1. Introduction
Nitroheterocycle benznidazole (BZN) and nifurtimox (NFX) continue to be, about 30 and 40 years after initial experimental assays, respectively, the most used drugs to treat Chagas disease [
1]. Chagas is lifelong and it is caused by the protozoan parasite Trypanosoma cruzi, being recognized by the World Health Organization [
2] as one of the thirteen most important neglected diseases worldwide. Both BZN and NFX are unable to provide a definitive cure for Chagas due to several limitations, such as strong adverse events, and both medicines are more effective only in the early stages of the disease but not in its chronic phase. Resistance of several Chagas strains against treatment was observed as well [
3], and the poor solubility of these drugs in water is a serious difficulty for their practical use [
4]. We focus on BZN in this work as it is the preferred choice due to the severe NFX gastrointestinal and neurological side effects. Traditional pharmaceutical [
4] as well as nanotechnological approaches [
5,
6] are being developed to enhance the BZN efficacy, improving its potency, reducing its toxicity, and increasing its solubility, which demands a deeper understanding of its molecular properties and protein targets to correlate the BZN structural characteristics with its trypanosomacidal effect.
Reviewed pediatric trials showed that antitrypanosomal drug treatment in children with chronic T. cruzi infection was accepted as the care standard throughout Latin America by the late 1990s [
7], followed by a crescent trend to offer treatment to older patients in recent years. Most experts now believe that the majority of patients up to 50 years of age who have chronic T. cruzi infection [
7], including those without symptoms and those with early manifestations of cardiomyopathy, must receive antitrypanosomal treatment. However, the recommended dosage of benznidazole is given in tablet form, which is inadequate for children, neonates and the elderly [
8]. The development of liquid benznidazole formulations, on the other hand, needs more physical-chemical data to allow for their characterization. Since liquid environments (mainly water) impose serious problems for the vibrational (principally the IR) characterization of solvated molecules, one has to rely on Nuclear Magnetic Resonance (NMR) and optical absorption spectroscopies to obtain details of the aqueous form of benznidazole. In particular, the optical absorption of solvated benznidazole could be very helpful as a characterization technique in a pharmaceutical production line.
Despite the Chagas disease recognition as a relevant social and economic problem in several Latin America countries [
9], it is surprising that very few data on the BZN properties are available in the vibrational [
8] and in the UV-vis optical optical absorption spectroscopy fields [
10,
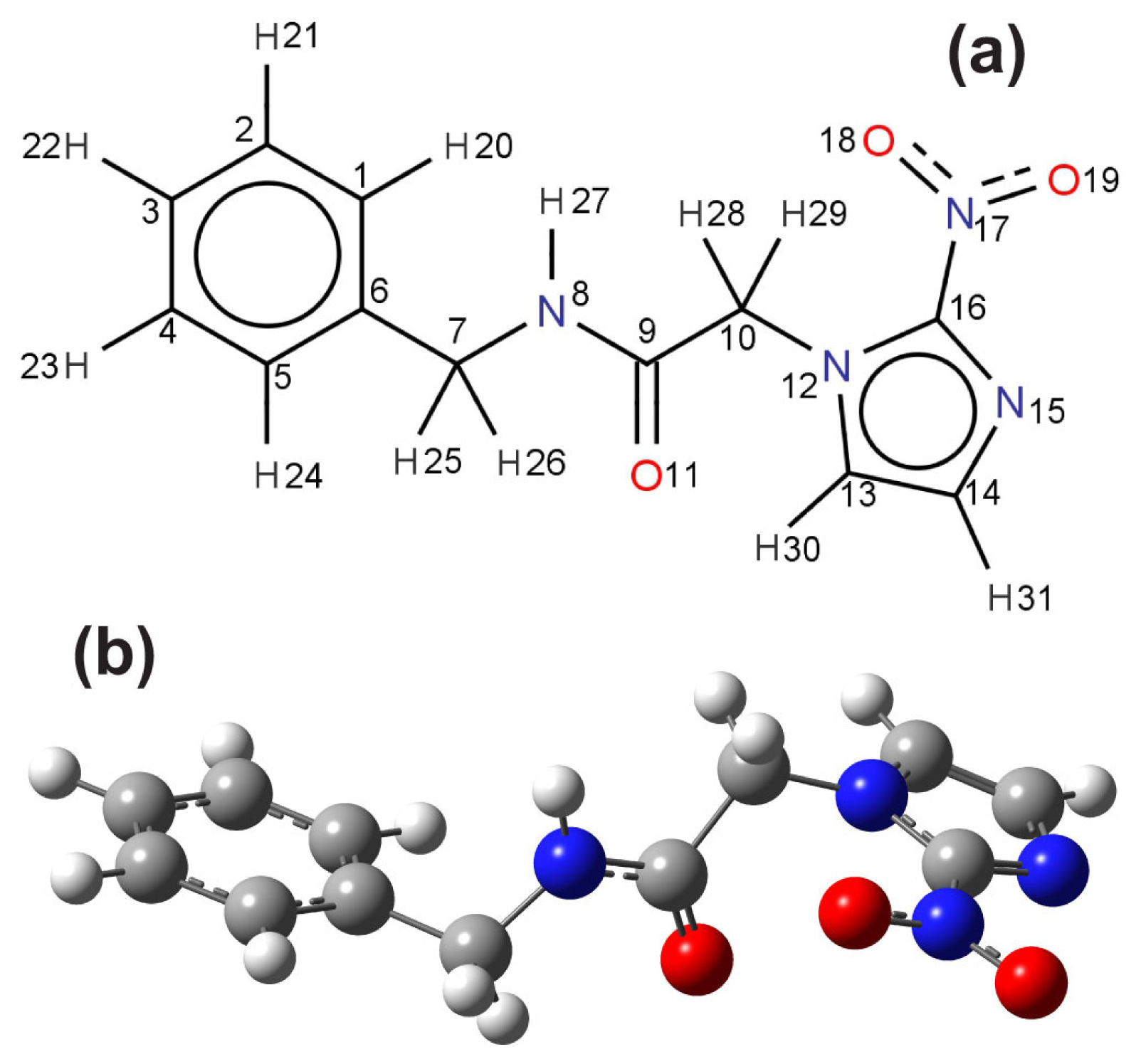
11]. As a matter of fact, even the benznidazole conformation in the solid state was resolved only recently through X-ray diffraction measurements carried out by Soares-Sobrinho
et al. [
12]. The benznidazole conformation in the crystalline state was described in terms of the relative orientation of three planar fragments,
i.e., the groups imidazol, benzyl, and acetamide (see
Figure 1 below). On the other hand, preliminary
1H Nuclear Magnetic Resonance (NMR) and vibrational (infrared and Raman) spectroscopic data in the 500–3500 cm
