Inhibition and Biochemical Characterization of Methicillin-Resistant Staphylococcus aureus Shikimate Dehydrogenase: An in Silico and Kinetic Study

Abstract
:
1. Introduction
2. Results and Discussion
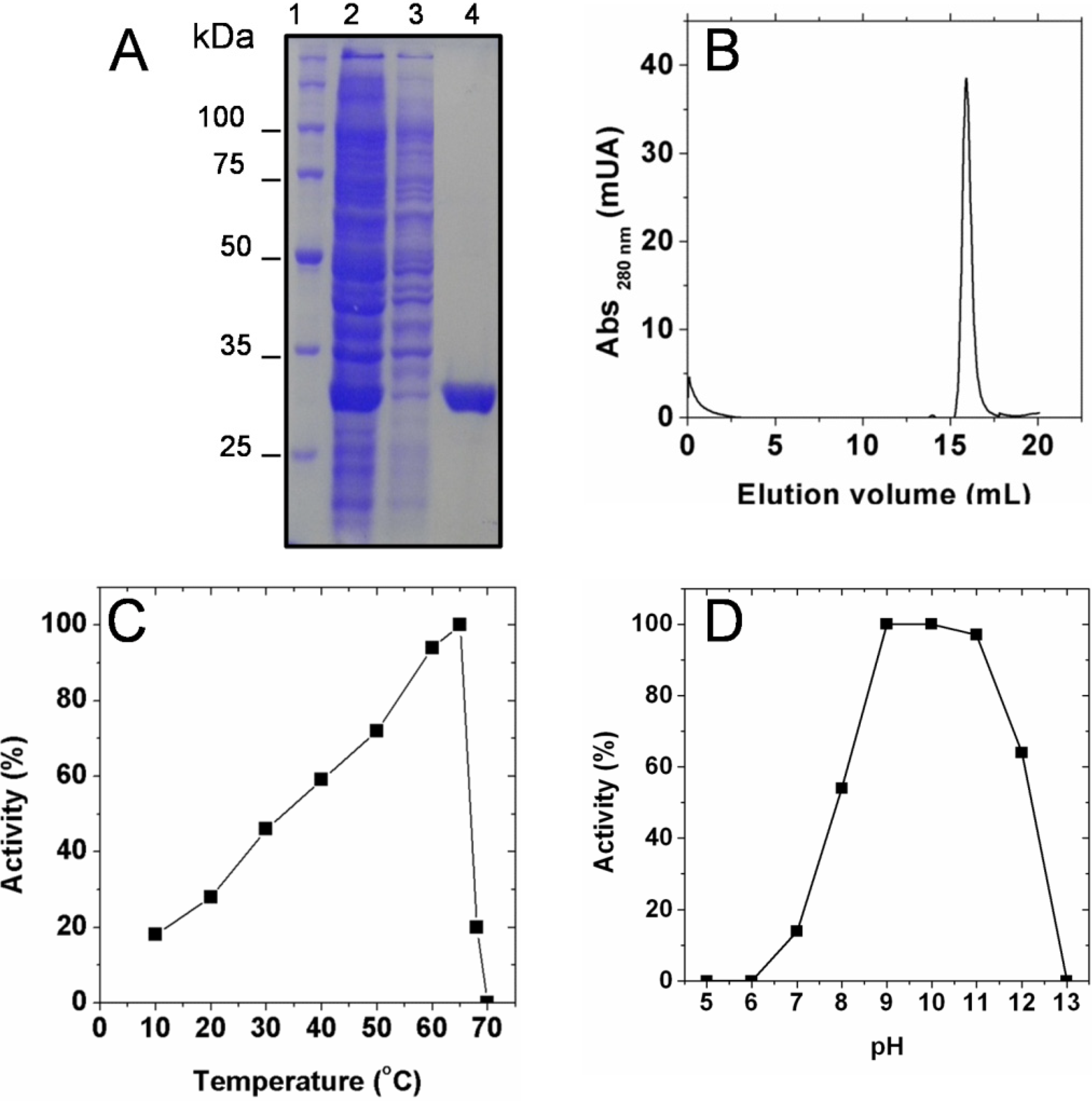
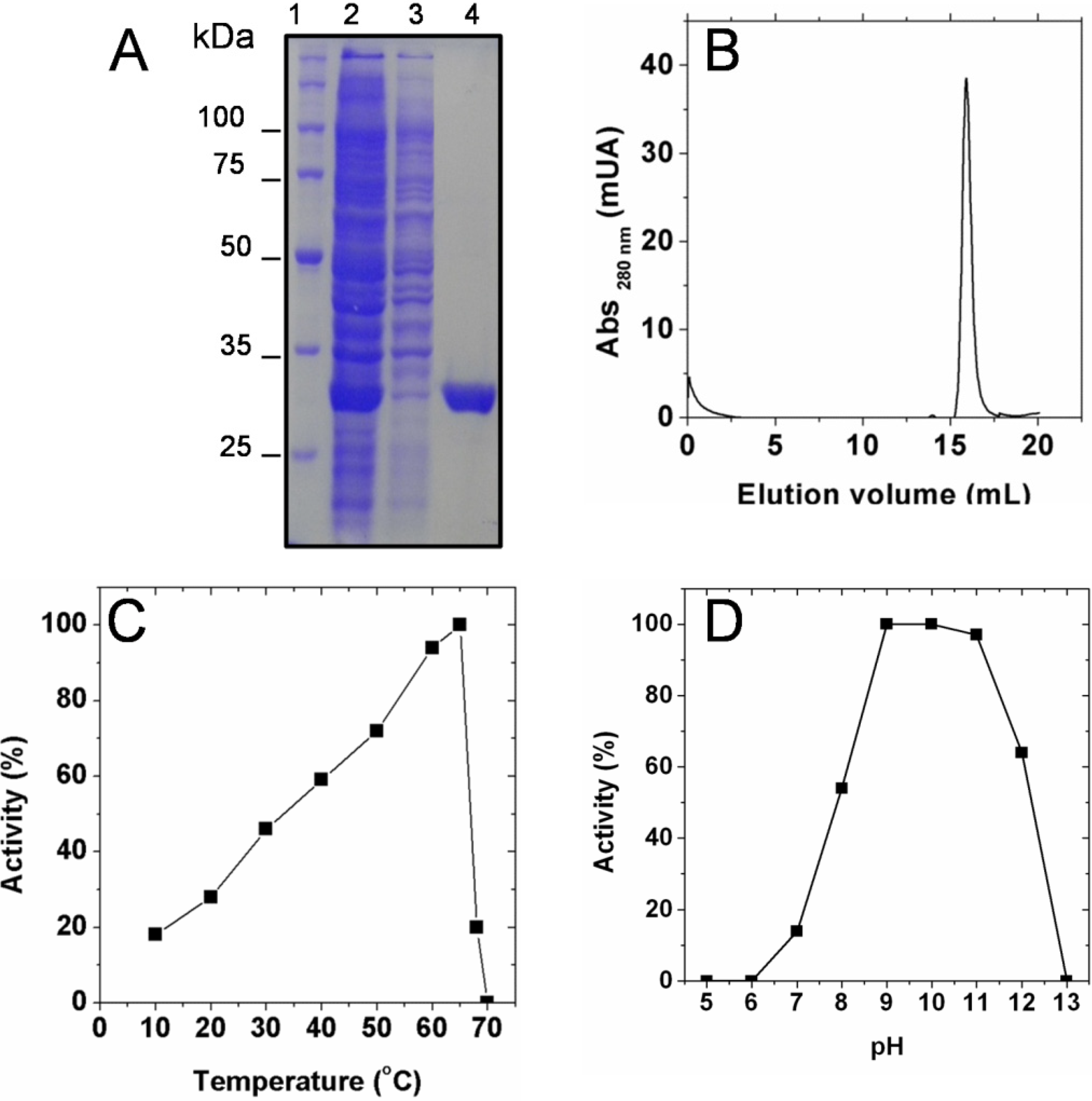
2.1. Biochemical Characterization of SaSDH
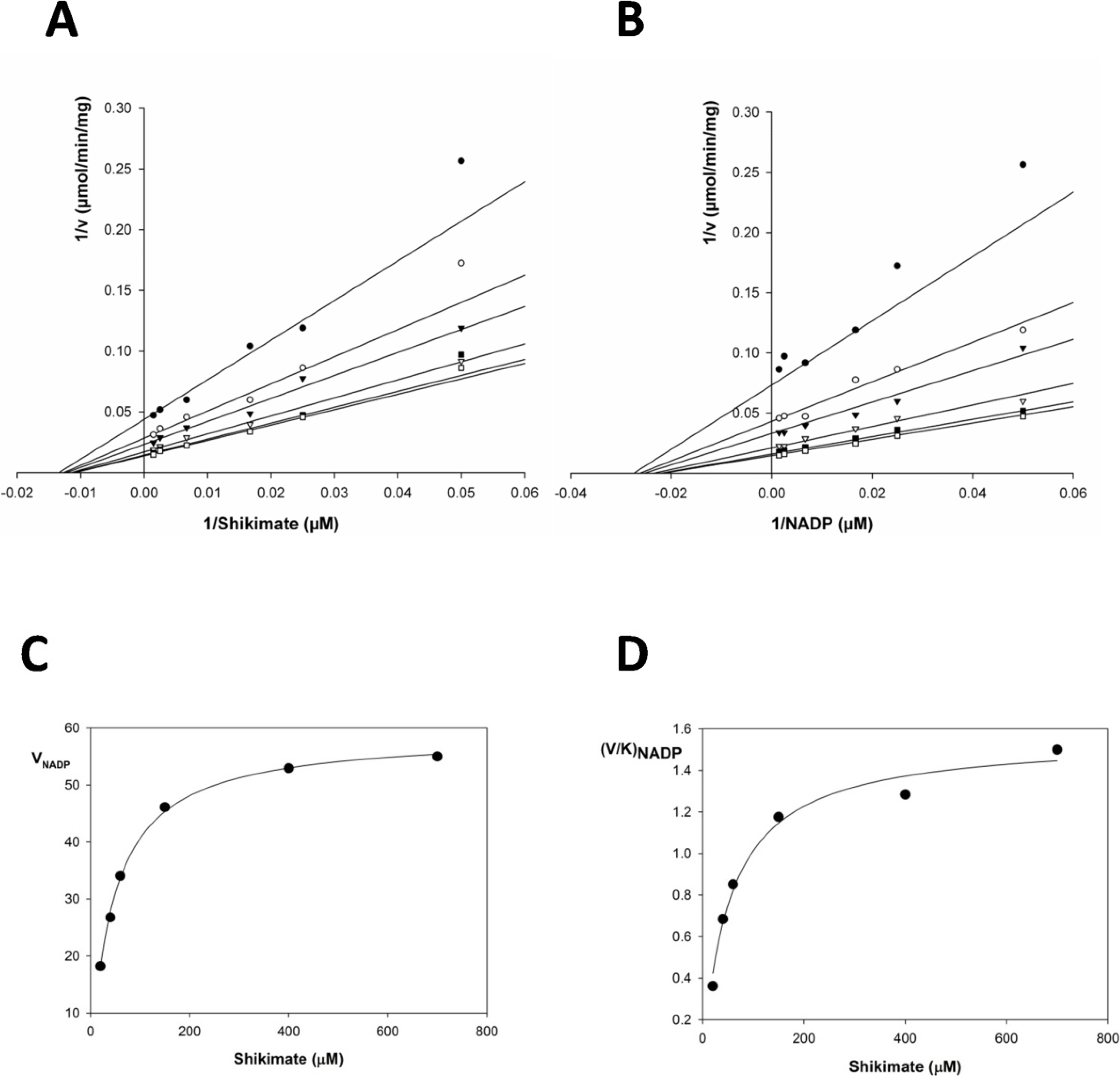
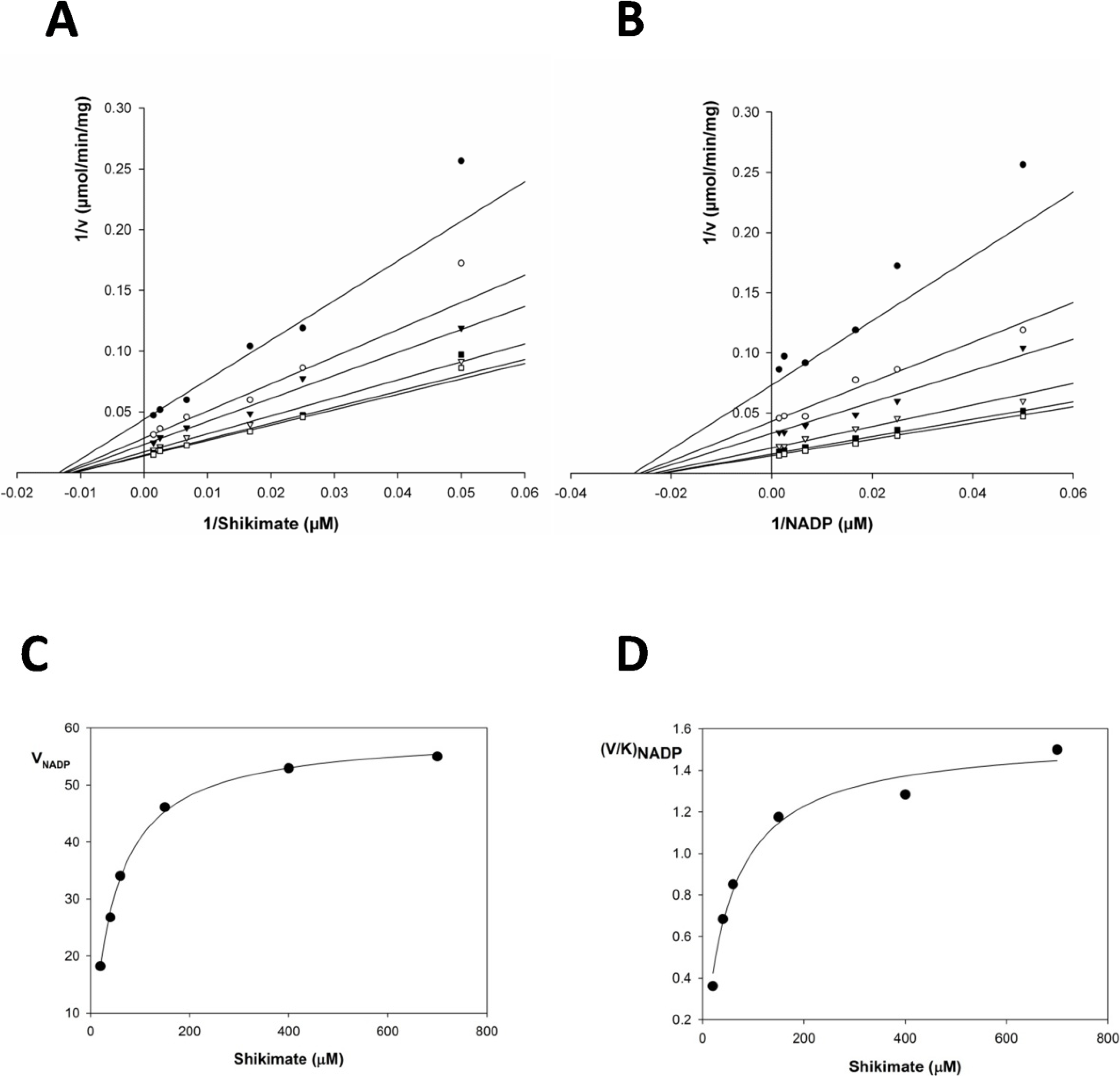
2.2. Kinetic Constants and Reaction Mechanism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Km (μM) | Vmax (μmol/min/mg) | Kcat (s−1) | Kcat/Km (M−1x s−1) | Kia | Kia/Km |
|---|---|---|---|---|---|---|
| Shikimate | 37.46 ± 3.7 | 233.6 ± 1.5 | 135.81 | 3.62 × 106 | 21.224 | 1.76 |
| NADP+ | 42.55 ± 9.9 | 251.41 ± 7.2 | 146.16 | 3.43 × 106 | 21.015 | 2.02 |
2.3. SaSDH Inhibition
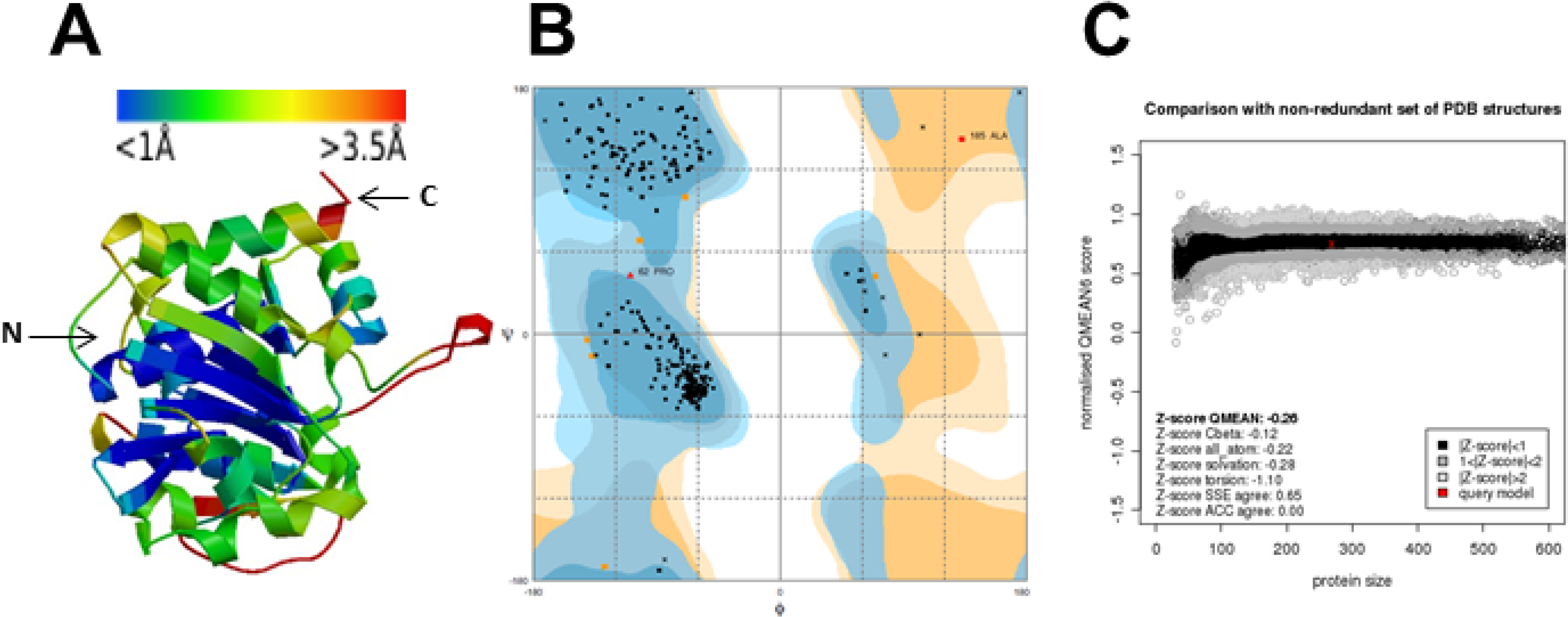
2.3.1. Homology Modeling and Virtual SCREENING

| Compound Structure | Molecular Weight a | H-bond Donor a | H-bond Acceptor a | LogP a | Drug Likeness a, b | Binding energy kcal/mol | % Inhibition 200 µM |
