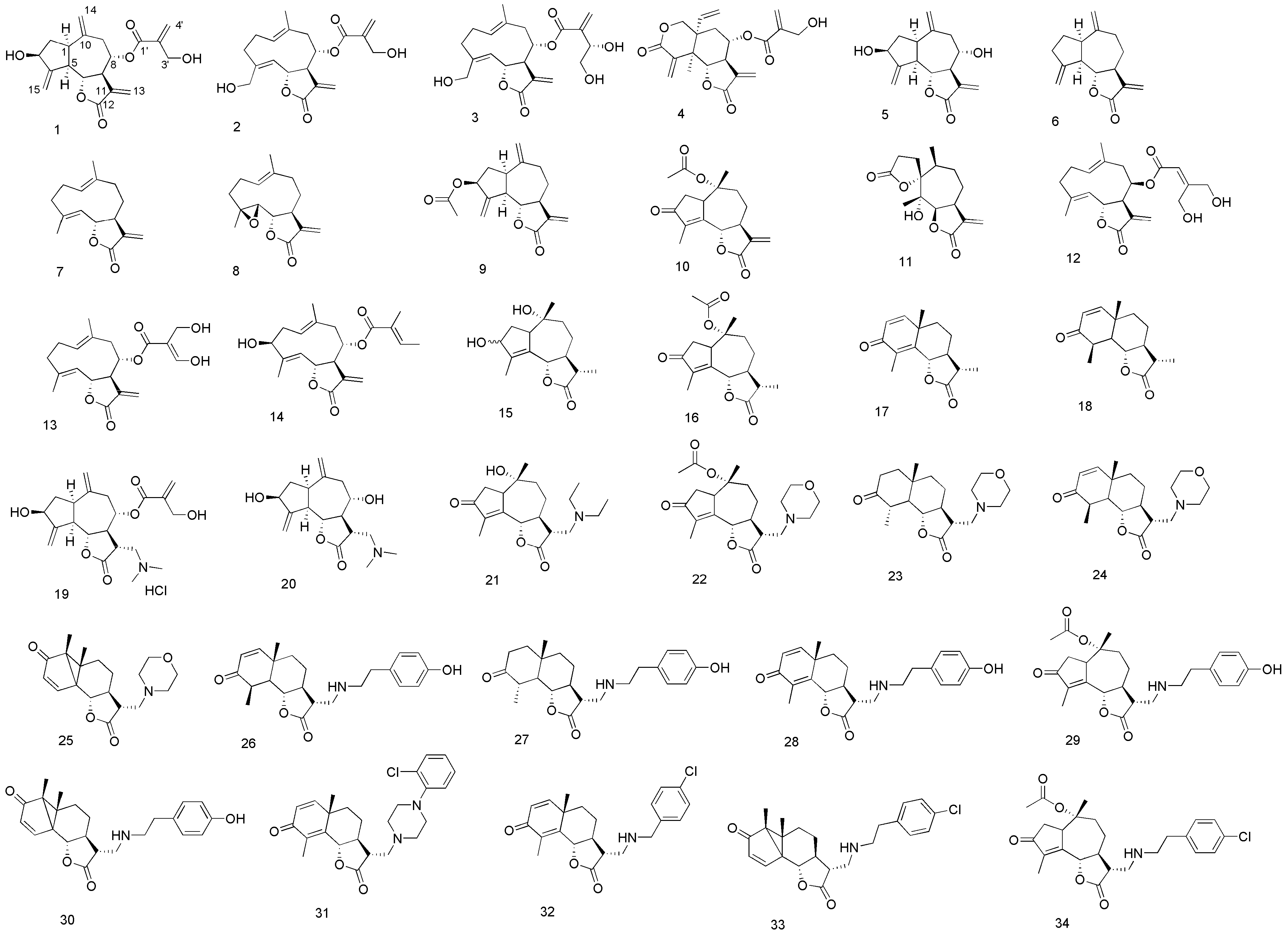
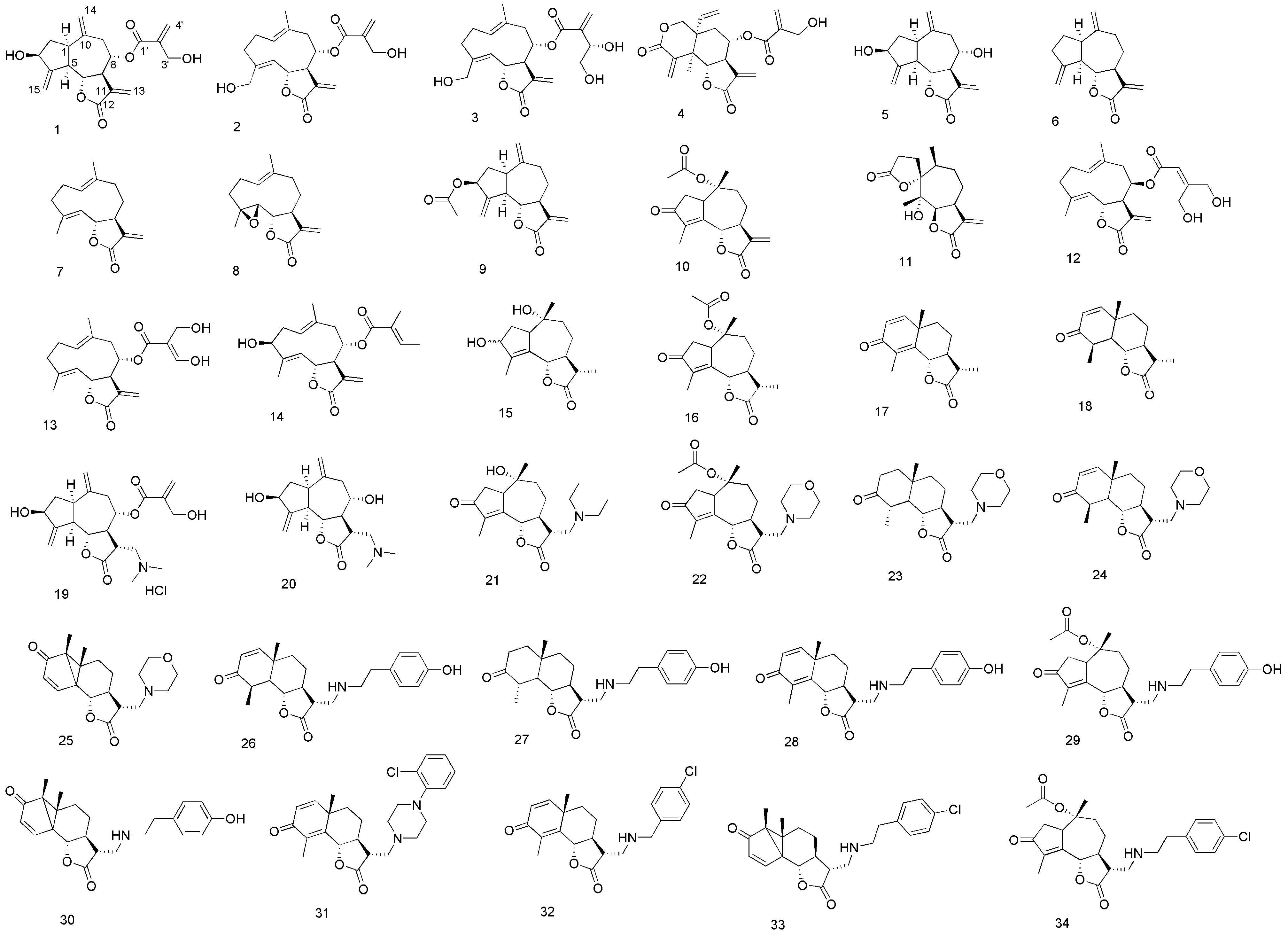
3.3. Syntheses and Spectral Data of Analogues 15, 21–34
3-Hydroxyisophotosantonin (15). A solution of O-acetylisophotosantonin (1.475 g, 4.813 mmol) in MeOH (49 mL) at 0 °C was treated with sodium borohydride (0.293 g, 7.750 mmol) carefully. The reaction was left at 0 °C for 3 h, then left to warm to room temperature overnight. The mixture was extracted from saturated aqueous NH4Cl (50 mL) with ethyl acetate (3 × 50 mL), the extracts pooled and dried (MgSO4). The dried filtrate was concentrated to a tacky white foam, then dissolved in absolute ethanol (16 mL). 5% Aqueous KOH (150 mL) was added and the mixture stirred for 18 h at room temperature. The mixture was acidified to pH < 2 with 18% aqueous HCl, stirred for 30 min, extracted with ethyl acetate (3 × 50 mL) and washed with saturated aqueous K2CO3. Concentration yielded a yellow solid, which was recrystallized (EtOAc/hexane) to a white amorphous powder (0.313 g, 36%). NMR showed an approximately 2.2:1 mixture of secondary alcohols had been isolated. Major isomer: 1H-NMR (400 MHz, CDCl3 + CD3OD): δ 4.73 (1H, d, J = 11.0 Hz, H-6), 4.49 (1H, t, J = 6.8 Hz, H-3), 2.90 (1H, ~ br td, J = 7.5 and 1.9 Hz, H-1), 2.45 (1H, dt, J = 13.8 and 8.0 Hz, H-2a), 2.34–2.12 (2H, m, H-9a and H-11), 2.03–1.91 (2H, m, H-7 