−1 range were published only two years ago by Soares-Sobrinho
et al. [
8]. For the latter, vibrational band assignments were performed without the aid of theoretical computations of the benznidazole molecular properties. It is worth to mention that Silva
et al. [
11] have optimized the BZN structure using density functional theory (DFT) calculations without publishing their structural data. They have also measured the UV-vis optical absorption spectra of BZN in water, ethanol and acetone, and have argued that their experimental electronic absorption spectra data are in agreement with their TD-DFT computations [
11], but without exhibiting any plot depicting their measured UV-vis and simulated spectra, and not giving a completely adequate description of their experimental and theoretical procedures.
It is quite regrettable that improved experimental or theoretical information on benznidazole structural conformers in vacuum or solvated are still missing nowadays. Our research group has recently measured the benznidazole vibrational properties in vacuum and at room temperature to allow for an improved DFT-based assignment of its vibrational modes. Through a scan of a large set of structural configurations, we have found two benznidazole conformers with very close total energy values: BZN1, which has the smallest energy, has the benzene and imidazole rings in opposite sites relative to the acetamide group, its structure resembling that of the crystal state [
12]; the second smaller energy conformation, BZN2, has the benzene and imidazole rings facing each other. A comparison between the solid-state measured and the BZN1 and BZN2 calculated spectra at 1350–1450 cm
−1 allowed us to confirm that the benznidazole samples used in the IR and Raman measurements have only the BNZ1 structure of the crystal. Besides, we performed a molecular docking study of both conformers in the sterol 14
α-demethylase CYP51 enzyme demonstrating that BZN1 has the best pose, suggesting not only the benznidazole mode of action at the proteic level, but also a smaller pharmacological activity of the BZN2 conformation (in comparison to BZN1) with respect to their role as an anti-fungal azole inhibitor of CYP51 for the treatment of Chagas disease [
13].
In this work we characterize the antitrypanocidal drug benznidazole in water through optical absorptions measurements and ab initio molecular computing, with both DFT and TD-DFT calculations being performed for an improved understanding of the experimental UV-vis optical absorption data. The measurements were performed for several BZN concentrations in water, with the obtained spectra showing a characteristic broad absorption peak around 3.80 eV. The spectra in the 3.0–6.0 eV energy range were deconvoluted into six Gaussian absorption components. A computational scan was performed to find out the possible benznidazole conformers in the framework of the polarized continuum model (PCM) to take into account the water environment. Besides, the DFT electronic structure of the lowest energy BZN conformer in water is calculated, as well as its HOMO, LUMO energy levels and orbitals, which are related to the main optical absorption transitions. Finally, an improved description of the optical absorption spectra through TD-DFT calculations is presented to explain the measured benznidazole UV-vis optical absorption data, with the most relevant optical transitions strongly correlated to the deconvoluted Gaussian peaks.