|---|---|---|---|---|---|---|---|
894 | 150.1 | 1 | 3 | 0.97 | 0.13 | −11.93 | 99 |
766 | 206.2 | 1 | 3 | 2.43 | −0.76 | −12.55 | 87 |
238 | 210.3 | 1 | 4 | 3.32 | −099 | −12.38 | 87 |
626 | 270.2 | 1 | 2 | 2.83 | −0.40 | −12.85 | 49 |
463 | 195.3 | 1 | 3 | 1.05 | 0.24 | −11.89 | 45 |
62 | 208.2 | 1 | 4 | 2.69 | −0.70 | −11.98 | 33 |
291 | 164.2 | 1 | 2 | 1.66 | −0.80 | −12.27 | 31 |
692 | 273.2 | 1 | 4 | 3.87 | −0.99 | −11.90 | 31 |
306 | 221.2 | 1 | 4 | 1.23 | 0.20 | −11.90 | 31 |
637 | 192.2 | 1 | 3 | 2.83 | −0.79 | −12.25 | 29 |
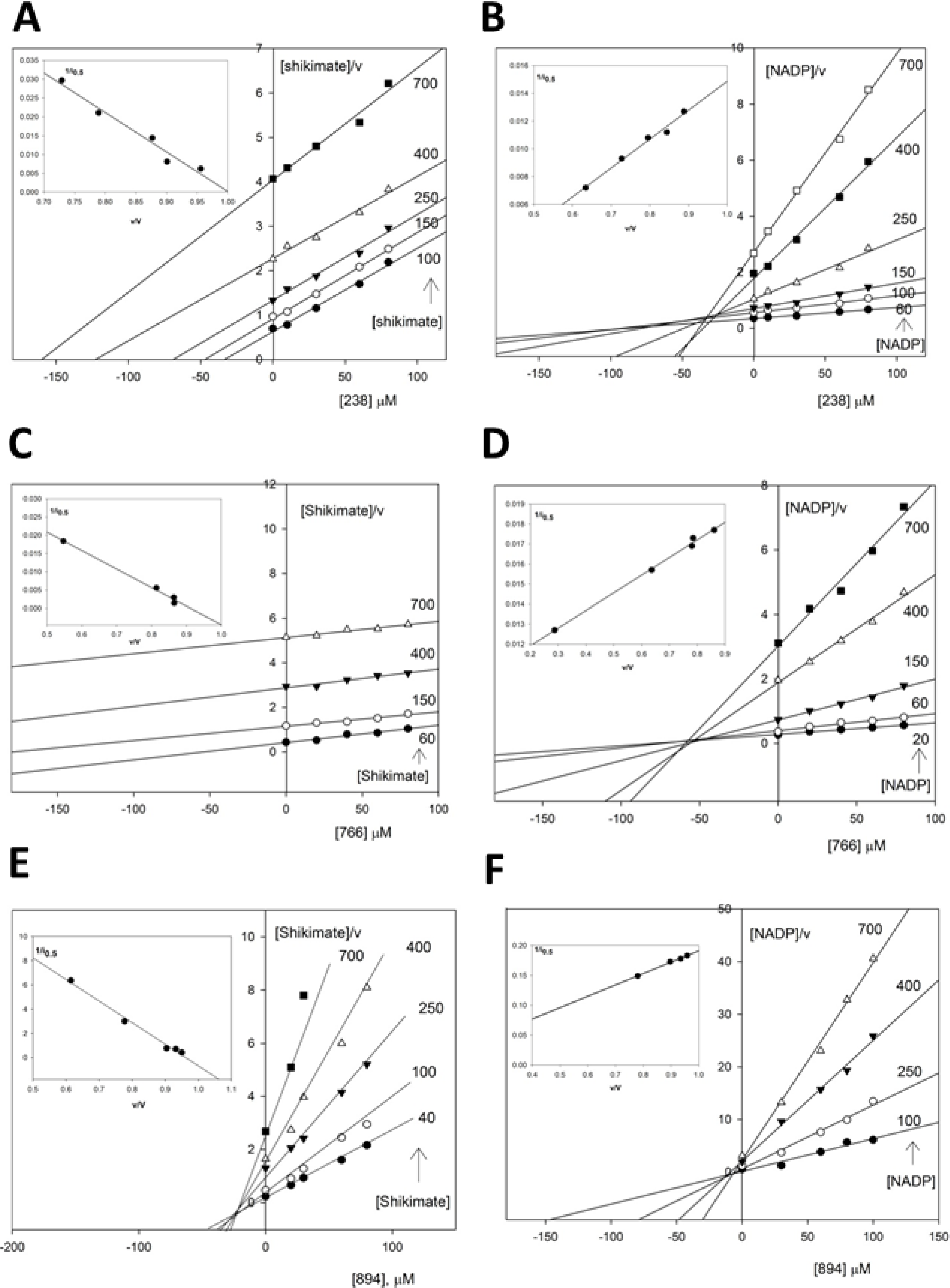
2.3.2. Kinetic Study of the SaSDH-Inhibitor Complex
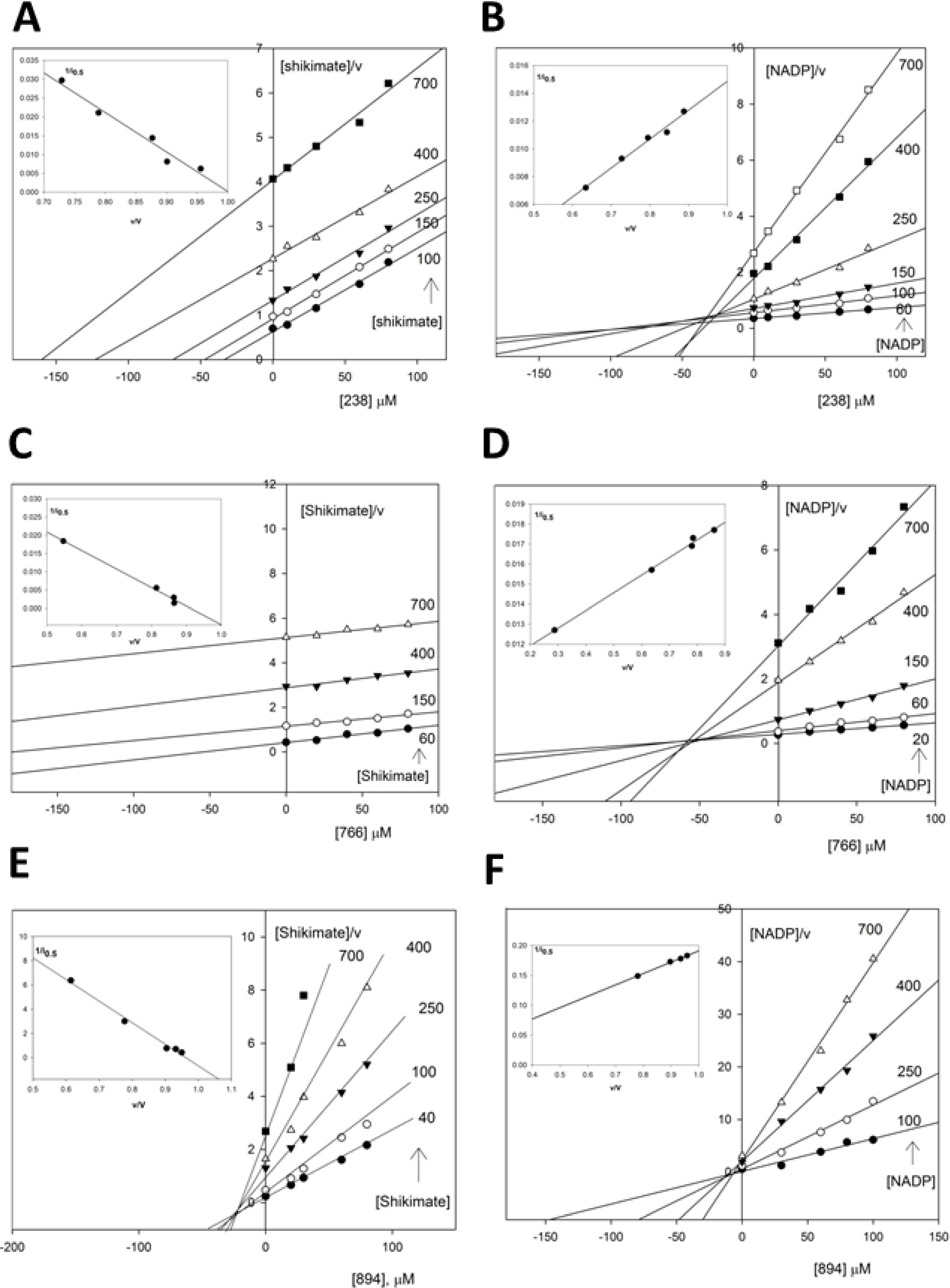
| Substrate | Inhibitor | Kic (μM) | Kiu (μM) | i0.5 at 400 μM of substrate | Pattern |
|---|---|---|---|---|---|
| Shikimate | 238 | 9.53 | NA | 122.94 | Competitive |
| NADP+ | 238 | ND | 48.3 | 107.52 | Uncompetitive |
| Shikimate | 766 | 19.76 | NA | 343.43 | Competitive |
