3. Experimental
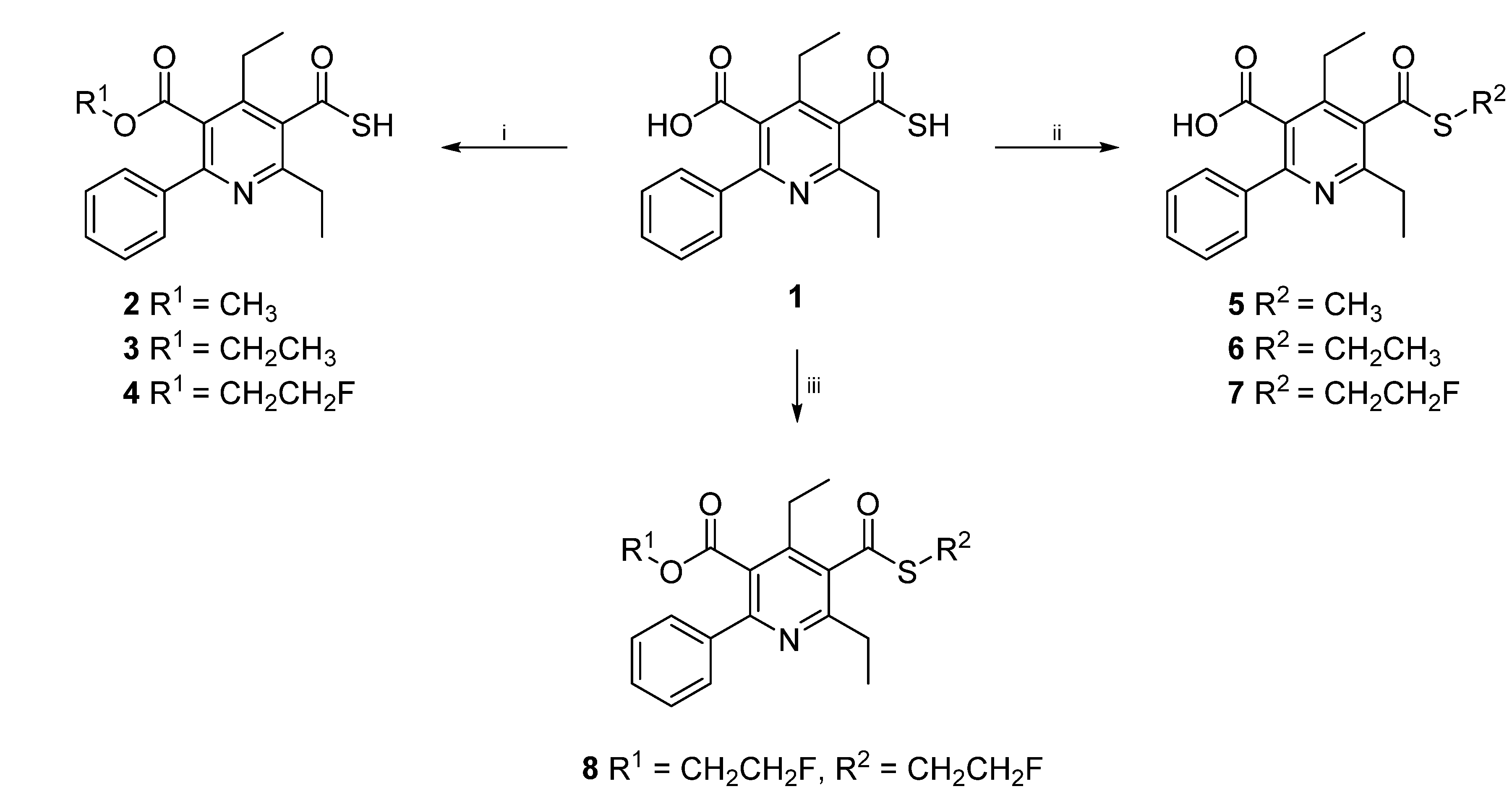
3.2. General Procedure for the Synthesis of Compounds 2–4
4,6-Diethyl-2-phenyl-5-(sulfanylcarbonyl)pyridine-3-carboxylic acid (0.10 g, 0.32 mmol), Cs2CO3 (0.22 g, 0.66 mmol) and the according trifluoromethanesulfonate (0.72 mmol) were suspended in CH3CN (10 mL). The reaction mixture was heated in the microwave oven to 150 °C at 300 W for 10 min (3, 4) or 15 min (2), respectively. Then the solvent was removed under reduced pressure and the product purified by column chromatography.
