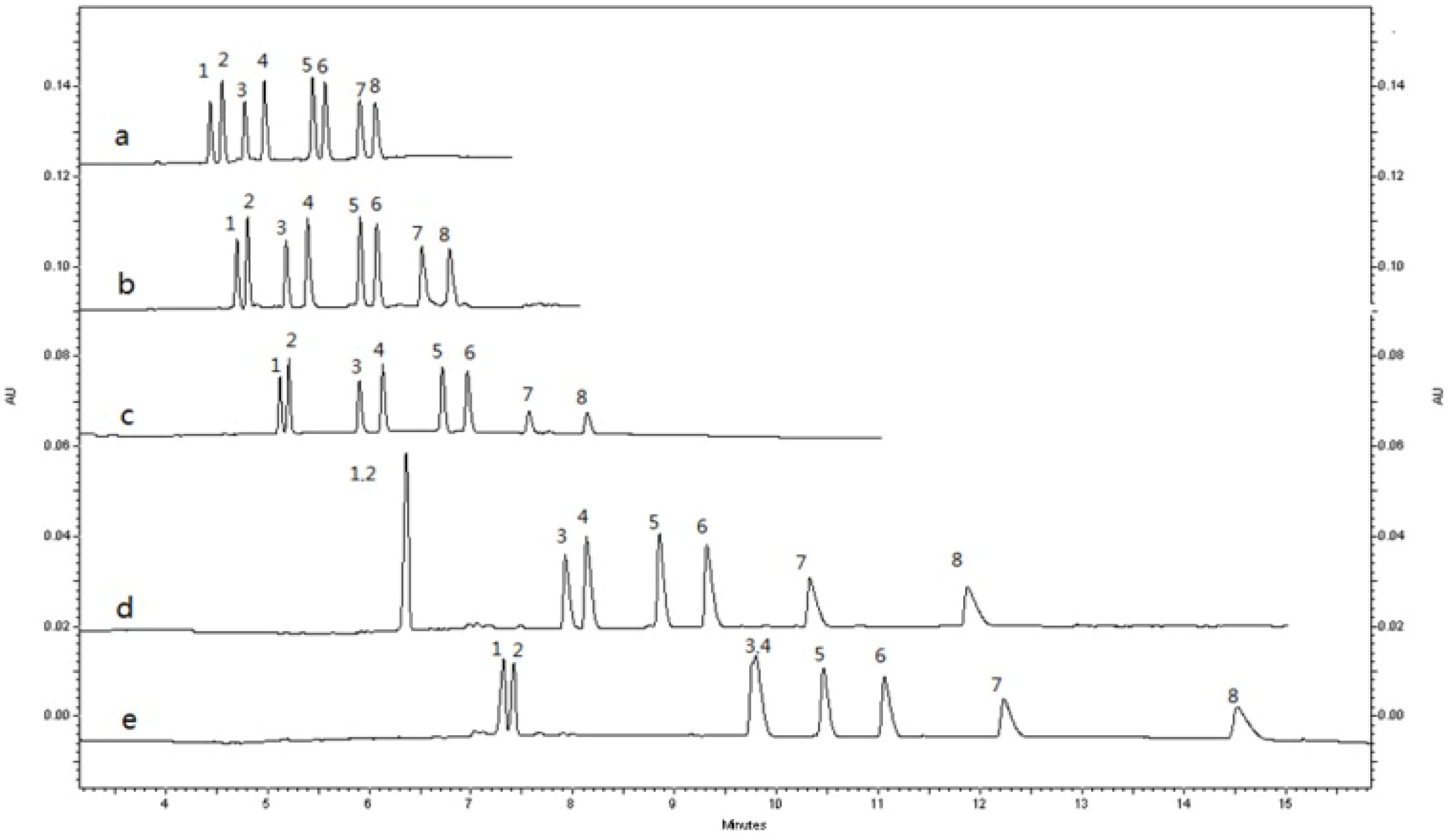
2.1.4. Sample Preparation
2.2. Method Validation
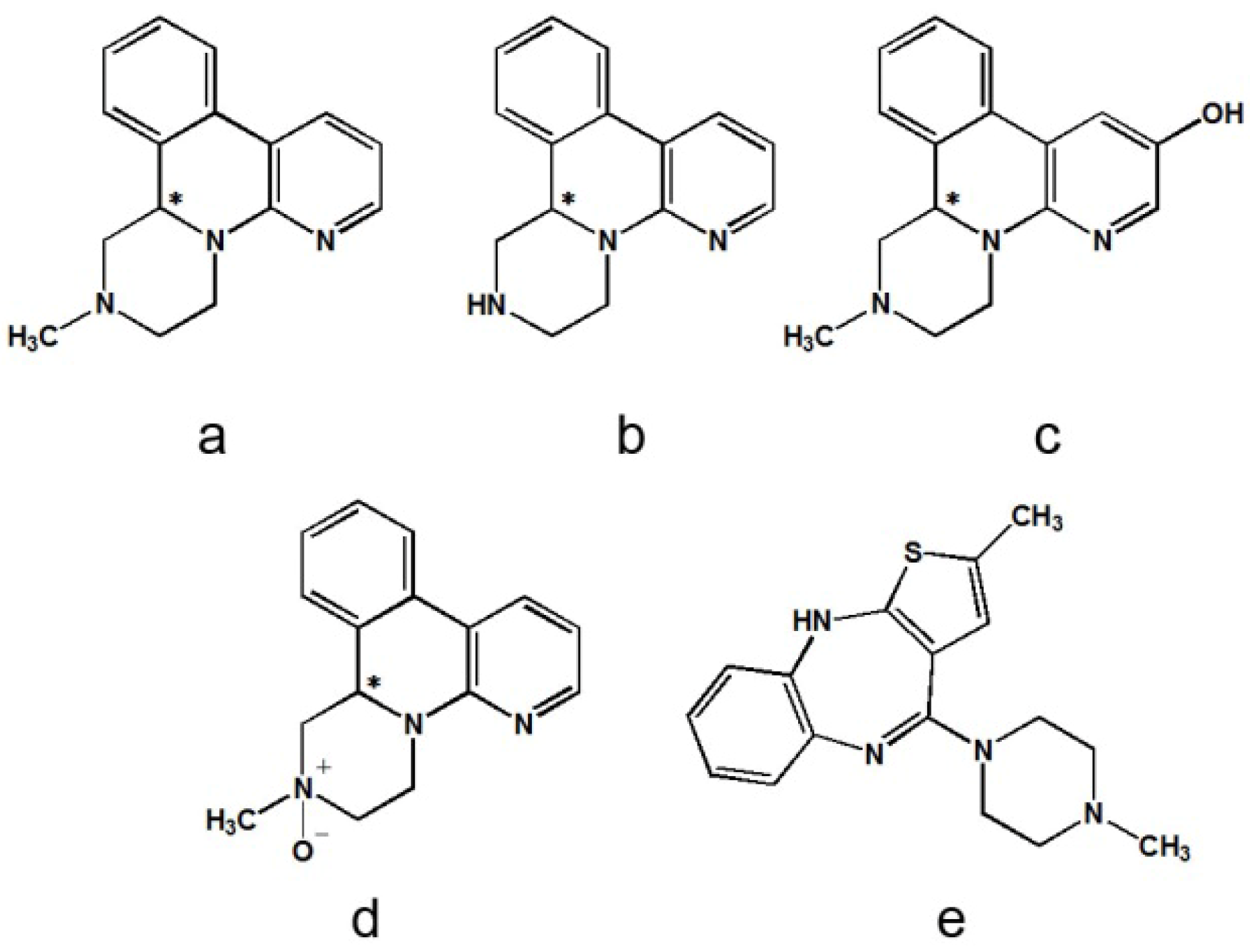
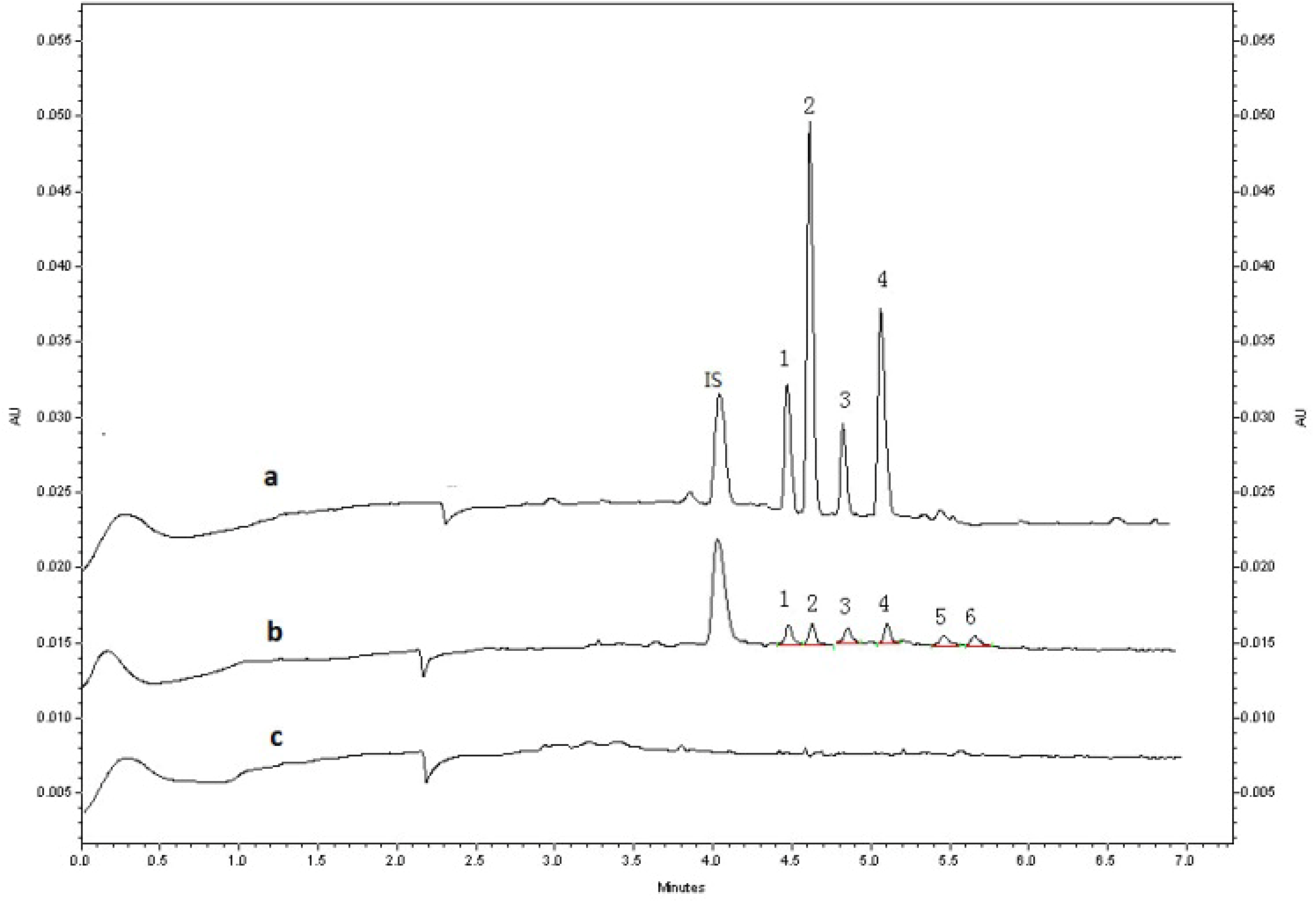
2.2.1. Specificity
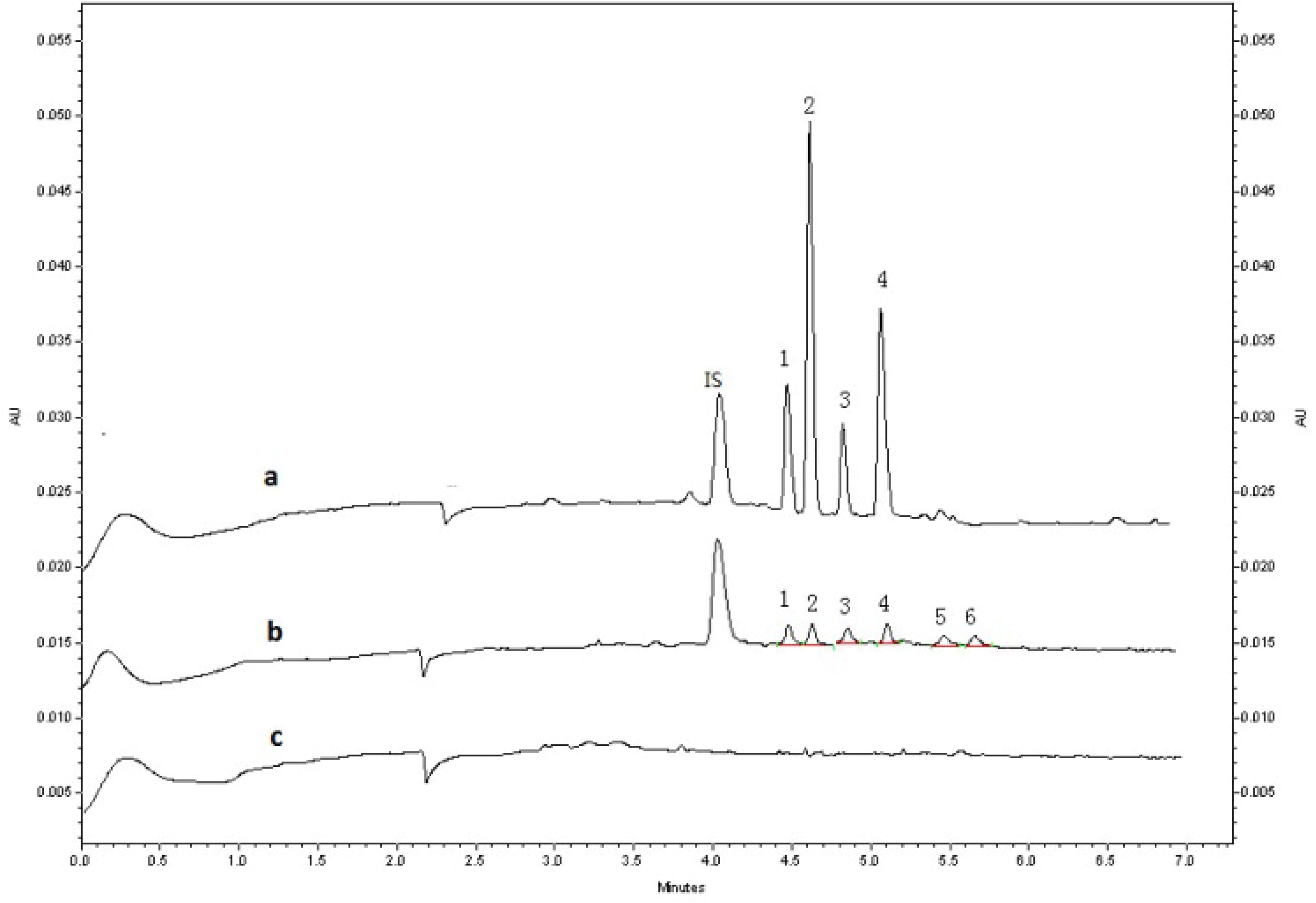
2.2.2. Linearity of Calibration Curves and Lower Limits of Quantification
2.2.3. Accuracy, Precision and Extraction Recovery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enantiomer | Range(ng/mL) | Regression Equations | Correlation Coefficient (R2) | SD for the Slope | SD for the Inercept |
|---|---|---|---|---|---|
| (−)-R-MRT | 0.5–50 | y = 0.256x + 0.0188 | 0.998 | 0.050 | 0.027 |
| (+)-S-MRT | 0.5–50 | y = 0.232x + 0.0156 | 0.992 | 0.048 | 0.015 |
| (−)-R-DMR | 0.5–50 | y = 0.315x − 0.0314 | 0.995 | 0.037 | 0.030 |
| (+)-S-DMR | 0.5–50 | y = 0.290x − 0.0290 | 0.993 | 0.041 | 0.020 |
| (−)-R-8-OHM | 0.5–50 | y = 0.145x − 0.0517 | 0.995 | 0.044 | 0.030 |
| (+)-S-8-OHM | 0.5–50 | y = 0.130x − 0.0444 | 0.994 | 0.039 | 0.033 |
| Enantiomer | LLOQ | Low QC | Medium QC | High QC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 ng/mL | 1 ng/mL | 5 ng/mL | 40 ng/mL | |||||||||
| Mean ± SD | RE | RSD | Mean ± SD | RE | RSD | Mean ± SD | RE | RSD | Mean ± SD | RE | RSD | |
| (ng/mL) | (%) | (%) | (ng/mL) | (%) | (%) | (ng/mL) | (%) | (%) | (ng/mL) | (%) | (%) | |
| (−)-R-MRT | 0.54 ± 0.041 | 8.0 | 8.2 | 1.08 ± 0.12 | 7.8 | 11.3 | 5.16 ± 0.36 | 3.3 | 7.0 | 40.34 ± 1.4 | 0.8 | 3.4 |
| (+)-S-MRT | 0.46 ± 0.060 | −8.1 | 14.0 | 0.99 ± 0.10 | −1.2 | 10.0 | 4.88 ± 0.33 | −2.4 | 6.7 | 39.32 ± 1.3 | −1.7 | 3.4 |
| (−)-R-DMR | 0.55 ± 0.069 | 10.4 | 13.2 | 1.03 ± 0.15 | 2.7 | 14.2 | 4.61 ± 0.27 | −7.9 | 6.0 | 39.57 ± 1.2 | −1.1 | 3.0 |
| (+)-S-DMR | 0.49 ± 0.052 | −2.2 | 9.2 | 1.04 ± 0.11 | 3.7 | 10.8 | 5.08 ± 0.25 | 1.6 | 4.9 | 39.28 ± 1.5 | −1.8 | 3.9 |
| (−)-R-8-OHM | 0.45 ± 0.041 | −10.8 | 7.9 | 0.97 ± 0.09 | −3.4 | 9.7 | 4.68 ± 0.30 | −6.3 | 6.3 | 43.68 ± 1.8 | 9.2 | 4.0 |