and H-9b), 1.89 (3H, s, C-10 Me), 1.65 (1H, dd, J = 16.0 and 2.3 Hz, H-8a), 1.61 (1H, td, J = 13.7 and 6.7 Hz, H-2b), 1.46–1.30 (1H, m, H-8b), 1.22 (3H, d, J = 6.9 Hz, C-11 Me), 1.03 (3H, s, C-4 Me); 13C-NMR (101MHz, CDCl3 + CD3OD): δ 178.82 (C, C-12), 143.96 (C, C-14), 131.28 (C, C-5), 82.04 (CH, C-6), 76.97 (C, C-10), 74.40 (CH, C-3), 54.31 (CH, C-1), 48.92 (CH, C-7), 44.87 (CH2, C-9), 41.34 (CH, C-11), 34.36 (CH2, C-2), 25.41 (CH2, C-8), 20.88 (CH3, C-4 Me), 13.23 (CH3, C-11 Me), 12.23 (CH3, C-10 Me); minor isomer: 1H-NMR (400MHz, CDCl3 + CD3OD): δ 4.64 (1H, d, J = 11.0 Hz, H-6), 4.54 (1H, d, J = 7.1 Hz, H-3), 1.92 (3H, s, C-10 Me), 1.22 (3H, d, J = 6.9 Hz, C-11 Me), 0.91 (3H, s, C-4 Me); 13C-NMR (101 MHz, CDCl3 + CD3OD): δ 178.71 (C, C-12), 143.79 (C, C-4), 133.32 (C, C-5), 81.71 (CH, C-6), 79.59 (C, C-10), 74.23 (CH, C-3), 55.57 (CH, C-1), 48.96 (CH, C-7), 44.27 (CH2, C-9), 41.51 (CH, C-11), 34.81 (CH2, C-2), 25.26 (CH2, C-8), 20.56 (CH3, C-4 Me), 13.23 (CH3, C-11 Me), 12.20 (CH3, C-10 Me); HRMS (ESI) calculated for C15H21O3 249.1491; found 249.1423 (MH+–H2O); and calculated for C15H21O4 265.1440; found 265.1392 (MH+–H2).
Cynaropicrin dimethylamine adduct hydrochloride 19. To a cold solution of 1 (0.50 g; 1.44 mmol) in absolute EtOH (15 mL), dimethylamine, (0.72 mL, 2.0 M solution in MeOH) was added under argon atmosphere. The solution