| NADP+ | 766 | ND | 11.35 | 564.2 | Uncompetitive |
| Shikimate | 894 | 58.3 | 1470 | 142.9 | Mixed-competitive |
| NADP+ | 894 | 900.9 | 5.2 | 614 | Mixed-uncompetitive |
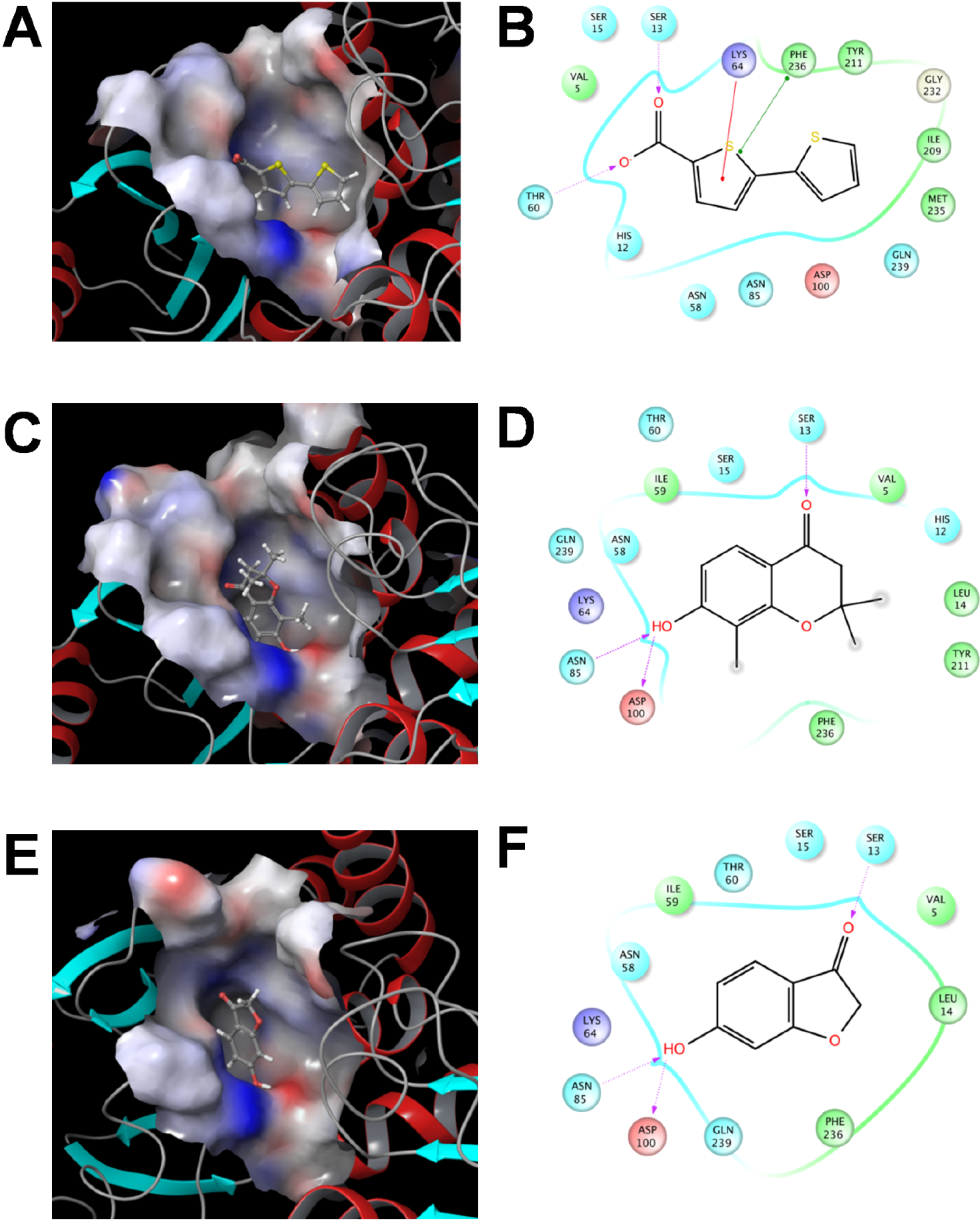
2.3.3 Flexible Docking of the SaSDH-Inhibitor Complex
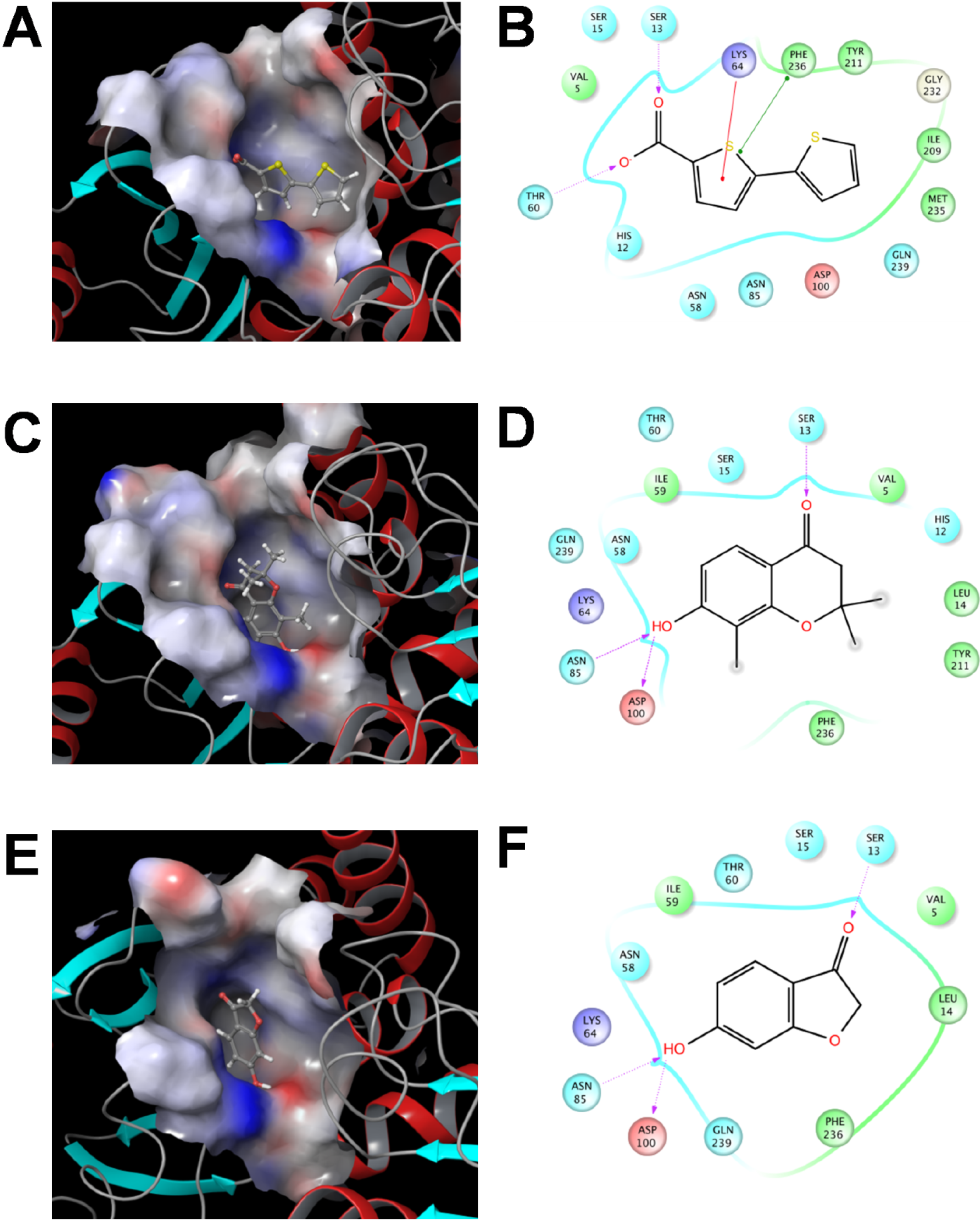
3. Experimental
3.1. SaSDH Gene Cloning
3.2. Enzyme Purification
3.3. Molecular Weight Determination
3.4. Enzyme Activity
3.5. Reaction Mechanism and Kinetic Parameters
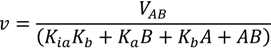
3.6. Inhibition Assays
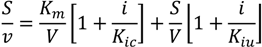
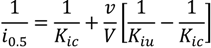
3.7. Biochemical Determinations
3.8. Homology Modeling
3.9. Virtual Screening
3.10. Induced Fit Docking (IFD)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Emori, T.G.; Gaynes, R.P. An overview of nosocomial infections, including the role of the microbiology laboratory. Clin. Microbiol. Rev. 1993, 6, 428–442. [Google Scholar]