| (+)-S-8-OHM | 0.54 ± 0.041 | 7.6 | 8.2 | 0.95 ± 0.08 | −4.8 | 8.8 | 5.35 ± 0.28 | 6.9 | 5.3 | 39.94 ± 0.95 | −0.2 | 2.4 |
| Enantiomer | Low QC | Medium QC | High QC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 ng/mL | 5 ng/mL | 40 ng/mL | |||||||
| Mean ± SD | RE | RSD | Mean ± SD | RE | RSD | Mean ± SD | RE | RSD | |
| (ng/mL) | (%) | (%) | (ng/mL) | (%) | (%) | (ng/mL) | (%) | (%) | |
| (−)-R-MRT | 1.04 ± 0.11 | 4.3 | 10.2 | 5.10 ± 0.36 | 2.1 | 7.1 | 41.26 ± 1.8 | 3.1 | 4.3 |
| (+)-S-MRT | 0.99 ± 0.10 | 1.0 | 9.5 | 4.98 ± 0.32 | −0.5 | 6.5 | 39.69 ± 1.4 | −0.8 | 3.6 |
| (−)-R-DMR | 1.07 ± 0.14 | 6.8 | 12.9 | 4.86 ± 0.33 | −2.6 | 6.8 | 39.70 ± 1.3 | −0.8 | 3.2 |
| (+)-S-DMR | 1.02 ± 0.10 | 1.6 | 9.6 | 5.05 ± 0.29 | 0.9 | 5.7 | 39.08 ± 1.5 | −2.2 | 3.9 |
| (−)-R-8-OHM | 0.93 ± 0.10 | −6.7 | 10.8 | 4.92 ± 0.35 | −1.6 | 7.2 | 42.83 ± 1.8 | 7.1 | 4.1 |
| (+)-S-8-OHM | 0.99 ± 0.09 | −0.6 | 9.0 | 5.14 ± 0.31 | 2.8 | 6.1 | 40.61 ± 1.5 | 1.5 | 3.6 |
| Enantiomer | Concentration (ng/mL) | Recovery (%) (mean ± SD) | RSD (%) |
|---|---|---|---|
| (−)-R-MRT | 1 | 86.7 ± 8.9 | 10.3 |
| 5 | 89.4 ± 5.7 | 6.4 | |
| 40 | 90.2 ± 4.8 | 5.3 | |
| (+)-S-MRT | 1 | 87.7 ± 7.3 | 8.4 |
| 5 | 90.1 ± 3.9 | 4.3 | |
| 40 | 92.0 ± 5.4 | 5.9 | |
| (−)-R-DMR | 1 | 82.8 ± 8.6 | 10.4 |
| 5 | 87.5 ± 7.6 | 8.7 | |
| 40 | 87.5 ± 3.0 | 3.4 | |
| (+)-S-DMR | 1 | 83.5 ± 6.8 | 8.2 |
| 5 | 85.8 ± 8.9 | 10.3 | |
| 40 | 88.9 ± 3.7 | 4.1 | |
| (+)-R-8-OHM | 1 | 81.8 ± 5.9 | 7.3 |
| 5 | 85.1 ± 8.7 | 10.2 | |
| 40 | 89.5 ± 2.3 | 2.5 | |
| (+)-S-8-OHM | 1 | 83.2 ± 9.6 | 11.5 |
| 5 | 86.1 ± 7.5 | 8.8 | |
| 40 | 87.8 ± 4.4 | 5.0 |
2.2.4. Stability
| Enantiomer | Nominal Concentration (ng/mL) | Freeze-Thaw Stability a | 30-Day Stability b | Bench Top Stability c | Autosampler Stability d | ||||
|---|---|---|---|---|---|---|---|---|---|
| Measured Concentration (ng/mL) (mean ± SD) | RSD (%) | Measured Concentration (ng/mL) (mean ± SD) | RSD (%) | Measured Concentration (ng/mL) (mean ± SD) | RSD (%) | Measured Concentration (ng/mL) (mean ± SD) | RSD (%) | ||