was stirred at 5 °C for 5 h, then, after concentration of the solvent under reduced pressure, the crude compound was recrystallized from acetone/Et2O. The amino adduct (0.222 g; 0.566 mmol) was then dissolved in MeOH (5 mL) and a solution of HCl (0.45 mL; 1.25 N solution in MeOH) was added dropwise. After evaporation of the solvent compound 19 was recovered as a brown solid (0.242 g; 40%). 1H-NMR (500 MHz, CD3OD): δ 6.28 (1H, br s, H-4'a), 5.97 (1H, br s, H-4'b), 5.35 (1H, br s, H-15a), 5.33 (1H, br s, H-15b), 5.15 (2H, m, H-8, H-14a), 5.00 (1H, br s, H-14b), 4.50 (1H, dddd, J = 9.0, 8.0, 2.2 and 2.2 Hz, H-3), 4.44 (1H, m, H-6), 4.30 (2H, s, H2-3'), 3.62 (1H, m, H-13a), 3.50–3.42 (2H, m, H-11, H-13b), 3.03–2.91 (8H, m, H-1, H-5, NH(CH3)2), 2.83 (1H, dd, J = 13.4 and 5.0 Hz, H-9a), 2.72 (1H, m, H-7), 2.33 (1H, dd, J = 13.4 and 7.0 Hz, H-9b), 2.24 (1H, m, H-2a), 1.75 (1H, m, H-2b); 13C-NMR (125 MHz, CD3OD): δ 177.55 (C, C-12), 166.72 (C, C-1'), 154.17 (C, C-4), 143.95 (C, C-10), 141.84 (C, C-2'), 127.82 (CH2, C-4'), 117.58 (CH2, C-14), 111.28 (CH2, C-15), 81.77 (CH, C-6), 77.26 (CH, C-8), 73.82 (CH, C-3), 61.98 (CH2, C-3'), 58.34 (CH2, C-13), 50.74 (CH, C-5), 49.85 (CH, C-7), 44.98 (CH, C-1), 44.77 (CH3, N(CH3)2), 42.02 (CH, C-11), 40.54 (CH, C-9), 39.51 (CH2, C-2).HRMS (ESI) calculated for C21H30NO6 [M+H]+, 392.2067; found 392.2062.