3. Experimental
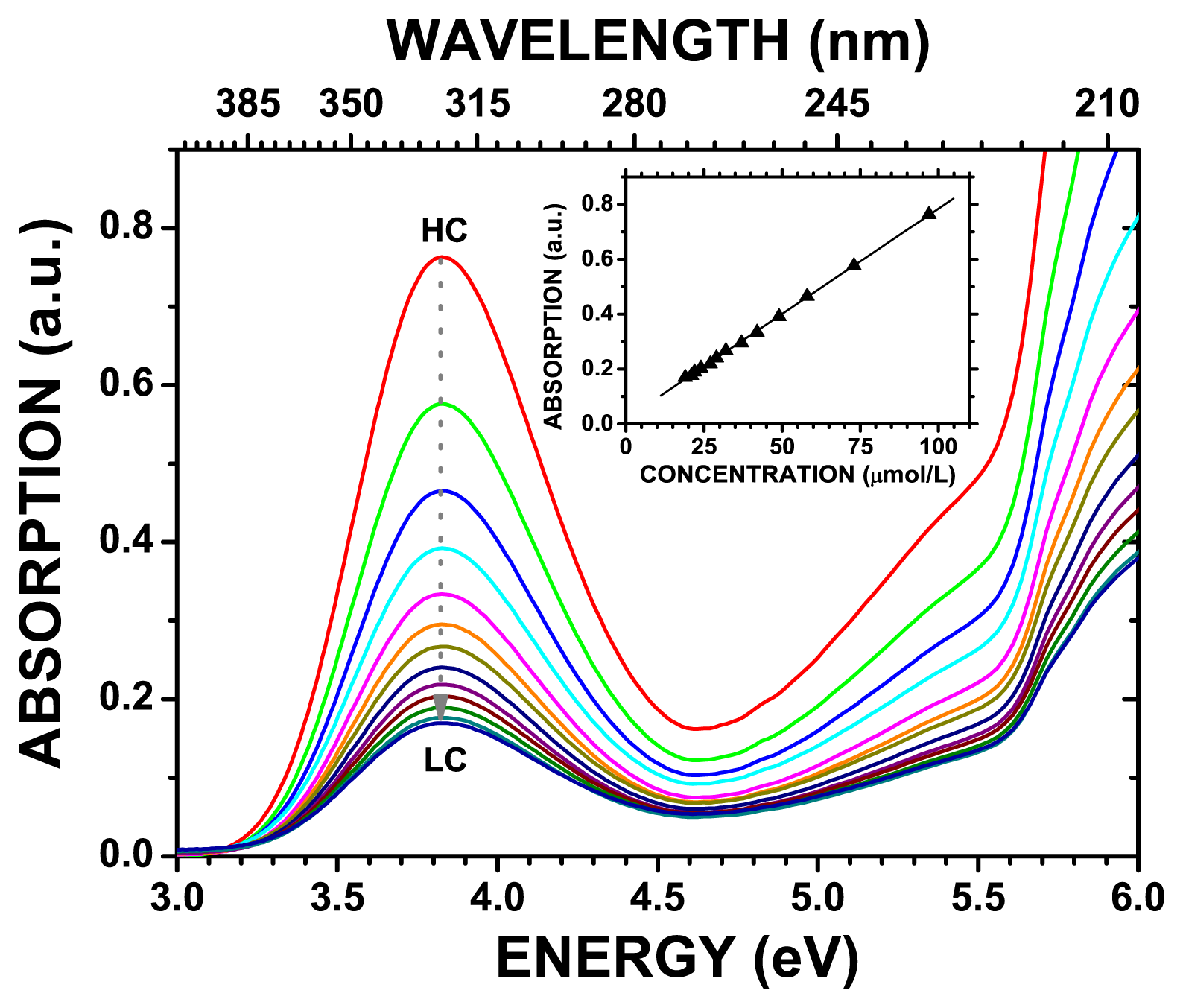
The measured 3.0–6.0 eV UV-vis optical absorption spectra of benznidazole in water for concentrations in the 19.5–146.0 μmol/L range are depicted in
Figure 1, where it is shown that the highest (smallest) absorption occurs for the 146.0 (19.5) μmol/L benznizadole concentration. One can observe a broad band centered around 3.8 eV whose intensity gradually diminishes when the benznidazole concentration in water becomes smaller. This decrease is practically linear, as shown in the inset of
Figure 1, being fitted by the equation
y =
b +
a ×
x = 0.0177 + 0.0077 ×
x, where
y is the intensity of the optical absorption and
x the benznidazole concentration in water (μmol/L). The
a and
b parameters have estimated uncertainties Δ
a = 0.0024 and Δ
b = 5.2
E − 5, with statistical deviation of 0.9997. The deconvolution of the UV-vis absorption spectra of benznidazole in water with Gaussian curves gives rise to peaks centered at energies which can be compared with the DFT calculated HOMO-LUMO transition energies, as well as with the TD-DFT excitations with more intense oscillator strengths.