| (−)-R-MRT | 1 | 0.99 ± 0.11 | 11.5 | 1.08 ± 0.12 | 10.8 | 1.04 ± 0.12 | 11.8 | 0.99 ± 0.10 | 9.7 |
| 5 | 4.70 ± 0.25 | 5.4 | 4.98 ± 0.29 | 5.8 | 5.33 ± 0.29 | 5.5 | 5.13 ± 0.31 | 5.9 | |
| 40 | 37.61 ± 2.0 | 5.4 | 41.96 ± 1.3 | 3.0 | 41.33 ± 2.3 | 5.5 | 38.34 ± 1.4 | 3.6 | |
| (+)-S-MRT | 1 | 0.95 ± 0.09 | 9.7 | 1.07 ± 0.12 | 1.6 | 1.02 ± 0.12 | 12.3 | 1.03 ± 0.12 | 11.8 |
| 5 | 5.06 ± 0.35 | 6.9 | 4.89 ± 0.28 | 5.8 | 5.08 ± 0.30 | 5.8 | 5.06 ± 0.30 | 5.9 | |
| 40 | 38.6 ± 1.8 | 4.7 | 42.5 ± 2.1 | 5.0 | 44.0 ± 1.6 | 3.7 | 36.3 ± 1.9 | 3.5 | |
| (−)-R-DMR | 1 | 0.91 ± 0.079 | 8.6 | 1.08 ± 0.12 | 10.8 | 1.02 ± 0.088 | 9.1 | 1.03 ± 0.078 | 8.2 |
| 5 | 4.98 ± 0.25 | 5.1 | 4.63 ± 0.23 | 4.9 | 5.33 ± 0.29 | 5.5 | 4.86 ± 0.26 | 5.4 | |
| 40 | 39.20 ± 2.1 | 5.2 | 37.26 ± 1.8 | 4.7 | 40.55 ± 2.6 | 6.3 | 39.00 ± 2.0 | 5.0 | |
| (+)-S-DMR | 1 | 1.03 ± 0.12 | 11.1 | 1.07 ± 0.12 | 11.6 | 1.00 ± 0.070 | 6.8 | 1.03 ± 0.12 | 11.7 |
| 5 | 4.65 ± 0.28 | 6.0 | 4.52 ± 0.26 | 5.7 | 5.40 ± 0.34 | 6.2 | 4.98 ± 0.25 | 4.9 | |
| 40 | 39.20 ± 2.1 | 5.2 | 39.98 ± 2.3 | 5.6 | 36.51 ± 1.4 | 3.9 | 40.61 ± 1.5 | 3.6 | |
| (−)-R-8-OHM | 1 | 0.91 ± 0.081 | 8.6 | 1.02 ± 0.11 | 10.5 | 1.13 ± 0.10 | 9.2 | 0.95 ± 0.09 | 9.4 |
| 5 | 5.12 ± 0.22 | 4.3 | 5.07 ± 0.18 | 3.6 | 4.99 ± 0.25 | 4.9 | 5.31 ± 0.36 | 6.8 | |
| 40 | 39.55 ± 0.93 | 2.4 | 40.4 ± 2.0 | 4.8 | 42.9 ± 1.8 | 4.1 | 40.0 ± 2.0 | 4.9 | |
| (+)-S-8-OHM | 1 | 0.97 ± 0.11 | 10.9 | 1.01 ± 0.09 | 8.5 | 1.02 ± 0.12 | 12.3 | 0.98 ± 0.11 | 10.8 |
| 5 | 5.10 ± 0.20 | 3.9 | 5.06 ± 0.19 | 3.7 | 5.04 ± 0.27 | 5.3 | 4.93 ± 0.34 | 6.8 | |
| 40 | 40.12 ± 2.0 | 5.0 | 40.71 ± 1.3 | 3.2 | 42.55 ± 1.8 | 4.1 | 38.72 ± 1.8 | 4.7 | |
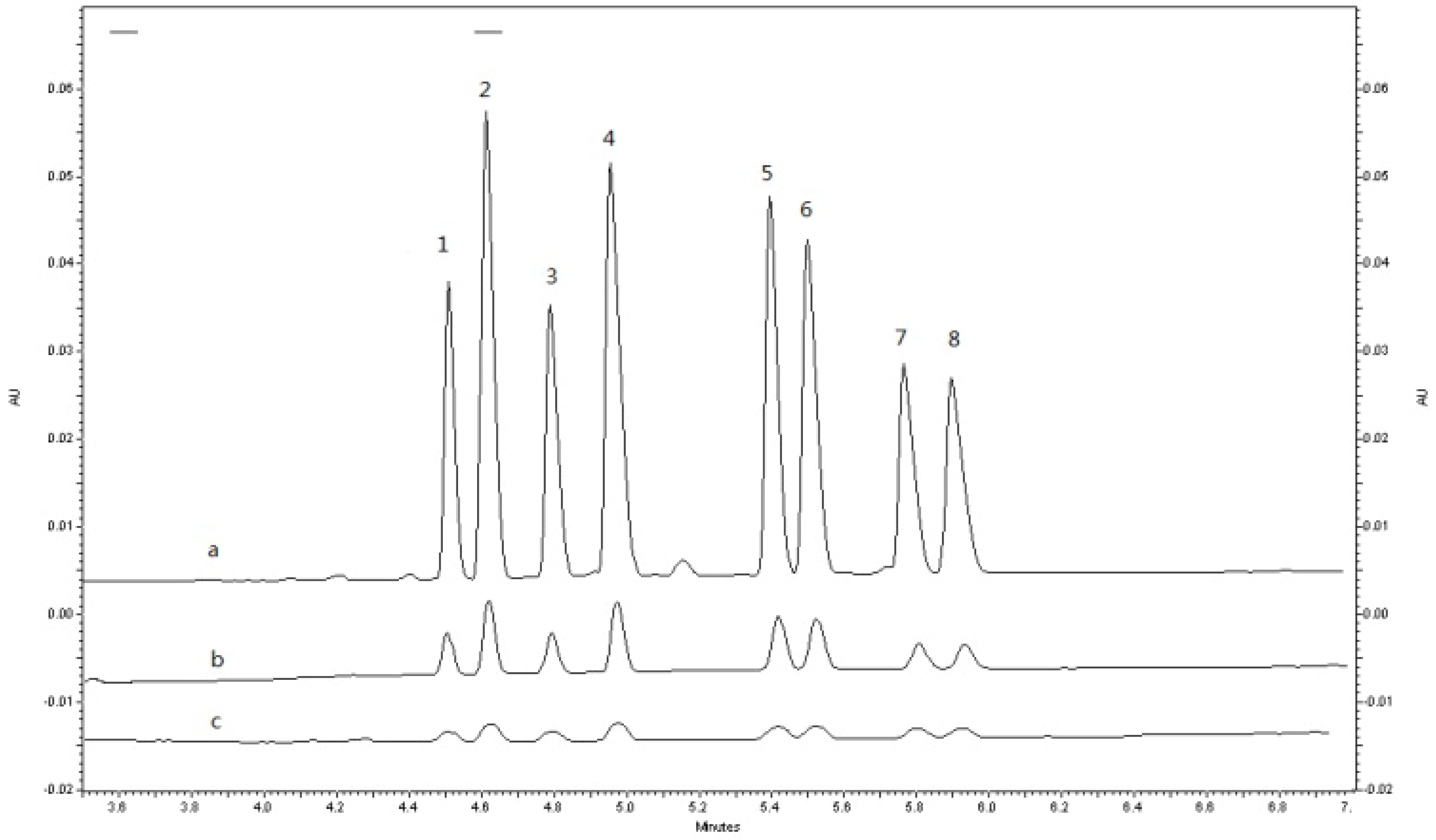
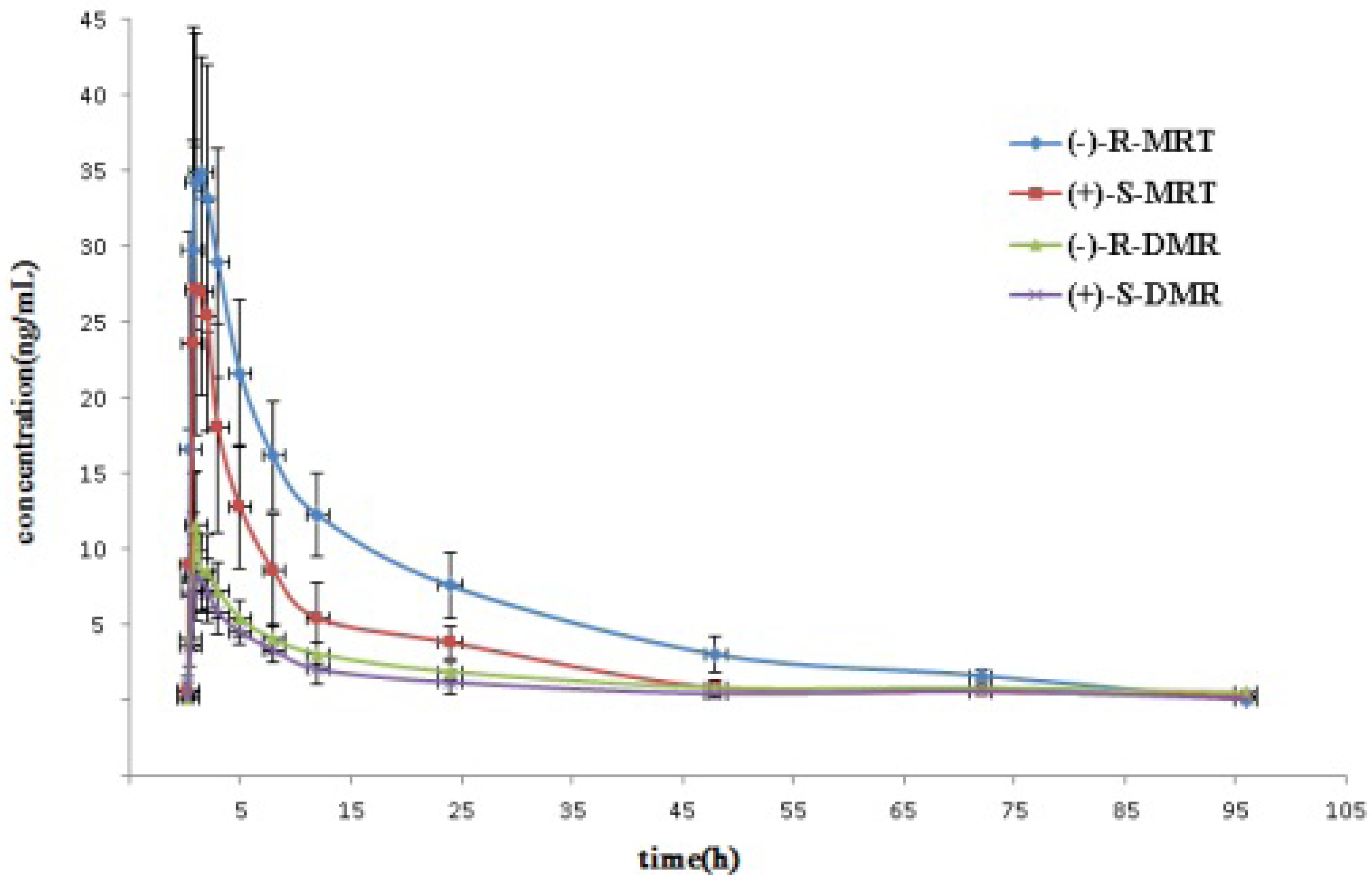
2.3. Application to Pharmacokinetic Studies
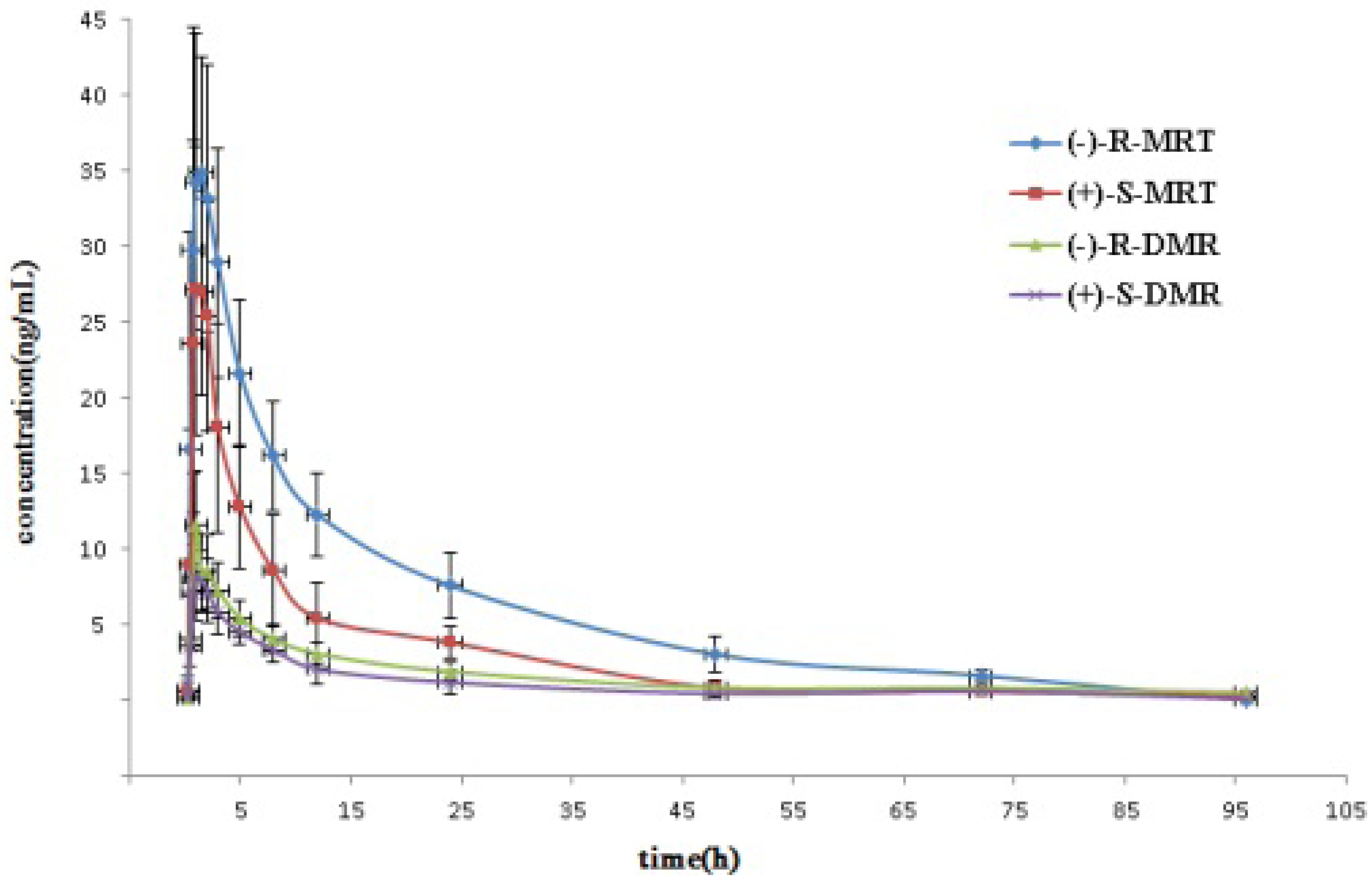
| Enantiomer | Cmax (ng/mL) | Tmax (h) | t1/2(h) | MRT * (h) | AUC0-96 (ng h/mL) | AUC0−∞ (ng h/mL) |