- Dickema, D.J.; Pfaller, M.A.; Schmitz, F.J.; Smayevsky, J.; Bell, J.; Jones, R.N.; Beach, M. Survey of infections due to Staphylococcus species: Frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western pacific region for the SENTRY antimicrobial surveillance program, 1997–1999. Clin. Infect. Dis. 2001, 32, S114–S132. [Google Scholar] [CrossRef]
- Picao, R.; Sader, H.; Jones, R.; Andrade, S.; Gales, A. Analysis of resistance and vancomycin “reverse creep” in Latin American Staphylococcus aureus: ten-year report of the SENTRY Antimicrobial Surveillance Program (1997–2006). Clin. Microbiol. Infect. 2008, 14, S173. [Google Scholar]
- Garza-González, E.; Dowzicky, M.J. Changes in Staphylococcus aureus susceptibility across Latin America between 2004 and 2010. Braz. J. Infect. Dis. 2013, 17, 13–19. [Google Scholar] [CrossRef]
- Welsh, K.J.; Skrobarcek, K.A.; Abbott, A.N.; Lewis, C.T.; Kruzel, M.C.; Lewis, E.M.; Gardiner, J.M.; Mohr, J.F.; Armitige, L.Y.; Wanger, A. Predictors of relapse of methicillin-resistant Staphylococcus aureus bacteremia after treatment with vancomycin. J. Clin. Microbiol. 2011, 49, 3669–3672. [Google Scholar] [CrossRef]