Deacylcynaropicrin dimethylamine adduct 20. To a cold solution of 5 (0.050 g; 0.191 mmol) in absolute EtOH (5 mL), dimethylamine, (0.1 mL, 2.0 M solution in methanol) was added under argon atmosphere. The solution was stirred at 5 °C for 5 h and then the mixture was concentrated under reduced pressure. The crude residue was then purified by column chromatography on silica gel (CH2Cl2/MeOH 9:1) to afford compound 20 as a yellow oil (0.052 g; 88%). 1H-NMR (500 MHz, CD3OD): δ 5.32 (1H, br s, H-15a), 5.28 (1H, br s, H-15b), 5.05 (1H, br s, H-14a), 5.00 (1H, br s, H-14b), 4.48 (1H, dddd, J = 9.0, 8.0, 2.2 and 2.2 Hz, H-3), 4.16 (1H, dd, J = 9.8 and 9.7 Hz, H-6), 3.67 (1H, ddd, J = 9.0, 7.3 and 5.0 Hz, H-8), 3.02–2.80 (3H, m, H-1, H-5, H-11), 2.76 (1H, dd, J = 12.7 and 2.7, H-13a), 2.68 (1H, m, H-9a), 2.60 (1H, m, H-13b), 2.34 (6H, s, N(CH3)2), 2.26–2.14 (3H, m, H-2a, H-7, H-9b), 1.70 (1H, ddd, J = 12.8, 9.7 and 8.8 Hz, H-2b); 13C-NMR (125 MHz, CD3OD): δ 177.71 (C, C-12), 154.33 (C, C-4), 145.14 (C, C-10), 116.01 (CH2, C-14), 111.70 (CH2, C-15), 80.53 (CH, C-6), 74.00 (CH, C-3 and C-8), 60.81 (CH2, C-13), 58.34 (CH, C-7), 50.53 (CH, C-5), 46.44 (CH, C-1), 44.89 (CH3, N(CH3)2), 44.82 (CH, C-11), 43.08 (CH2, C-9), 39.49 (CH2, C-2). HRMS (ESI) calculated for C17H26NO4 [M+H]+, 308.1856; found 308.1862.
General Procedure for Preparing Lactone Methylamines: A solution of the appropriate enoate (1 eq.) in ethanol (0.1M) containing the required volatile amine (2.5 eq.) or non-volatile amine (0.6 eq.) and triethylamine (1.1–2.5 eq., for the appropriate hydrochloride salt) was heated at 85 °C under microwave irradiation set at 30 W for 30 min to 1 h, depending on the amine. All were prepared on a sufficiently small scale that the solutions could simply be concentrated and purified by column chromatography. The following compounds were produced this way:
Deacylated diethylamine adduct 21. A mixture of 15 and its lumisantonin equivalent (~2:1 mixture, 0.128 g, 0.325 mmol) and diethylamine (68 μL, 0.663 mmol) in ethanol (3.3 mL) was stirred at room temperature for 18 h, and then concentrated to a brown oil. Column chromatography (10% ethanol/ethyl acetate as eluent) afforded an orange oil (48.9 mg, 45%); Rf 0.11 (10% ethanol/ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ 4.76 (1H, br d, J = 9.6), 3.31–3.12 (1H, m), 2.82 (1H, dd, J = 3.9, 13.8), 2.68 (1H, dd, J = 6.0, 13.7), 2.61–2.48 (4H, m), 2.48–2.31 (5H, m), 2.09 (1H, d, J = 12.1), 1.97 (1H, dt, J = 3.5, 13.6), 1.82 (3H, br t), 1.79 (1H, td, J = 3.9, 13.4), 1.44–1.28 (1H, m), 0.91 (6H, t, J = 7.1), 0.91 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 208.14, 176.47, 162.00, 161.98, 142.69, 81.45, 74.35, 52.04, 50.40, 47.18, 45.76, 45.17, 44.97, 37.15, 26.11, 21.18, 11.63, 9.41; HRMS (ESI) calculated C19H30NO4 336.2175, found 336.2135 (MH+).
Morpholine adduct 23. α-Methylenesantonin (51.7 mg, 0.213 mmol), morpholine (46.5 μL, 0.534 mmol) and ethanol (2 mL) afforded, after column chromatography (2%–4% methanol/chloroform as eluent), a yellow solid (45.7 mg, 65%); Rf 0.24 (4% methanol/chloroform). Recrystallisation yielded orange needles, mp. 165–167 °C (ethyl acetate/hexane); 1H-NMR (400 MHz, CDCl3) δ 6.71 (1H, d, J = 9.9), 6.26 (1H, d, J = 9.9), 4.82 (1H, dd, J = 1.3, 11.5), 3.89–3.46 (4H, m), 2.95–2.81 (1H, m), 2.71–2.57 (2H, m), 2.57–2.47 (2H, m), 2.47–2.35 (2H, m), 2.34–2.25 (1H, m), 2.12 (3H, d, J = 1.2), 2.05 (1H, qd, J = 3.5, 11.6), 1.89 (1H, ddd, J = 2.2, 3.6, 13.4), 1.74 (1H, ddd, J = 3.8, 12.9, 25.1), 1.55 (1H, td, J = 4.5, 13.2), 1.33 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 186.22, 176.14, 154.93, 150.92, 128.52, 125.73, 81.35, 66.73, 57.70, 53.72, 51.82, 43.46, 41.12, 37.89, 25.00, 23.75, 10.80; HRMS (ESI) calculated C19H26NO4 332.1862, found 332.1835 (MH+). The resultant dienone (0.434 g, 1.334 mmol), 5% Pd-C (0.470 g), 32% hydrochloric acid (0.5 mL) and ethanol (10 mL) afforded, after column chromatography (30%–50% acetone/hexane as eluent), an orange foam (0.209 g, 48%); Rf 0.68 (20% ethanol/ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ 3.91 (1H, t, J = 10.6), 3.76–3.60 (4H, m), 2.82 (1H, dd, J = 4.3, 12.7), 2.65–2.44 (6H, m), 2.44–2.32 (3H, m), 2.19–2.08 (2H, m), 1.92–1.53 (5H, m), 1.42–1.28 (1H, m), 1.25 (3H, d, J = 6.6), 1.18 (3H, s); 13C-NMR (101MHz, CDCl3) δ 211.44, 177.55, 83.11, 66.85, 57.72, 53.88, 53.47, 51.11, 44.93, 43.36, 40.65, 40.28, 37.29, 36.32, 23.77, 18.39, 13.91; HRMS (ESI) calculated C19H29NaNO4 358.1994, found 358.2048 (M++Na+) and calculated C19H30NO4 336.2175, found 336.2152 (MH+).