|---|---|---|---|---|---|---|
| (−)-R-MRT | 41.81 ± 4.1 | 1.42 ± 0.66 | 23.22 ± 4.9 | 27.37 ± 5.5 | 570.94 ± 98 | 613.84 ± 103 |
| (+)-S-MRT | 34.15 ± 5.9 | 1.30 ± 0.71 | 15.54 ± 4.4 | 17.43 ± 4.0 | 276.94 ± 76 | 284.14 ± 74 |
| (−)-R-DMR | 12.73 ± 2.5 | 1.32 ± 0.54 | 19.80 ± 4.3 | 22.55 ± 5.7 | 133.75 ± 36 | 149.83 ± 39 |
| (+)-S-DMR | 10.02 ± 1.8 | 1.29 ± 0.73 | 11.45 ± 3.3 | 17.90 ± 4.5 | 96.10 ± 34 | 109.84 ± 36 |
3. Experimental
3.1. Chemicals and Reagents
3.2. Apparatus and Capillary Electrophoretic Conditions
3.3. Preparation of Stock Solutions, Calibration Samples and Quality Control Samples
3.4. Preparation of Human Plasma Sample
3.5. Clinical Pharmacokinetic Study
4. Conclusion
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Anttila, S.A.; Leinonen, E.V. A review of the pharmacological and clinical profile of mirtazapine. CNS. Drug. Rev. 2001, 7, 249–264. [Google Scholar] [CrossRef]
- Montgomery, S. Safety of mirtazapine: A review. Int. Clin. Psychopharmacol. 1995, 10, 37–45. [Google Scholar] [CrossRef]
- Fawcett, J.; Barkin, R.L. Review of the results from clinical studies on the efficacy, safety and tolerability of mirtazapine for the treatment of patients with major depression. J. Affect. Disord. 1998, 51, 267–285. [Google Scholar] [CrossRef]
- Timmer, C.J.; Sitsen, J.A.; Delbressine, L.P. Clinical pharmacokinetics of mirtazapine. Clin. Pharmacokinet. 2000, 38, 461–474. [Google Scholar] [CrossRef]
- De Boer, T. The pharmacologic profile of mirtazapine. J. Clin. Psychiat. 1996, 57, 19–25. [Google Scholar]
- Quimby, J.M.; Gustafson, D.L.; Lunn, K.F. The pharmacokinetics of mirtazapine in cats with chronic kidney disease and in age-matched control cats. J. Vet. Intern. Med. 2011, 25, 985–989. [Google Scholar] [CrossRef]
- Giorgi, M.; Yun, H. Pharmacokinetics of mirtazapine and its main metabolites in Beagle dogs: A pilot study. Vet. J. 2012, 192, 239–241. [Google Scholar] [CrossRef]