2,4-Diethyl-5-(methoxycarbonyl)-6-phenylpyridine-3-carbothioic S-acid (2). Purification: RP-18 silica gel, CH3CN/H2O 9/1. Yield: 30 mg (29%), white crystals, m.p. 161–162 °C; 1H-NMR (200 MHz, CDCl3) δ (ppm): 7.66 (q, 2H, J = 3.9 Hz), 7.45 (t, 3H, J = 3.42 Hz), 3.66 (s, 3H), 3.06 (t, 2H, J = 7.46 Hz), 2.92 (m, 2H), 1.40 (t, 3H, J = 7.44 Hz), 1.28 (t, 3H, J = 7.32 Hz); 13C-NMR (50 MHz, CDCl3) δ (ppm): 189.4, 168.6, 160.1, 157.9, 149.2, 139.3, 130.5, 129.2, 128.4, 128.2, 126.5, 52.4, 29.4, 15.8, 14.1; IR (KBr): v (cm−1) 3427, 2974, 2940, 2877, 1728, 1559, 1439, 1403; MS: m/z (%): 328 (M+, 1.29), 327 (5), 297 (21), 296 (100), 264 (5), 236 (10), 208 (5), 77 (5); HRMS: m/z calculated for C18H18NO3S (M+–SH): 296.1287. Found: 296.1289; CHN: Anal. calculated for C18H19NO3S: C, 65.63; H, 5.81; N, 4.25. Found: C, 65.37; H, 5.50; N, 4.41.
5-(Ethoxycarbonyl)-2,4-diethyl-6-phenylpyridine-3-carbothioic S-acid (3). Purification: RP-18 silica gel, CH3CN/H2O 9/1. Yield: 120 mg (50%), yellow oil; 1H-NMR (200 MHz, CDCl3): δ (ppm) 7.63 (m, 2H), 7.44 (m, 3H), 4.14 (q, 2H, J = 7.20 Hz), 3.05 (q, 2H, J = 7.44 Hz), 2.93 (q, 2H, J = 7.44 Hz), 1.40 (t, 3H, J = 7.32 Hz), 1.29 (t, 3H, J = 7.46 Hz), 1.03 (t, 3H, J = 7.20 Hz); 13C-NMR (50 MHz, CDCl3): δ (ppm) 189.3, 167.9, 160.0, 158.0, 149.5, 139.1, 130.6, 129.2, 128.4, 127.0, 61.8, 29.3, 24.4, 15.9, 14.3, 13.6; IR (KBr): ν (cm−1) 3419, 3058, 2971, 2939, 2875, 1724, 1559, 1475, 1458, 1445, 1403, 1374, 1344, 1282, 1250, 1215, 1170, 1145, 1092, 1077, 1016, 963; MS: m/z (%) 344 (2), 343 (3), 341 (43), 312 (19), 311 (17), 310 (100), 282 (29), 254 (13), 236 (11), 77 (14); HRMS: m/z calculated for C19H21NO3S: 343.1242. Found: 343.1235.
2,4-Diethyl-5-[(2-fluoroethoxy)carbonyl]-6-phenylpyridine-3-carbothioic S-acid (4). Purification: RP-18 silica gel, CH3CN. Yield: 113 mg (95%), colorless crystals, m.p. 45–47 °C; 1H-NMR (300 MHz, CDCl3): δ (ppm) 7.64 (m, 2H, Ph H-2/H-6), 7.46 (m, 3H, Ph H-3/H-4/H-5), 4.39/4.29 (m, 2H, FCH2), 4.34/4.29 (m, 2H, CH2O), 3.05 (q, 2H, J = 7.50 Hz, C-2CH2), 2.93 (q, 2H, J = 7.50 Hz, C-4CH2) 1.40 (t, 3H, J = 7.50 Hz, C-2CH2CH3), 1.29 (t, 3H, J = 7.50 Hz, C-4CH2CH3); 13C-NMR (75 MHz, CDCl3): δ (ppm) 189.3 (COSH), 167.9 (COO), 160.4 (C-2), 158.3 (C-6), 149.5 (C-4), 139.3 (Ph C-1), 130.6 (C-3), 129.3 (Ph C-4), 128.5 (Ph C-3/C-5), 128.3 (Ph C-2/ C-6), 126.2 (C-5), 80.5 (d, FCH2, 1JCF = 172.0 Hz), 64.5 (d, CH2O 2JCF = 20.2 Hz), 29.5 (C-2CH2), 24.4 (C-4CH2), 15.9 (C-4CH2CH3), 14.3 (C-2CH2CH3); IR (KBr): ν (cm−1) 3439, 2975, 2938, 2877, 1731, 1556, 1495, 1448, 1403, 1377, 1278, 1250, 1166, 1144, 1077, 1060, 1033, 960; MS: m/z (%) 362 (1), 361 (3), 359 (31), 329 (21), 328 (100), 312 (14), 254 (15), 236 (12), 77 (20), 47 (48); CHN: Anal. calculated for C19H20FNO3S∙0.2 H2O: C, 62.52; H, 5.63; N, 3.84. Found: C, 62.45; H, 5.44; N, 3.73.