Morpholine adduct 24. The appropriate enoate [
20,
21] (53.8 mg, 0.218 mmol), morpholine (17.7 μL, 0.203 mmol) and ethanol (2 mL) were mixed at 0 °C, then left to warm to room temperature for 72 h. The mixture was concentrated to afforded, after column chromatography (30% acetone/hexane as eluent), a pale yellow oil (66.0 mg, 985%);
Rf 0.25 (30% acetone/hexane);
1H-NMR (400 MHz, CDCl
3) δ 6.65 (1H, d,
J = 9.9), 5.83 (1H, d,
J = 9.9), 3.94 (1H, t,
J = 10.7), 3.73–3.46 (4H, m), 2.76 (1H, dd,
J = 3.6, 12.1), 2.62–2.39 (4H, m), 2.39–2.26 (2H, m), 2.14 (1H, dt,
J = 5.7, 8.6), 2.13–2.05 (1H, m), 1.97–1.88 (1H, m), 1.80 (1H, td,
J = 5.8, 11.5), 1.70–1.46 (3H, m), 1.29 (3H, d,
J = 6.9), 1.11 (3H, s);
13C-NMR (101 MHz, CDCl
3) δ 200.54, 177.32, 158.13, 126.55, 81.77, 66.70, 57.61, 53.77, 51.62, 51.08, 43.14, 42.09, 38.14, 37.45, 23.38, 19.18, 14.53; HRMS (ESI) calculated C
19H
28NO
4 334.2018, found 334.1974 (MH
+).
Tyramine adduct 26. The appropriate enoate [
20,
21] (49.7 mg, 0.202 mmol), tyramine (27.9 mg, 0.203 mmol) and ethanol (2 mL) were mixed at 0 °C, then left to warm to room temperature for 72 h. The mixture was concentrated to afforded, after column chromatography (70% acetone/hexane as eluent), a beige foam (65.5 mg, 85%);
Rf 0.40 (70% acetone/hexane);
1H-NMR (400 MHz, CDCl
3) δ 6.95 (2H, d,
J = 8.4), 6.65 (2H, d,
J = 8.4), 6.60 (1H, d,
J = 9.9), 5.81 (1H, d,
J = 9.9), 4.93 (1H, s), 3.91 (1H, t,
J = 10.5), 2.91–2.72 (4H, m), 2.67 (2H, t,
J = 7.0), 2.55–2.40 (2H, m), 1.91–1.69 (3H, m), 1.67–1.57 (1H, m), 1.57–1.36 (2H, m), 1.24 (3H, d,
J = 6.8), 1.06 (3H, s);
13C-NMR (101 MHz, CDCl
3) δ 200.76, 177.96, 158.26, 155.00, 130.33, 129.72, 126.54, 115.65, 82.15, 51.55, 51.47, 48.88, 47.26, 45.29, 42.07, 38.26, 37.22, 34.73, 29.18, 22.75, 19.16, 14.49; HRMS (ESI) calculated C
23H
30NO
4 384.2175, found 384.2130 (MH
+).