- Giorgi, M.; Owen, H. Mirtazapine in veterinary medicine a pharmacological rationale for its application in chronic pain. Am. J. Anim. Vet. Sci. 2012, 7, 42–47. [Google Scholar] [CrossRef]
- Rouini, M.R.; Lavasani, H.; Sheikholeslami, B.; Nikoui, V.; Bakhtiarian, A.; Sgorbini, M.; Giorgi, M. Pharmacokinetics of mirtazapine and its main metabolites after single oral administration in fasting/Fed horses. J. Equine Vet. Sci. 2013, 33, 410–414. [Google Scholar] [CrossRef]
- Dahl, M.L.; Voortman, G.; Alm, C.; Elwin, C.E.; Delbressine, L.; Vos, R.; Bogaards, J.; Bertilsson, L. In vitro and in vivo studies on the disposition of mirtazapine in humans. Clin. Drug Invest. 1997, 13, 37–46. [Google Scholar] [CrossRef]
- Grasmäder, K.; Verwohlt, P.L.; Kühn, K.U.; Dragicevic, A.; von Widdern, O.; Zobel, A.; Hiemke, C.; Rietschel, M.; Maier, W.; Jaehde, U. Population pharmacokinetic analysis of mirtazapine. Eur. J. Clin. Pharmacol. 2004, 60, 473–480. [Google Scholar] [CrossRef]
- Delbressine, L.; Moonen, M.; Kaspersen, F.; Wagenaars, G.; Jacobs, P.; Timmer, C.; Paanakker, J.; van Hal, H.; Voortman, G. Pharmacokinetics and biotransformation of mirtazapine in human volunteers. Clin. Drug Invest. 1998, 15, 45–55. [Google Scholar] [CrossRef]
- Dodd, S.; Boulton, D.W.; Burrows, G.D.; Vane, D.; Lindsay, C.; Norman, T.R. In vitro metabolism of mirtazapine enantiomers by human cytochrome P450 enzymes. Hum. Psychopharmacol. Clin. Exp. 2001, 16, 541–544. [Google Scholar] [CrossRef]
- De Santana, F.J.M.; de Oliveira, A.R.M.; Bonato, P.S. Chiral liquid chromatographic determination of mirtazapine in human plasma using two-phase liquid-phase microextraction for sample preparation. Anal. Chim. Acta 2005, 549, 96–103. [Google Scholar] [CrossRef]
- Dodd, S.; D Burrows, G.; Norman, T.R. Chiral determination of mirtazapine in human blood plasma by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 2000, 748, 439–443. [Google Scholar] [CrossRef]