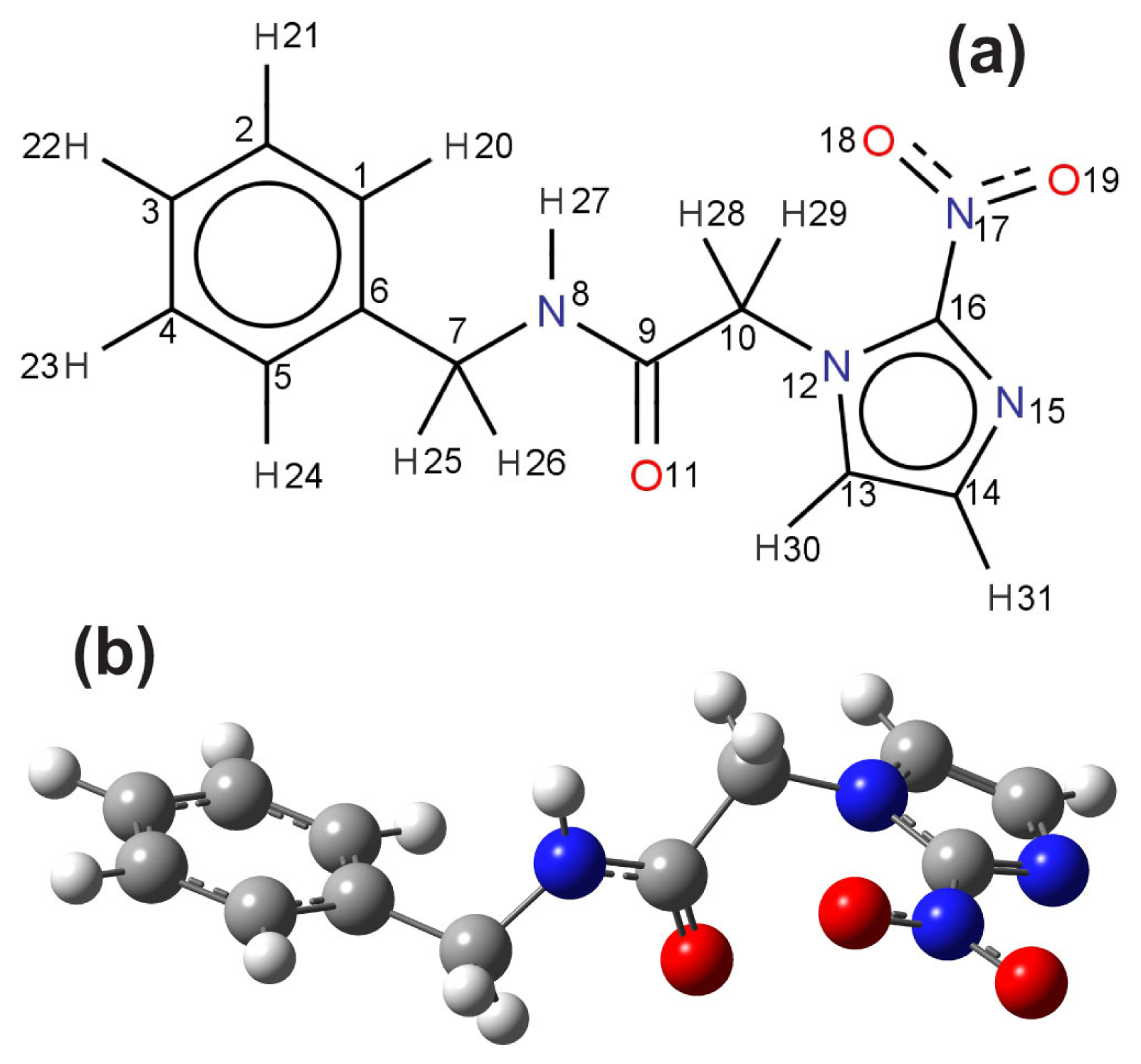
The planar molecular structure of benzonidazole and its atomic labels are shown in
Figure 3a. As in the vacuum case, a scan of a multitude of benznidazole conformations in water (described within the scope of the PCM model) suggests the existence of two conformers with formation energies differing by less than
kT at 300 K. However, only the lowest energy conformer was considered in this work for the UV-vis optical absorption calculations, which is depicted in
Figure 3b.