- Marty, F.M.; Yeh, W.W.; Wennersten, C.B.; Venkataraman, L.; Albano, E.; Alyea, E.P.; Gold, H.S.; Baden, L.R.; Pillai, S.K. Emergence of a clinical daptomycin-resistant Staphylococcus aureus isolate during treatment of methicillin-resistant Staphylococcus aureus bacteremia and osteomyelitis. J. Clin. Microbiol. 2006, 44, 595–597. [Google Scholar] [CrossRef]
- Ikeda-Dantsuji, Y.; Hanaki, H.; Nakae, T.; Takesue, Y.; Tomono, K.; Honda, J.; Yanagihara, K.; Mikamo, H.; Fukuchi, K.; Kaku, M.; et al. Emergence of linezolid-resistant mutants in a susceptible-cell population of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 2466–2468. [Google Scholar] [CrossRef]
- Mir, R.; Jallu, S.; Singh, T.P. The shikimate pathway: Review of amino acid sequence, function and three-dimensional structures of the enzymes. Crit. Rev. Microbiol. 2013, 6, 1–18. [Google Scholar]
- Kapnick, S.M.; Zhang, Y. New tuberculosis drug development: targeting the shikimate pathway. Expert Opin. Drug Discov. 2008, 3, 565–577. [Google Scholar] [CrossRef]
- Parish, T.; Stoker, N.G. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology 2002, 148, 3069–3077. [Google Scholar]
- Han, C.; Hu, T.; Wu, D.; Qu, S.; Zhou, J.; Ding, J.; Shen, X.; Qu, D.; Jiang, H. X-ray crystallographic and enzymatic analyses of shikimate dehydrogenase from Staphylococcus epidermidis. FEBSJ. 2009, 276, 1125–1139. [Google Scholar]
- Han, C.; Wang, L.; Yu, K.; Chen, L.; Hu, L.; Chen, K.; Jiang, H.; Shen, X. Biochemical characterization and inhibitor discovery of shikimate dehydrogenase from Helicobacter pylori. FEBS J. 2006, 273, 4682–4692. [Google Scholar]
- Padyana, A.K.; Burley, S.K. Crystal structure of shikimate 5-dehydrogenase (SDH) bound to NADP: insights into function and evolution. Structure 2003, 11, 1005–1013. [Google Scholar] [CrossRef]