- De Santana, F.J.M.; Cesarino, E.J.; Bonato, P.S. New method for the chiral evaluation of mirtazapine in human plasma by liquid chromatography. J. Chromatogr. B 2004, 809, 351–356. [Google Scholar] [CrossRef]
- Fanali, S.; Aturki, Z.; Raggi, M.A.; D’Orazio, G. Enantiomeric separation of mirtazapine and its metabolites by nano-liquid chromatography with UV-absorption and mass spectrometric detection. J. Sep. Sci. 2005, 28, 1719–1728. [Google Scholar] [CrossRef]
- Rao, R.N.; Kumar, K.N.; Ramakrishna, S. Enantiomeric separation of mirtazapine and its metabolite in rat plasma by reverse polar ionic liquid chromatography using fluorescence and polarimetric detectors connected in series. J. Chromatogr. B 2011, 879, 1911–1916. [Google Scholar] [CrossRef]
- Paus, E.; Jonzier-Perey, M.; Cochard, N.; Eap, C.B.; Baumann, P. Chirality in the new generation of antidepressants: Stereoselective analysis of the enantiomers of mirtazapine, N-demethylmirtazapine, and 8-hydroxymirtazapine by LC-MS. Ther. Drug Monit. 2004, 26, 366–374. [Google Scholar] [CrossRef]
- De Santana, F.J.M.; Jabor, V.A.P.; Cesarino, E.J.; Lanchote, V.L.; Bonato, P.S. Enantioselective analysis of mirtazapine, demethylmirtazapine and 8-hydroxy mirtazapine in human urine after solid-phase microextraction. J. Sep. Sci. 2010, 33, 268–276. [Google Scholar] [CrossRef]
- De Santana, F.J.M.; Bonato, P.S. Enantioselective analysis of mirtazapine and its two major metabolites in human plasma by liquid chromatography-mass spectrometry after three-phase liquid-phase microextraction. Anal. Chim. Acta 2008, 606, 80–91. [Google Scholar] [CrossRef]
- Mandrioli, R.; Pucci, V.; Sabbioni, C.; Bartoletti, C.; Fanali, S.; Raggi, M.A. Enantioselective determination of the novel antidepressant mirtazapine and its active demethylated metabolite in human plasma by means of capillary electrophoresis. J. Chromatogr. A 2004, 1051, 253–260. [Google Scholar]