3.3. Alternative Method for the Synthesis of 2,4-Diethyl-5-(methoxycarbonyl)-6-phenylpyridine-3-carbothioic S-acid (2)
To a stirred solution of 4,6-diethyl-2-phenyl-5-(sulfanylcarbonyl)pyridine-3-carboxylic acid (0.20 g, 0.63 mmol) in toluene/MeOH (9/6), trimethylsilyldiazomethane (0.4 mL, 0.63 mmol) was added dropwise until the yellow color of the mixture persisted. Thereafter, the solution was stirred for another 30 min at room temperature. The solvent was removed by reduced pressure and product 2 purified by reversed phase column chromatography (RP-18 silica gel, CH3CN/H2O 9/1). Yield: 141 mg (68%).
3.4. General Procedure for the Synthesis of Compounds 5‒7
4,6-Diethyl-2-phenyl-5-(sulfanylcarbonyl)pyridine-3-carboxylic acid (0.10 g, 0.32 mmol), NaI (0.48 g, 3.17 mmol) and the according trifluoromethanesulfonate (0.72 mmol) were dissolved in DMF (10 mL). The reaction mixture was heated in the microwave oven at 300 W and 100 °C for 10 min (6, 7) or 15 min (5), respectively. The solvent was removed under reduced pressure and the crude product purified by column chromatography.
4,6-Diethyl-5-[(methylsulfanyl)carbonyl]-2-phenylpyridine-3-carboxylic acid (5). Purification: silica gel 60, petrol ether/EtOAc 8/2. Yield: 52 mg (49%), colorless crystals, m.p. 243–244 °C; 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 13.63 (s, 1H), 7.55 (q, 5H, J = 3.54 Hz), 2.69 (m, 7H), 1.19 (m, 6H); 13C-NMR (50 MHz, DMSO-d6) δ (ppm): 195.3, 169.3, 157.8, 155.0, 146.4, 139.1, 132.7, 129.0, 128.3, 127.9, 28.3, 23.8, 15.6, 13.8, 12.4; IR (KBr): v (cm−1) 3441, 2983, 2938, 2868, 2492, 1921, 1712, 1667, 1554; MS: m/z (%): 330 (M+, 0.12), 298 (1), 283 (17), 282 (100), 236 (6), 180 (5), 127 (7), 77 (10), 47 (10), 45 (6); HRMS: m/z calculated for C18H19NO3S: 329.1086. Found: 329.1076; CHN: Anal. calculated for C18H19NO3S: C, 65.63; H, 5.81; N, 4.25. Found: C, 65.64; H, 5.81; N, 4.06.
4,6-Diethyl-5-[(ethylsulfanyl)carbonyl]-2-phenylpyridine-3-carboxylic acid (6). Purification: silica gel 60, petrol ether/EtOAc 8/2.Yield: 94 mg (85%), colorless crystals, m.p. 200–202 °C; 1H-NMR (300 MHz, DMSO- d6): δ (ppm) 13.20 (br s, 1H), 7.65 (m, 2H), 7.45 (m, 3H), 3.12 (q, 2H, J = 7.30 Hz), 2.76 (q, 2H, J = 7.50 Hz), 2.66 (q, 2H, J = 7.50 Hz), 1.32 (t, 3H, J = 7.50 Hz), 1.24 (t, 3H, J = 7.50 Hz), 1.17 (t, 3H, J = 7.50 Hz); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 194.8, 169.2, 155.0, 146.2, 139.1, 132.6, 132.6, 128.3, 128.2, 127.9, 28.2, 24.2, 23.6, 15.5, 14.4, 13.7; IR (KBr): v (cm−1) 3443, 2977, 2491, 1925, 1711, 1661, 1555, 1453, 1413, 1376, 1260, 1184, 1149, 1078; MS: m/z (%) 344 (1), 343 (1), 327 (1), 326 (4), 283 (18), 282 (100), 236 (5), 180 (5, 126 (7), 91 (7), 77 (11); CHN: Anal. calculated for C19H21NO3S: C, 66.45; H, 6.16; N, 4.08. Found: C, 66.18; H, 6.21; N, 3.96.