Tables S1–S3 of the Supplementary Material show that the bond lengths, angles and dihedral angles of the X-ray benznidazole crystal structure are not very distinct from the DFT-calculated corresponding values for the water solvated benznidazole conformer we have considered for the TD-DFT UV-vis optical absorption calculations.
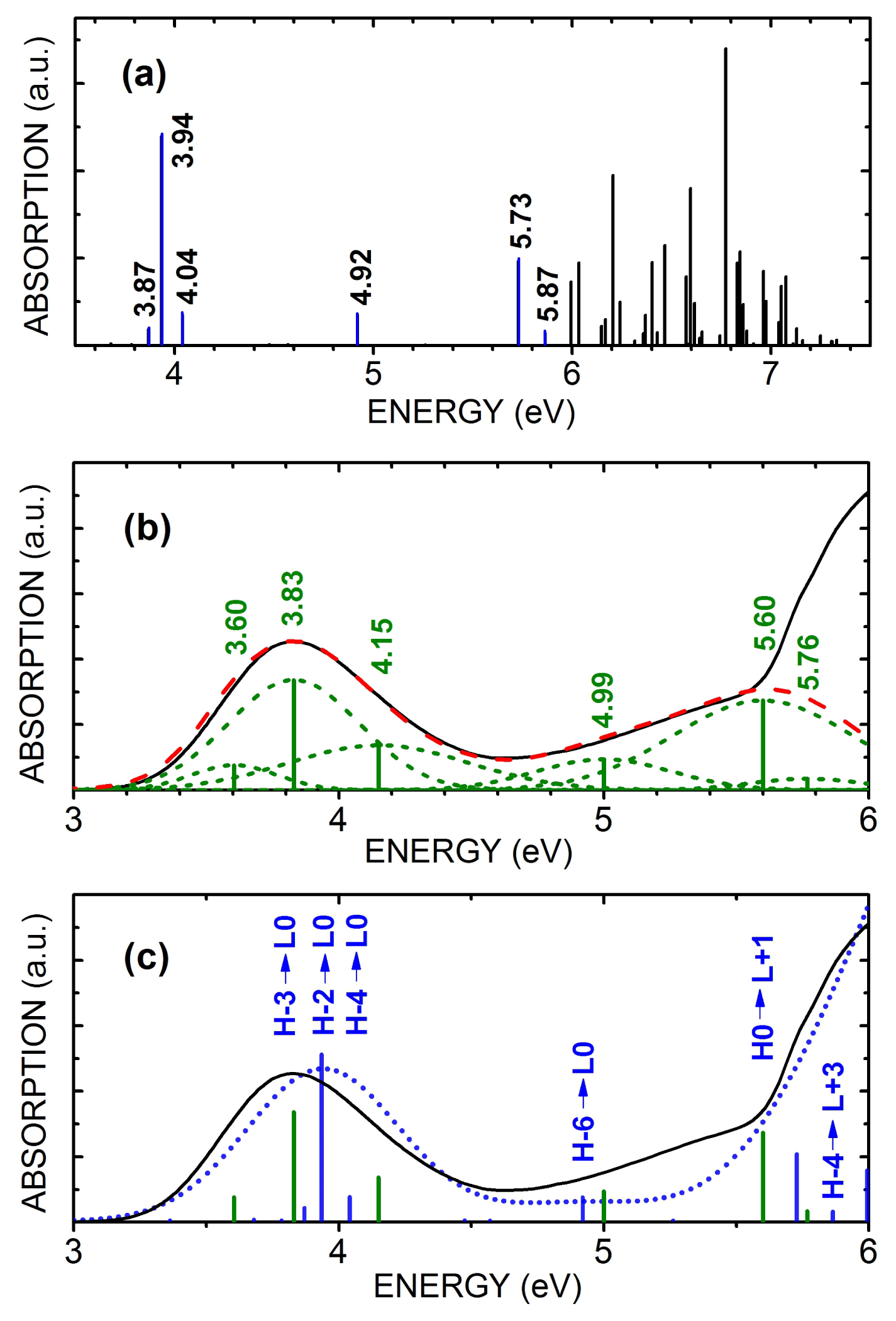
Figure 2a depicts the TD-DFT calculated excited states oscillator strengths for benznidazole within the water PCM model. There is a cluster of optically active transitions around 3.94 eV, a single active transition at 4.921 eV, and a broad set of optical transitions within the 5.0–7.5 eV range. The optical transitions around 3.94 eV involves eight molecular states, according with
Table 1, but only three terms contribute significantly to the oscillator strength: H-3→L0, with 67.4% of the 0.0191 oscillator strength and transition energy of 3.87 eV; H-2→L0, with 85.5% of the 0.0349 oscillator strength and transition energy of 3.94 eV; and H-4→L0, with 63.9% of the 0.0349 oscillator strength and transition energy of 4.04 eV The single state at 4.92 eV has contributions from two transitions, the most important being H-6→L0, with 92.5% of the 0.0343 oscillator strength and transition energy of 4.92 eV The most active states in the 5.0–6.0 eV range have two significant transitions: H0→L+1, with 63.7% of the 0.0963 oscillator strength and transition energy of 5.73 eV, and H-4→L+3, with 18.2% of the 0.0138 oscillator strength and transition energy of 5.87 eV.