- De Santana, F.J.M.; Lanchote, V.L.; Bonato, P.S. Capillary electrophoretic chiral determination of mirtazapine and its main metabolites in human urine after enzymatic hydrolysis. Electrophoresis 2008, 29, 3924–3932. [Google Scholar] [CrossRef]
- Aturki, Z.; Scotti, V.; D’Orazio, G.; Rocco, A.; Raggi, M.A.; Fanali, S. Enantioselective separation of the novel antidepressant mirtazapine and its main metabolites by CEC. Electrophoresis 2007, 28, 2717–2725. [Google Scholar] [CrossRef]
- Everaerts, F.M.; Verheggen, T.P.; Mikkers, F.E. Determination of substances at low concentrations in complex mixtures by isotachophoresis with column coupling. J. Chromatogr. A 1979, 169, 21–38. [Google Scholar] [CrossRef]
- Shihabi, Z.K. Organic solvent high-field amplified stacking for basic compounds in capillary electrophoresis. J. Chromatogr. A 2005, 1066, 205–210. [Google Scholar]
- Li, J.; Li, Y.; Zhang, W.; Chen, Z.; Fan, G. Glucose-β-CD interaction assisted ACN field-amplified sample stacking in CZE for determination of trace amlodipine in beagle dog plasma. J. Sep. Sci. 2013, 36, 1817–1825. [Google Scholar] [CrossRef]
- Li, J.; Bi, Y.; Wang, L.; Sun, F.; Chen, Z.; Xu, G.; Fan, G. β-Cyclodextrin enhanced on-line organic solvent field-amplified sample stacking in capillary zone electrophoresis for analysis of ambroxol in human plasma, following liquid-liquid extraction in the 96-well format. J. Pharm. Biomed. Anal. 2012, 66, 218–224. [Google Scholar] [CrossRef]
- Chen, C.C.; Wu, S.M.; Huang, Y.H.; Ko, W.K.; Kou, H.S.; Wu, H.L. On-line field-amplified sample stacking in capillary electrophoresis for analysis of amitriptyline and its metabolite nortriptyline in plasma. Anal. Chim. Acta 2004, 517, 103–110. [Google Scholar] [CrossRef]
- Tsioupi, D.A.; Stefan-vanStaden, R.I.; Kapnissi-Christodoulou, C.P. Chiral selectors in CE: Recent developments and applications. Electrophoresis 2013, 34, 178–204. [Google Scholar] [CrossRef]
- Quirino, J.P.; Terabe, S. Approaching a million-fold sensitivity increase in capillary electrophoresis with direct ultraviolet detection: Cation-selective exhaustive injection and sweeping. Anal. Chem. 2000, 72, 1023–1030. [Google Scholar] [CrossRef]