Tyramine adduct 27. Adduct 28 (0.355 g, 0.945 mmol), 5% Pd-C (0.266 g), 32% hydrochloric acid (0.5 mL) and ethanol (10 mL) afforded, after column chromatography (30%–50% acetone/hexane as eluent), a pale orange foam (0.144 g, 40%); Rf 0.44 (20% ethanol/ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ 7.03 (2H, d, J = 8.1), 6.72 (2H, d, J = 8.3), 3.97 (2H, br s), 3.91 (1H, t, J = 10.4), 3.66 (1H, td, J = 0.7, 6.6), 3.03–2.80 (4H, m), 2.74 (2H, t, J = 6.9), 2.58–2.37 (3H, m), 1.86–1.69 (3H, m), 1.69–1.46 (3H, m), 1.33–1.23 (2H, m), 1.21 (3H, d, J = 6.5), 1.15 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 211.56, 178.11, 154.76, 130.65, 129.75, 115.57, 83.44, 53.37, 51.48, 48.84, 47.26, 45.41, 44.85, 40.62, 39.98, 37.32, 36.39, 34.79, 23.07, 18.34, 13.81; HRMS (ESI) calculated C23H31NaNO4 408.2151, found 408.2169 (M++Na+) and calculated C23H32NO4 386.2331, found 386.2299 (MH+).
Tyramine adduct 28. α-Methylenesantonin (0.220 g, 0.910 mmol), tyramine (77.1 mg, 0.560 mmol) and ethanol (2 mL) afforded, after column chromatography (10%–20% ethanol/ethyl acetate as eluent), a yellow foam (89.7 mg, 44%); Rf 0.26 (20% ethanol/ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ 7.00 (2H, d, J = 8.5), 6.74 (2H, d, J = 8.5), 6.69 (1H, d, J = 9.9), 6.24 (1H, d, J = 9.9), 4.78 (1H, dd, J = 1.1, 11.4), 4.72 (2H, br s), 2.98–2.88 (2H, m), 2.88–2.80 (2H, m), 2.72 (2H, t, J = 7.0), 2.63 (1H, dt, J = 6.0, 12.2), 2.08 (3H, d, J = 0.9), 1.98 (2H, ddd, J = 7.0, 15.9, 31.8), 1.88–1.76 (1H, m), 1.66 (1H, qd, J = 3.4, 12.8), 1.40 (1H, td, J = 4.2, 13.1), 1.27 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 186.57, 176.76, 155.42, 155.07, 151.32, 130.17, 129.60, 128.39, 125.49, 115.52, 81.52, 58.08, 51.30, 49.55, 47.17, 45.61, 41.22, 37.51, 34.73, 24.86, 22.92, 18.20, 10.81; HRMS (ESI) calculated C23H28NO4 382.2018, found 382.1970 (MH+).
1-(2-Chlorophenyl)piperazine adduct 31. α-Methylenesantonin (49.0 mg, 0.202 mmol), 1-(2-chlorophenyl)piperazine (0.120 g, 0.516 mmol) and ethanol (2 mL) afforded, after column chromatography (2%–4% methanol/chloroform as eluent), a beige solid (65.3 mg, 73%); Rf 0.41 (4% methanol/chloroform). Recrystallisation yielded a white powder, mp >190 °C (ethyl acetate/hexane); 1H-NMR (400 MHz, CDCl3) δ 7.35 (1H, dd, J = 1.2, 7.9), 7.27–7.15 (1H, m), 7.06–7.01 (1H, m), 7.01–6.92 (1H, m), 6.71 (1H, d, J = 9.9), 6.26 (1H, d, J = 9.9), 4.83 (1H, d, J = 11.4), 2.99 (4H, s), 2.95 (1H, t, J = 8.4), 2.81–2.64 (4H, m), 2.60 (2H, d, J = 5.5), 2.33 (1H, d, J = 12.8), 2.14 (3H, s), 2.05 (1H, ddd, J = 4.2, 7.6, 17.3), 1.89 (1H, d, J = 13.4), 1.76 (1H, ddd, J = 3.7, 12.8, 25.3), 1.56 (1H, td, J = 4.3, 13.1), 1.34 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 186.24, 176.26, 154.97, 151.03, 148.93, 130.53, 128.58, 128.49, 127.46, 125.72, 123.65, 120.19, 81.36, 57.26, 53.40, 51.87, 51.00, 43.68, 41.14, 37.92, 25.00, 23.77, 10.82; HRMS (ESI) calculated C25H29ClN2O3Na 463.1764, found 463.1759 (M++Na+), and calculated C25H30ClN2O3 441.1945, found 441.1870 (MH+).
4-Chlorobenzylamine adduct 32. α-Methylenesantonin (0.218 g, 0.902 mmol), 4-chlorobenzylamine (72.0 μL, 0.589 mmol) and ethanol (2 mL) afforded, after column chromatography (10%–20% ethanol/ethyl acetate as eluent), a yellow oil (0.156 g, 69%); Rf 0.52 (20% ethanol/ethyl acetate); 1H-NMR (400 MHz, CDCl3) δH 7.28 (2H, d, J = 8.7), 7.24 (2H, d, J = 8.7), 6.71 (1H, d, J = 9.9), 6.24 (1H, d, J = 9.9), 4.83 (1H, dd, J = 1.4, 11.5), 3.79 (1H, d, J = 13.6), 3.74 (1H, d, J = 13.6), 2.94 (1H, dd, J = 4.9, 12.3), 2.82 (1H, dd, J = 6.1, 12.3), 2.60 (1H, ddd, J = 5.0, 6.0, 12.2), 2.17 (1H, dd, J = 3.5, 12.1), 2.14–2.06 (1H, m), 2.12 (3H, d, J = 1.3), 2.02–1.93 (1H, m), 1.88 (1H, ddd, J = 2.2, 3.6, 13.4), 1.70 (1H, ddd, J = 3.8, 12.9, 25.4), 1.48 (1H, td, J = 4.5, 13.2), 1.32 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 186.15, 176.47, 154.92, 150.91, 138.22, 132.46, 129.23, 128.39, 128.31, 125.59, 81.41, 53.00, 49.16, 46.32, 46.09, 41.10, 37.58, 24.93, 23.02, 10.79; HRMS (ESI) calculated C22H25ClNO3 386.1523, found 386.1474 (MH+).