The high concentration (HC) water solvated benznidazole has its optical absorption spectrum depicted in
Figure 2b. Six Gaussian peaks were used to fit the spectral curve in the 3.0–6.0 eV range, with the best adjust being observed by centering the peaks at 3.60, 3.83, 4.15, 4.99, 5.60, and 5.76 eV. The curve resulting from the addition of the Gaussian curves exhibited a 0.999 mean error deviation from experimental data, and it is shown in
Figure 2b as a dashed red line; the theory-experiment shifts obtained for the TD-DFT calculated transition energies are −6.9%, −2.2%, +3.5%, +1.4%, −0.23%, and −1.9%. Thus, the TD-DFT calculated transition energies are in remarkable agreement with those obtained from the Gaussian deconvolution of the measured UV-vis optical absorption of the water dissolved antitrypanocidal drug benznidazole at the 146.0 μmol/L concentration. This is easily seen by contrasting the experimental (solid black) and theoretical (short-dotted blue) curves depicted in
Figure 2c, together with the center-peak energy values of the Gaussian curves fitted to experiment in
Figure 2b (green vertical bars). In
Figure 2c, one can see also the six most important TD-DFT calculated contributions for the excited states involved in the optically active transitions (blue vertical bars).
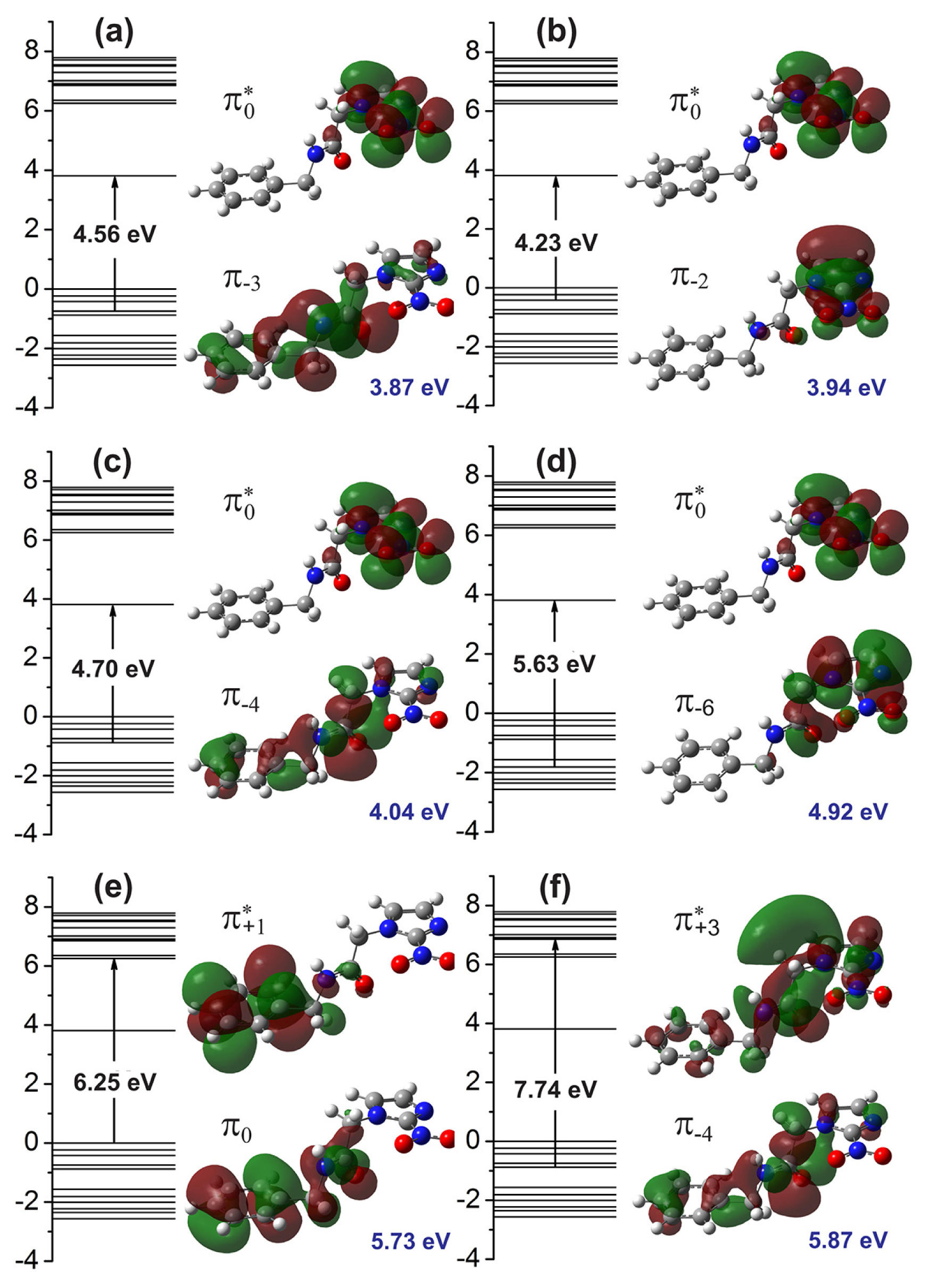
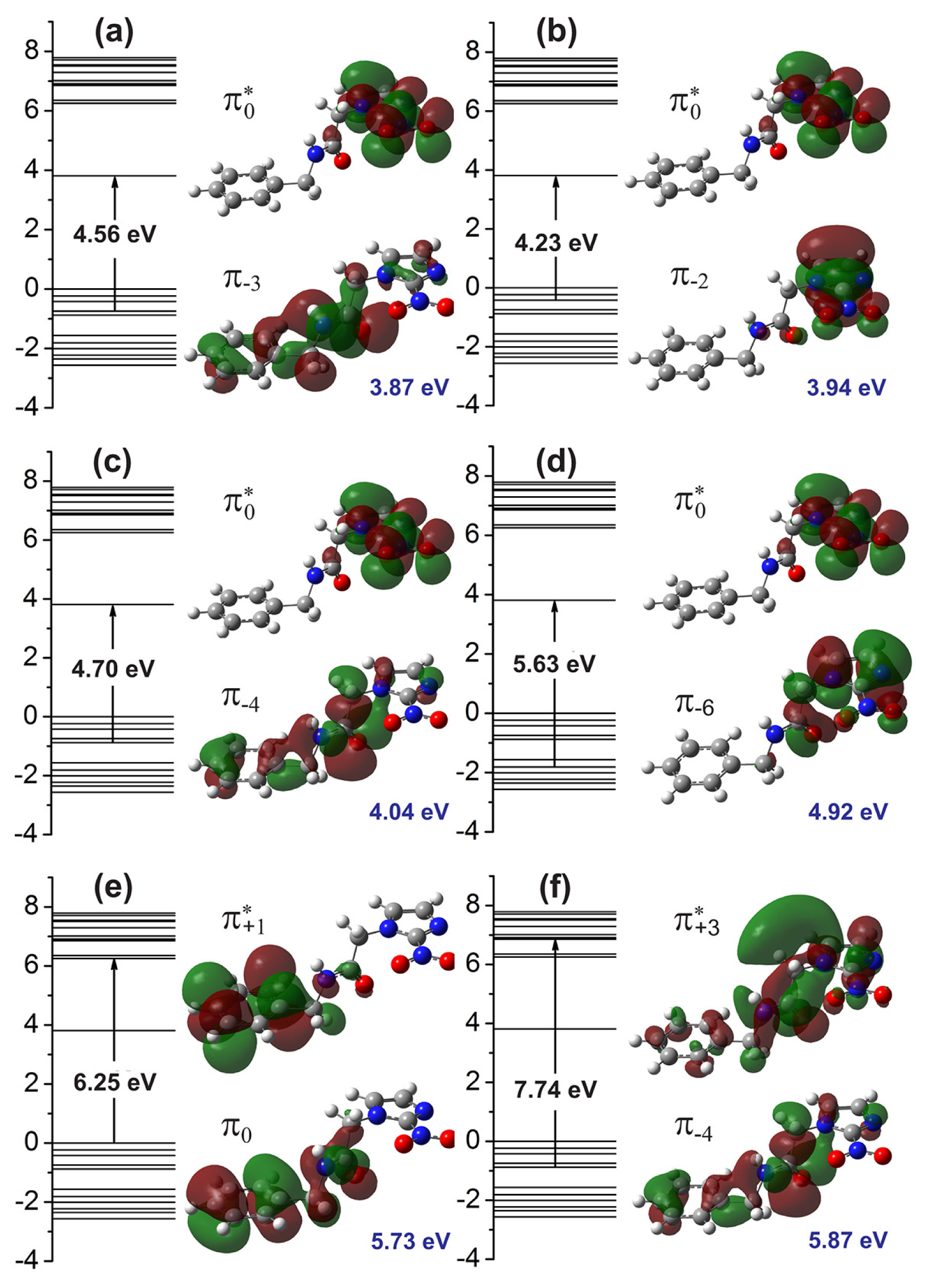
Figure 4 presents the molecular orbitals involved in the main optical absorption bands. For the 3.87 eV excitation energy (
Figure 4a), we have a strong contribution from the
transition (4.56 eV), where the
π−3 orbital is delocalized across the benzene and acetamide regions, while the
orbital (LUMO) is localized at the imidazole ring. The excitations to 3.94 eV and 4.92 eV, on the other hand (
Figure 4b,d), originates mainly from
and
transitions, where the
π−2 and
π−6 molecular orbitals are strongly localized at the imidazole group. The excitation energies of 4.04 eV is mainly assigned to a
configuration (4.70 eV), with the