- Simpson, S.L., Jr; Quirino, J.P.; Terabe, S. On-line sample preconcentration in capillary electrophoresis: Fundamentals and applications. J. Chromatogr. A 2008, 1184, 504–541. [Google Scholar]
- Quirino, J.P.; Terabe, S. Sample stacking of cationic and anionic analytes in capillary electrophoresis. J. Chromatogr. A 2000, 902, 119–135. [Google Scholar] [CrossRef]
- Chien, R.L.; Helmer, J.C. Electroosmotic properties and peak broadening in field-amplified capillary electrophoresis. Anal. Chem. 1991, 63, 1354–1361. [Google Scholar] [CrossRef]
- Liao, H.W.; Lin, S.W.; Wu, U.I.; Kuo, C.H. Rapid and sensitive determination of posaconazole in patient plasma by capillary electrophoresis with field-amplified sample stacking. J. Chromatogr. A 2012, 1226, 48–54. [Google Scholar]
- Sample Availability: Samples of the compounds MRT and DMR are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wen, J.; Zhang, W.-T.; Cao, W.-Q.; Li, J.; Gao, F.-Y.; Yang, N.; Fan, G.-R. Enantioselective Separation of Mirtazapine and Its Metabolites by Capillary Electrophoresis with Acetonitrile Field-Amplified Sample Stacking and Its Application. Molecules 2014, 19, 4907-4923. https://doi.org/10.3390/molecules19044907
Wen J, Zhang W-T, Cao W-Q, Li J, Gao F-Y, Yang N, Fan G-R. Enantioselective Separation of Mirtazapine and Its Metabolites by Capillary Electrophoresis with Acetonitrile Field-Amplified Sample Stacking and Its Application. Molecules. 2014; 19(4):4907-4923. https://doi.org/10.3390/molecules19044907
Chicago/Turabian StyleWen, Jun, Wen-Ting Zhang, Wei-Qun Cao, Ji Li, Fang-Yuan Gao, Nan Yang, and Guo-Rong Fan. 2014. "Enantioselective Separation of Mirtazapine and Its Metabolites by Capillary Electrophoresis with Acetonitrile Field-Amplified Sample Stacking and Its Application" Molecules 19, no. 4: 4907-4923. https://doi.org/10.3390/molecules19044907