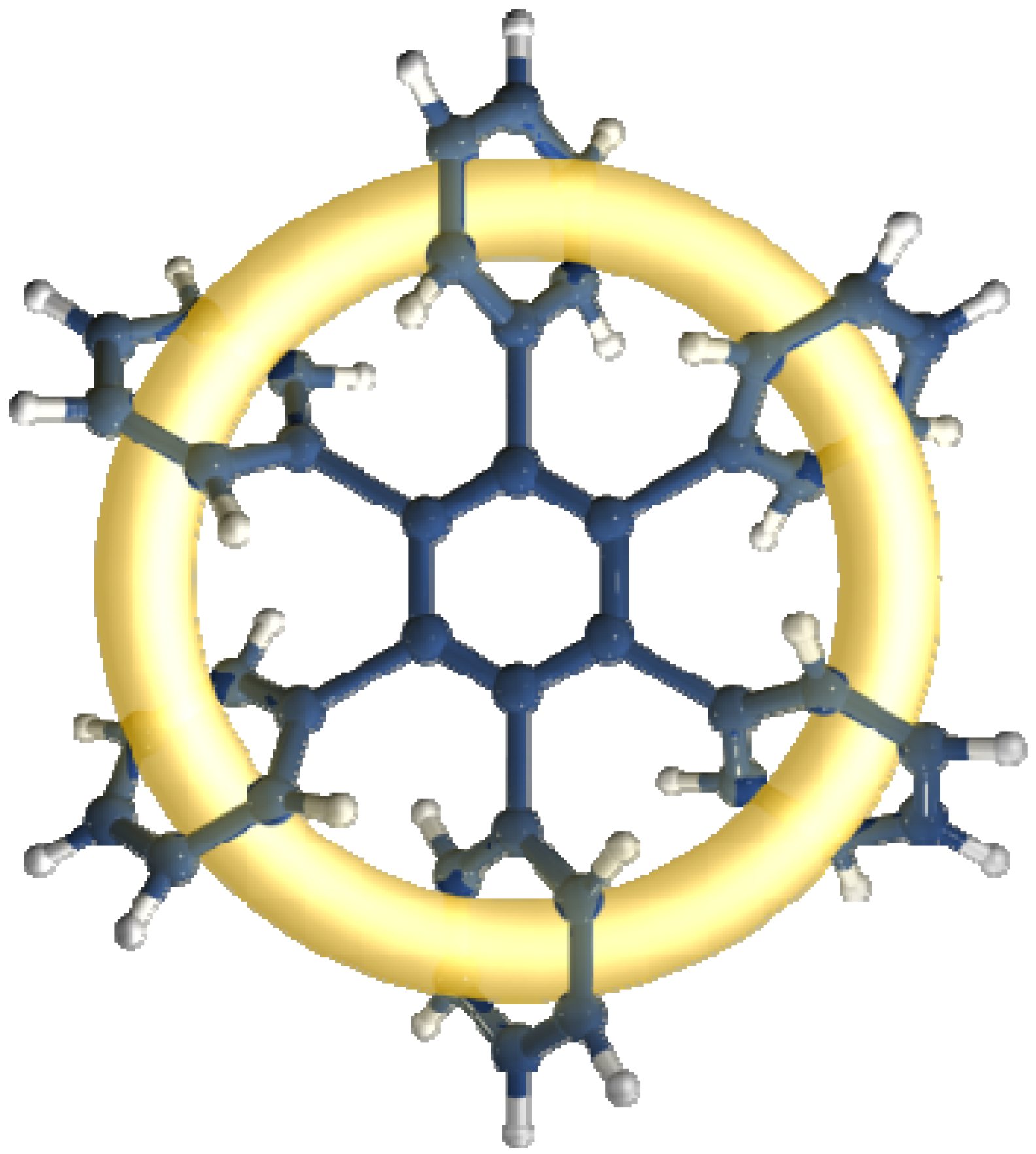
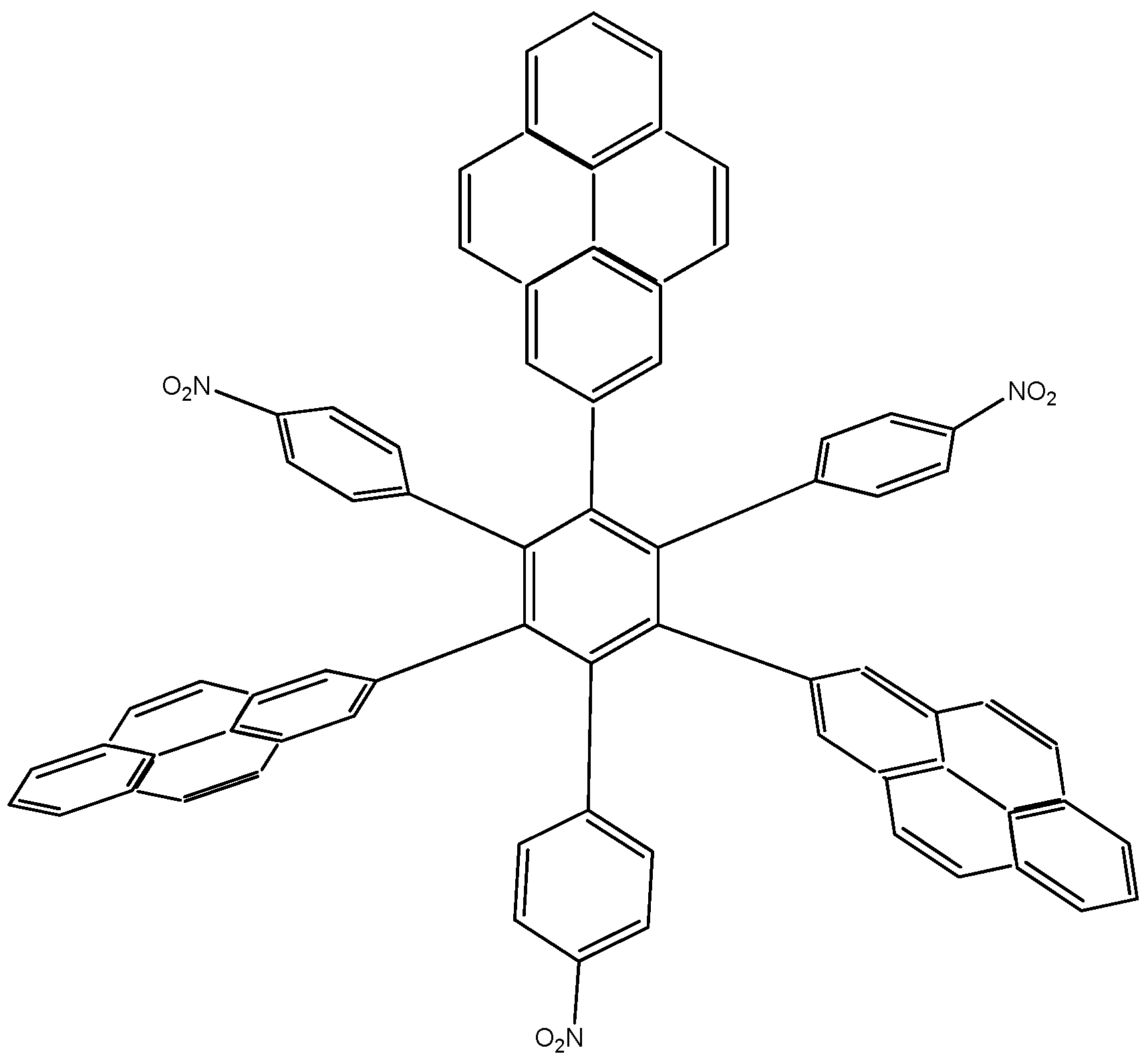
3.2.1. Molecules with Marked Toroidal Delocalization