4,6-Diethyl-5-{[(2-fluoroethyl)sulfanyl]carbonyl}-2-phenylpyridine-3-carboxylic acid (7). Purification: RP-18 silica gel, CH3CN/H2O 9/1. Yield: 30 mg (52%), colorless crystals, m.p. 195–196 °C; 1H-NMR (200 MHz, DMSO-d6): δ (ppm) 13.67 (s, 1H), 7.65 (m, 2H), 7.46 (t, 3H, J = 3.42 Hz), 4.79 (t, 1H, J = 5.68 Hz), 4.55 (t, 1H, J = 5.68 Hz), 3.58 (t, 1H, J = 5.68 Hz), 3.46 (t, 1H, J = 5.54 Hz), 2.71 (m, 4H), 1.20 (m, 6H); 13C-NMR (50 MHz, DMSO-d6): δ (ppm) 194.2, 169.2, 157.7, 155.2, 146.4, 139.1, 132.4, 129.0, 128.3, 128.0, 81.6 (d, 1JCF = 166.5 Hz), 29.9 (d, 2JCF = 20.7 Hz), 28.3, 23.7, 15.6, 13.8; IR (KBr): v (cm−1) 3446, 2974, 2454, 1966, 1708, 1671, 1555, 1412, 1287, 1183; MS: m/z (%) 283 (16), 282 (100), 77 (29), 69 (14), 59 (20), 45 (15), 43 (20), 41 (16); CHN: Anal. calculated for C19H20FNO3S: C, 63.14; H, 5.58; N, 3.88. Found: C, 63.04; H, 5.54; N, 3.73.
3.5. Synthesis of 2-Fluoroethyl 4,6-Diethyl-5-{[(2-fluoroethyl)sulfanyl]carbonyl}-2-phenylpyridine-3-carboxylate (8)
4,6-Diethyl-2-phenyl-5-(sulfanylcarbonyl)pyridine-3-carboxylic acid (0.073 g, 0.23 mmol), NaI (0.348 g, 2.3 mmol) and 2-fluoroethyl trifluoromethanesulfonate (0.11 g, 0.56 mmol) were dissolved in DMF (10 mL). The mixture was heated in the microwave oven at 600 W and 170 °C for 5 min. The solvent was then removed under reduced pressure and the crude product purified by RP-18 column chromatography (CH3CN/H2O 9/1). Yield: 67 mg (72%), orange oil; 1H-NMR (200 MHz, CDCl3) δ (ppm): 7.60 (m, 2H), 7.43 (m, 3H), 4.77 (t, 1H, J = 5.99 Hz), 4.54 (t, 1H, J = 5.94 Hz), 4.42 (m, 1H), 4.34 (q, 1H, J = 2.48 Hz), 4.18 (d, 2H, J = 5.18 Hz), 3.52 (t, 1H, J = 5.99 Hz), 3.41 (t, 1H, J = 5.94 Hz), 2.87 (q, 2H, J= 7.49 Hz), 2.73 (q, 2H, J = 7.53 Hz), 1.34 (t, 3H, J = 7.51 Hz), 1.24 (t, 3H, J = 7.57 Hz); 13C-NMR (50 MHz, CDCl3) δ (ppm): 194.3, 168.1, 159.6, 157.5, 148.2, 139.6, 132.6, 129.0, 128.4, 128.3, 126.0, 81.3 (d, FCH2CH2O, 1JCF = 170.5 Hz), 80.5 (d, FCH2CH2S, 1JCF = 170.2 Hz), 64.1 (d, CH2O 2JCF = 20.3 Hz), 30.0 (d, CH2S 2JCF = 22.2 Hz), 29.2, 24.2, 15.7, 14.0; IR (KBr): v (cm−1) 3447, 2977, 2879, 1734, 1675, 1558, 1496; MS: m/z (%): 407 (M+, 0.59), 329 (21), 328 (100), 282 (6), 254 (6), 236 (5), 47 (6); HRMS: m/z calculated for C21H24O3F2NS: 408.1445. Found: 408.1450.

 and
and {kind=link}
{kind=link}