- Bagautdinov, B.; Kunishima, N. Crystal structures of shikimate dehydrogenase AroE from Thermus thermophilus HB8 and its cofactor and substrate complexes: Insights into the enzymatic mechanism. J. Mol. Biol. 2007, 373, 424–438. [Google Scholar] [CrossRef]
- Ye, S.; von Delft, F.; Brooun, A.; Knuth, M.W.; Swanson, R.V.; McRee, D.E. The crystal structure of shikimate dehydrogenase (AroE) reveals a unique NADPH binding mode. J. Bacteriol. 2003, 185, 4144–4151. [Google Scholar] [CrossRef]
- Gan, J.; Wu, Y.; Prabakaran, P.; Gu, Y.; Li, Y.; Andrykovitch, M.; Liu, H.; Gong, Y.; Yan, H.; Ji, X. Structural and biochemical analyses of shikimate dehydrogenase AroE from Aquifex aeolicus: Implications for the catalytic mechanism. Biochemistry 2007, 46, 9513–9522. [Google Scholar] [CrossRef]
- Hammami, R.; Fliss, I. Current trends in antimicrobial agent research: Chemo- and bioinformatics approaches. Drug Discov. Today 2010, 15, 540–546. [Google Scholar] [CrossRef]
- Science Gateway. Available online: http://www.sciencegateway.org/ (accessed on 1 January 2014).