π−4 wavefunction being spread across the whole extension of the benznidazole molecule. The 5.73 eV absorption peak, on the other hand, involves the HOMO (
π0) orbital, mainly localized at the benzene and acetamide regions, and the
orbital, mainly localized at the benzene ring (Kohn-Sham orbital energy gap of 6.25 eV). Finally, the 5.87 eV transition is originated from a
configuration (7.74 eV), where the
state is mostly delocalized at the imidazole and acetamide groups.
In truth, benznidazole has low solubility in water (400 mg/L at 37 degrees Celsius), which precludes the preparation of liquid dosage forms and contributes to increase its toxicity. Several studies have been published proposing new forms to improve its solubility and, consequently, its bioavailability. For example, Lamas
et al. [
18] have investigated the cosolvation of BZN with ethyl alcohol, propylene glycol, polyethylene glycol (PEG) 400, benzyl alcohol, diethylene glycol monoethyl ether and some surfactants, obtaining formulations which remained stable for at least 1.5 years. A study on the release mechanism of BZN in solid dispersions with PEG 6000 and polyvinylpirrolydone K-30 (PVP K-30) [
19] revealed a strong interaction between BZN and the polymers, especially for PVP, which has improved BZN solubility in water. Complexation of BZN with ruthenium has lead to a compound with increased activity and lower acute toxicity
in vitro and
in vivo [
11]. It was shown that Hydroxypropyl-
β-cyclodextrin (CD) produces a great improvement (20×) in the aqueous solubility of BZN, with solid systems prepaired with BNZ and CD exhibiting physical-chemical reactions which help to promote the dissolution rate [
20]. Notwithstanding that, ordinary benznidazole administration is nowadays performed using 100 mg tablets which are rapidly absorbed by the gastrointestinal tract into the bloodstream, where the drug circulates in its pure form. As a matter of fact, our research group has performed biological essays testing the action of BZN on T. Cruzi using BZN concentrations in water similar to the values used to perform