General Procedure Used for the Conjugate Addition of Amines to Unsaturated Isophotosantonin Derivatives: Solutions of the enoate in absolute ethanol were dosed into 8 mL ChemSpeed reaction vessels, warmed to 30 °C and then treated with the appropriate amine in ethanol. The mixtures were agitated at 600 rpm for 18 h, concentrated to gums and purified by column chromatography. In this fashion the following were prepared:
Morpholine adducts 22 and 25. A mixture of 15 and its lumisantonin equivalent (~2:1 mixture, 0.100 g, 0.331 mmol), morpholine (57.3 μL, 0.657 mmol) and ethanol (5 mL) were treated as per the general procedure. The resultant orange gum afforded, after column chromatography (50% ethyl acetate/hexane – ethyl acetate as eluent), 22 (50.8mg, 39%); Rf 0.45 (ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ 4.77 (1H, d, J = 10.5), 4.12 (1H, dq, J = 2.2, 6.3), 3.72–3.51 (5H, m), 2.79 (1H, dd, J = 4.1, 13.3), 2.68–2.59 (1H, m), 2.56 (1H, dd, J = 4.5, 13.6), 2.52–2.29 (10H, m), 2.09 (1H, dt, J = 3.6, 13.6), 1.94 (3H, s), 1.83 (3H, dd, J = 1.6, 2.2), 1.41 (1H, tdd, J = 3.4, 10.8, 14.0), 1.01 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 206.91, 175.75, 170.33, 160.68, 143.13, 85.51, 81.19, 66.61, 57.11, 53.97, 47.13, 45.81, 43.80, 37.89, 36.70, 25.41, 22.23, 19.97, 9.45; HRMS (ESI) calculated C21H30NO6 392.2073, found 392.2047 (MH+). Also isolated was 25 slightly contaminated with the former product, as an orange gum (34.4mg, 32%); Rf 0.33 (ethyl acetate). An analytical sample was purified by preparative TLC (ethyl acetate): 1H-NMR (400MHz, CDCl3) δ 6.70 (1H, d, J = 9.9), 6.27 (1H, d, J = 9.9), 4.80 (1H, dq, J = 1.3, 11.5), 3.80–3.50 (4H, m), 2.98–2.75 (1H, m), 2.70–2.57 (2H, m), 2.57–2.48 (2H, m), 2.48–2.36 (2H, m), 2.36–2.25 (1H, m), 2.13 (3H, d, J = 1.4), 2.05 (1H, qd, J = 3.5, 11.7), 1.88 (1H, ddd, J = 2.2, 3.6, 13.4), 1.72 (1H, ddd, J = 3.8, 12.9, 25.0), 1.55 (1H, td, J = 4.5, 13.2), 1.33 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 186.29, 186.28, 176.15, 154.88, 150.82, 128.75, 125.89, 81.45, 66.82, 57.79, 53.82, 51.90, 43.60, 41.17, 37.99, 25.10, 23.88, 10.88; HRMS (ESI) calculated C19H26NO4 332.1862, found 332.1836 (MH+).