- Lim, S.; Schröder, I.; Monbouquette, H.G. A thermostableshikimate 5-dehydrogenase from the archaeon Archaeoglobus. fulgidus. FEMS Microbiol. Lett. 2004, 238, 101–106. [Google Scholar]
- Zhang, X.; Zhang, S.; Hao, F.; Lai, X.; Yu, H.; Huang, Y.; Wang, H. Expression, purification and properties of shikimate dehydrogenase from Mycobacterium tuberculosis. J. Biochem. Mol. Biol. 2005, 38, 624–631. [Google Scholar] [CrossRef]
- Kubota, T.; Tanaka, Y.; Hiraga, K.; Inui, M.; Yukawa, H. Characterization of shikimate dehydrogenase homologues of Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2013, 97, 8139–8149. [Google Scholar] [CrossRef]
- Balinsky, D.; Dennis, A.W.; Cleland, W.W. Kinetic and isotope-exchange studies on shikimate dehydrogenase from Pisum. sativum. Biochemistry 1971, 10, 1947–1952. [Google Scholar] [CrossRef]
- Fonseca, I.O.; Silva, R.G.; Fernandes, C.L.; de Souza, O.N.; Basso, L.A.; Santos, D.S. Kinetics and chemical mechanisms of shikimate dehydrogenase from Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2007, 457, 123–133. [Google Scholar] [CrossRef]
- Dowsett, J.R.; Corbett, J.R. The purification and properties of shikimate dehydrogenase. Biochem. J. 1971, 123, 23P. [Google Scholar]
- Cleland, W.W. The kinetics of enzyme-catalyzed reactions with two or more substrates or products I. Nomenclature and rate equations. Biochim. Biophys. Acta 1963, 67, 104–137. [Google Scholar] [CrossRef]
- Segel, I.H. Enzyme Kinetics. Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, 1st ed.Wiley-Interscience: New York, NY, USA, 1975; p. 957. [Google Scholar]
- Burnham, K.P.; Anderson, D.R. Multimodel inference, understanding AIC and BIC in Model selection. Sociol. Methods Res. 2004, 33, 261–304. [Google Scholar] [CrossRef]
- Michel, G.; Roszak, A.W.; Sauvé, V.; Maclean, J.; Matte, A.; Coggins, J.R.; Cygler, M.; Lapthorn, A.J. Structures of shikimate dehydrogenase AroE and its paralog YdiB. A common structural framework for different activities. J. Biol. Chem. 2003, 278, 19463–19472. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010, D66, 12–21. [Google Scholar]
- Benkert, P.; Tosatto, S.C.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Benkert, P.; Künzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef]
- Molsoft. Available online: http://www.molsoft.com/ (accessed on 1 January 2014).
- Cortés, A.; Cascante, M.; Cárdenas, M.L.; Cornish-Bowden, A. Relationship between inhibition constants, inhibitor concentration for 50% inhibition and type of inhibition: New ways of analyzing data. Biochem. J. 2001, 357, 263–268. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Saucedo-Mendiola, M.L.; Salas-Pacheco, J.M.; Nájera, H.; Rojo-Domínguez, A.; Yépez-Mulia, L.; Avitia-Domínguez, C.; Téllez-Valencia, A. Discovery of Entamoeba. histolytica hexokinase 1 inhibitors through homology modeling and virtual screening. J. Enzyme Inhib. Med. Chem. 2013. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada, 2013.
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Rost, B.; Yachdav, G.; Liu, J. The PredictProtein Server. Nucleic Acids Res. 2004, 32, W321–W326. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a Restrained Electrostatic Potential (RESP) model perform in calculating con- formational energies of organic and biological molecules. J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Maybridge. Available online: http://www.maybridge.com/ (accessed on 1 January 2014).
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular forcefield. J. Comput. Chem. 1996, 17, 490–641. [Google Scholar] [CrossRef]
- Maestro. version 9.6; Schrödinger, LLC: New York, NY, USA, 2013.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichment. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- LigPrep. version 2.8; Schrödinger, LLC: New York, NY, USA, 2013.
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchiyama, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of crystal packing forces in determining protein sidechain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Avitia-Domínguez, C.; Sierra-Campos, E.; Salas-Pacheco, J.M.; Nájera, H.; Rojo-Domínguez, A.; Cisneros-Martínez, J.; Téllez-Valencia, A. Inhibition and Biochemical Characterization of Methicillin-Resistant Staphylococcus aureus Shikimate Dehydrogenase: An in Silico and Kinetic Study. Molecules 2014, 19, 4491-4509. https://doi.org/10.3390/molecules19044491
Avitia-Domínguez C, Sierra-Campos E, Salas-Pacheco JM, Nájera H, Rojo-Domínguez A, Cisneros-Martínez J, Téllez-Valencia A. Inhibition and Biochemical Characterization of Methicillin-Resistant Staphylococcus aureus Shikimate Dehydrogenase: An in Silico and Kinetic Study. Molecules. 2014; 19(4):4491-4509. https://doi.org/10.3390/molecules19044491
Chicago/Turabian StyleAvitia-Domínguez, Claudia, Erick Sierra-Campos, José Manuel Salas-Pacheco, Hugo Nájera, Arturo Rojo-Domínguez, Jorge Cisneros-Martínez, and Alfredo Téllez-Valencia. 2014. "Inhibition and Biochemical Characterization of Methicillin-Resistant Staphylococcus aureus Shikimate Dehydrogenase: An in Silico and Kinetic Study" Molecules 19, no. 4: 4491-4509. https://doi.org/10.3390/molecules19044491