the optical absorption measurements presented in this work, which motivated us to consider only water as a solvent in the TD-DFT computations. It would be interesting to implement similar measurements and theoretical calculations for BZN in other solvents than water, and we are taking the first steps to carry out a more detailed study testing the effect of different solvation environments on the optical absorption spectrum of BZN (the results thus obtained will be published in the near future). In particular, preliminary data on the optical absorption of BZN in a solution of acetonitrile were shown to be almost identical to the case of BZN in aqueous solution, indicating that a solvation change does not necessarily modify the optical absorption by a significant amount.
4. Conclusions
In summary, we have presented the results of optical absorption measurements carried out for water solvated benznidazole (BNZ), the drug of choice for the treatment of Chagas disease, as well as the results of time-dependent density functional theory (TD-DFT) calculations to interpret them. An excellent agreement between theory and experiment data was achieved, with the measured absorption peaks being fitted by six Gaussian curves centered at 3.60, 3.83, 4.15, 4.99, 5.60, and 5.76 eV. The TD-DFT oscillator strengths predicted the smallest energy absorption bands at 3.87, 3.94, 4.04, 4.92, 5.73, and 5.87 eV. The main experimental absorption peak at 3.83 eV is assigned mainly to a
transition involving an orbital delocalized along the benzene and acetamide regions and another one (LUMO) located mainly at the benzene ring of benznidazole. One conclude that the TD-DFT is able to reproduce very satisfactorily the experimental UV-vis optical absorption band shape of benznidazole in water. The results are useful for the characterization of benznidazole dissolved in water, an aid to the development of liquid benznidazole formulations mainly indicated for children and the elderly suffering with Chagas disease.



{kind=link}
{kind=link}
{kind=link}
{kind=link}