Tyramine adducts 29 and 30. A mixture of 15 and its lumisantonin equivalent (~2:1 mixture, 0.101 g, 0.331 mmol), tyramine (90.2 mg, 0.657 mmol) and ethanol (5 mL) were treated as per the general procedure. The resultant brown gum afforded, after column chromatography (50% ethyl acetate:hexane – ethyl acetate as eluent), 29 (60.3 mg, 41.3%); Rf 0.25 (ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ 7.13–6.98 (2H, m), 6.84–6.68 (2H, m), 4.98 (1H, d, J = 10.7), 4.16 (1H, br m), 3.00–2.80 (4H, m), 2.76 (2H, d, J = 6.7), 2.65–2.46 (4H, m), 2.46–2.37 (1H, m), 2.18 (1H, dd, J = 3.4, 13.5), 2.09 (1H, s), 2.03 (3H, s), 1.89 (3H, d, J = 1.5), 1.53 (1H, ddd, J = 3.2, 10.5, 14.3), 1.37–1.19 (1H, m), 1.11 (3H, s); 13C-NMR (101 MHz, CD3OD) δ 208.56, 177.44, 171.36, 162.40, 155.73, 143.28, 130.61, 130.15, 130.03, 116.02, 115.80, 86.24, 82.10, 51.93, 47.90, 47.30, 46.40, 44.54, 38.21, 37.29, 35.31, 25.52, 22.54, 20.16, 9.64; HRMS (ESI) calculated C25H32NO6 442.2230, found 442.2229 (MH+). Also isolated was 30 slightly contaminated with the former product, as an orange gum (21.9 mg, 17.4%); Rf 0.14 (ethyl acetate). An analytical sample was purified by preparative TLC (ethyl acetate): 1H-NMR (400 MHz, CD3OD) δH 7.04 (2H, d, J = 8.5), 6.73 (2H, d, J = 8.5), 6.69 (1H, d, J = 9.9), 6.26 (1H, d, J = 9.9), 4.79 (1H, dd, J = 1.4, 11.4), 2.99 (1H, dd, J = 5.0, 12.4), 2.84 (4H, m), 2.72 (2H, t, J = 6.8), 2.63–2.54 (1H, m), 2.11 (3H, d, J = 1.3), 2.10–2.03 (1H, m), 2.03–1.94 (2H, m), 1.88 (1H, ddd, J = 2.1, 3.7, 13.4), 1.74 (1H, ddd, J = 3.9, 13.1, 25.8), 1.50 (1H, td, J = 4.1, 13.1), 1.33 (3H, s); 13C-NMR (101 MHz, CD3OD) δ 186.43, 176.61, 154.93, 154.15, 150.86, 131.40, 129.73, 128.72, 125.84, 115.34, 81.57, 51.51, 49.41, 47.11, 46.15, 41.20, 37.76, 35.09, 25.04, 23.23, 10.90; HRMS (ESI) calculated C23H28NO4 382.2018, found 382.1986 (MH+).
2-(4-Chlorophenyl)ethylamine adducts 33 and 34. A mixture of 15 and its lumisantonin equivalent (~2:1 mixture, 0.114 g, 0.376 mmol), 2-(4-chlorophenyl)ethylamine (91.4 μL, 0.657 mmol) and ethanol (5 mL) were treated as per the general procedure. The resultant orange gum afforded, after column chromatography (50% ethyl acetate/hexane – ethyl acetate as eluent), 33 (60.8 mg, 35%); Rf 0.27 (ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ H 7.19 (2H, d, J = 8.4), 7.07 (2H, d, J = 8.5), 4.76 (1H, d, J = 10.5), 4.10–4.04 (1H, m), 2.93 (1H, dd, J = 3.9, 12.4), 2.87–2.72 (3H, m), 2.69 (2H, t, J = 6.9), 2.55–2.36 (4H, m), 2.36–2.27 (1H, m), 2.11–1.98 (2H, m), 1.94 (3H, s), 1.81 (3H, dd, J = 1.6, 2.1), 1.37 (1H, tdd, J = 3.4, 10.7, 14.1), 1.00 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 206.90, 176.18, 170.23, 160.70, 143.02, 138.14, 131.82, 129.98, 128.44, 85.40, 81.46, 51.29, 47.15, 46.62, 46.50, 43.48, 37.71, 36.71, 35.48, 25.29, 22.21, 19.89, 9.41; HRMS (ESI) calculated C25H31ClNO5 460.1891, found 460.1866 (MH+). Also isolated was 34 slightly contaminated with the former product, as an orange gum (28.6mg, 19.0%); Rf 0.15 (ethyl acetate). An analytical sample was purified by preparative TLC (ethyl acetate): 1H-NMR (400 MHz, CDCl3) δ 7.25 (2H, d, J = 8.5), 7.13 (2H, d, J = 8.5), 6.70 (1H, d, J = 9.9), 6.27 (1H, d, J = 9.9), 4.80 (1H, dd, J = 1.4, 11.4), 2.96 (1H, dd, J = 5.3, 12.3), 2.90 (1H, dd, J = 5.8, 11.9), 2.89–2.80 (3H, m), 2.80–2.72 (2H, m), 2.62–2.52 (1H, m), 2.13 (3H, d, J = 1.3), 2.02 (1H, ddd, J = 3.7, 12.1, 23.8), 2.02–1.94 (1H, m), 1.86 (1H, ddd, J = 2.2, 3.7, 13.5), 1.68 (1H, ddd, J = 3.8, 12.9, 25.3), 1.47 (1H, td, J = 4.5, 13.3), 1.32 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 186.28, 176.57, 154.83, 150.72, 138.21, 131.91, 130.01, 128.74, 128.50, 125.88, 81.56, 51.28, 49.51, 47.30, 46.35, 41.18, 37.72, 35.63, 25.12, 23.30, 10.93; HRMS (ESI) calculated C23H27ClNO3 400.1679, found 400.1645 (MH+).


 ,
, 

{kind=link}