The first studied case was
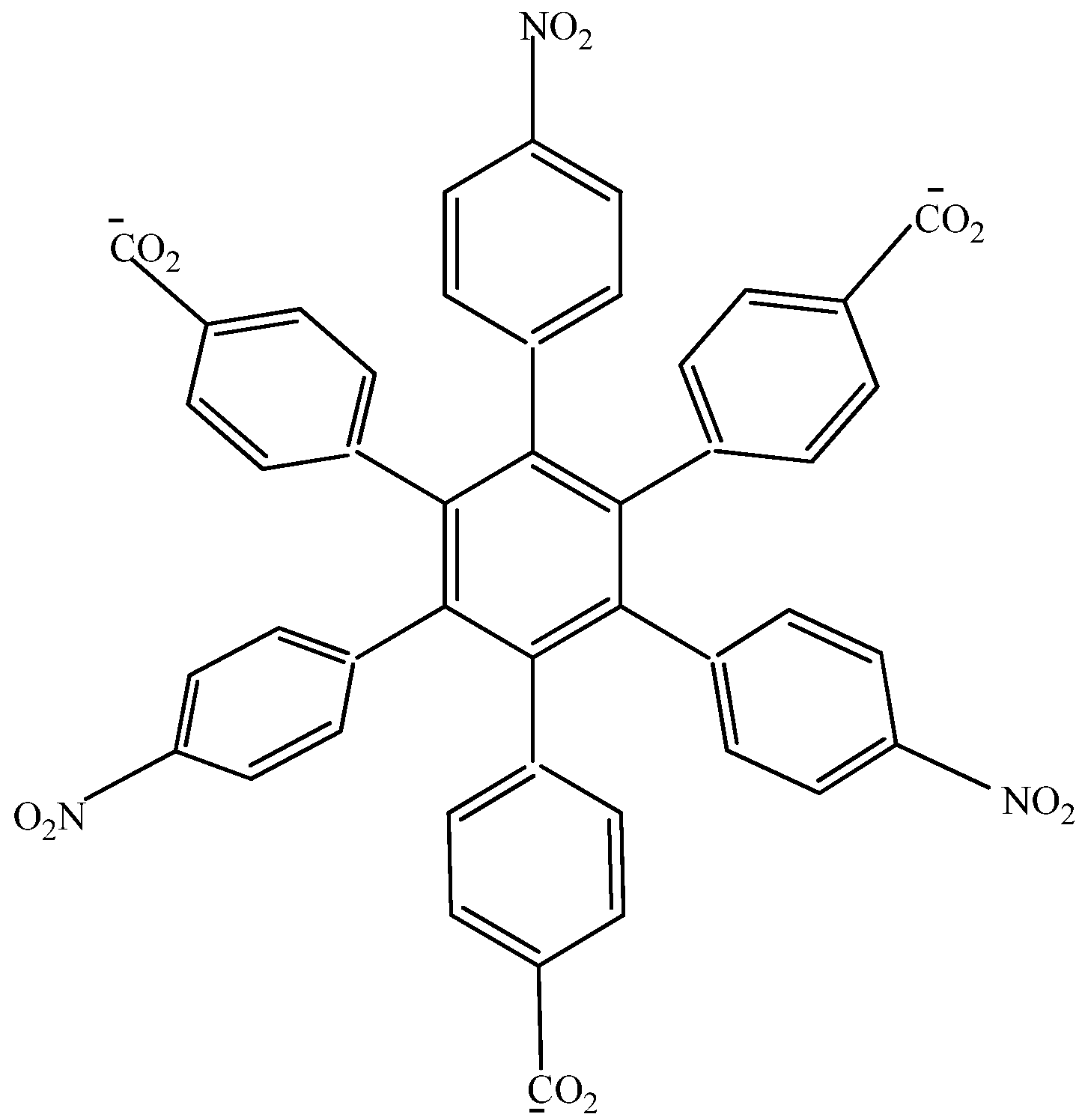
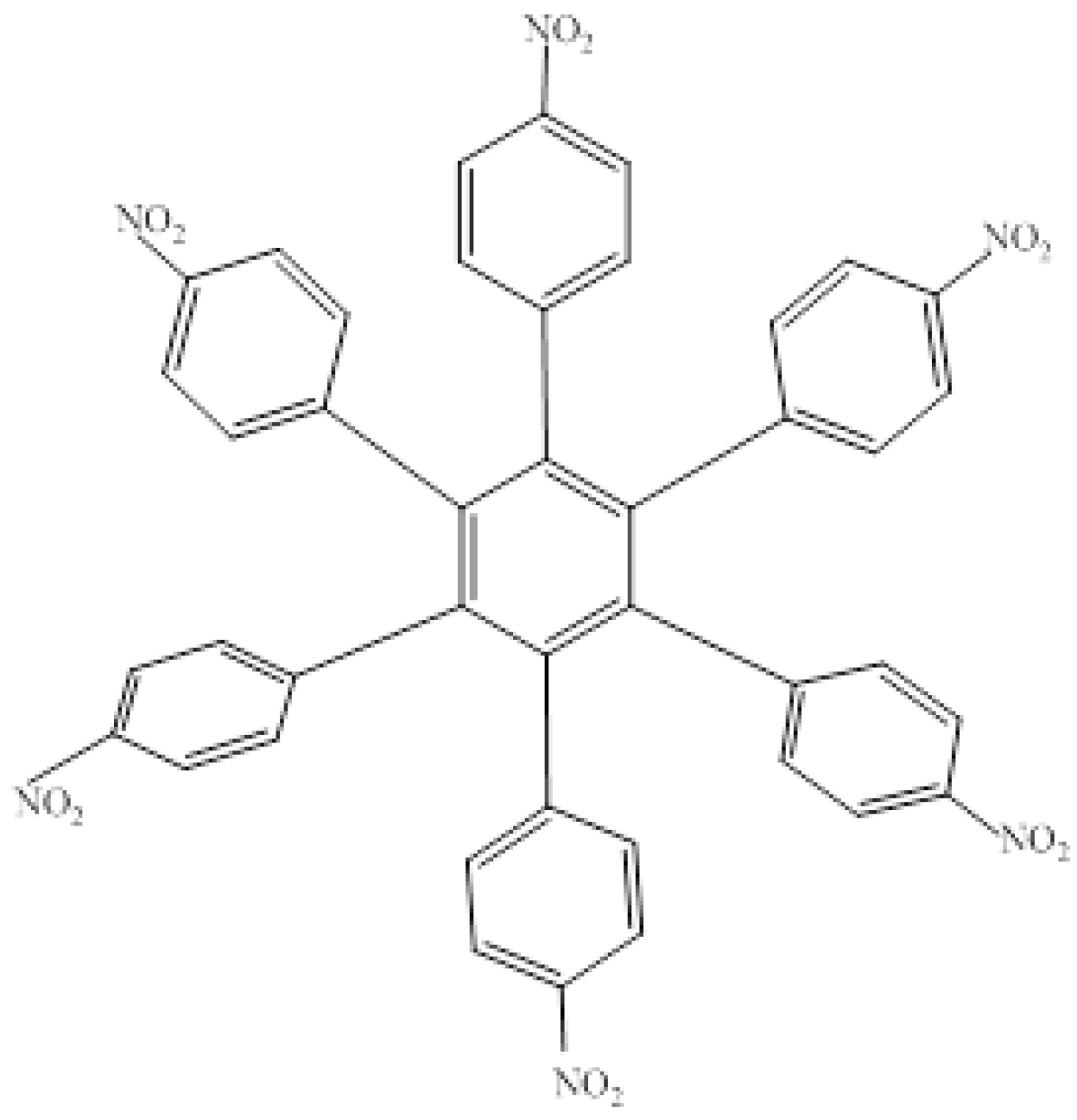
HAB1 (
Figure 2) containing three 4-nitrophenyl and three (4-carboxylate) phenyl (benzoate) fragments.
Figure 2.
Structure of HAB1.
Figure 2.
Structure of HAB1.
The corresponding isodesmic reaction for
HAB1 is:
The corresponding isodesmic reaction for benzoate anion is:
Whereas the same for nitrobenzene is:
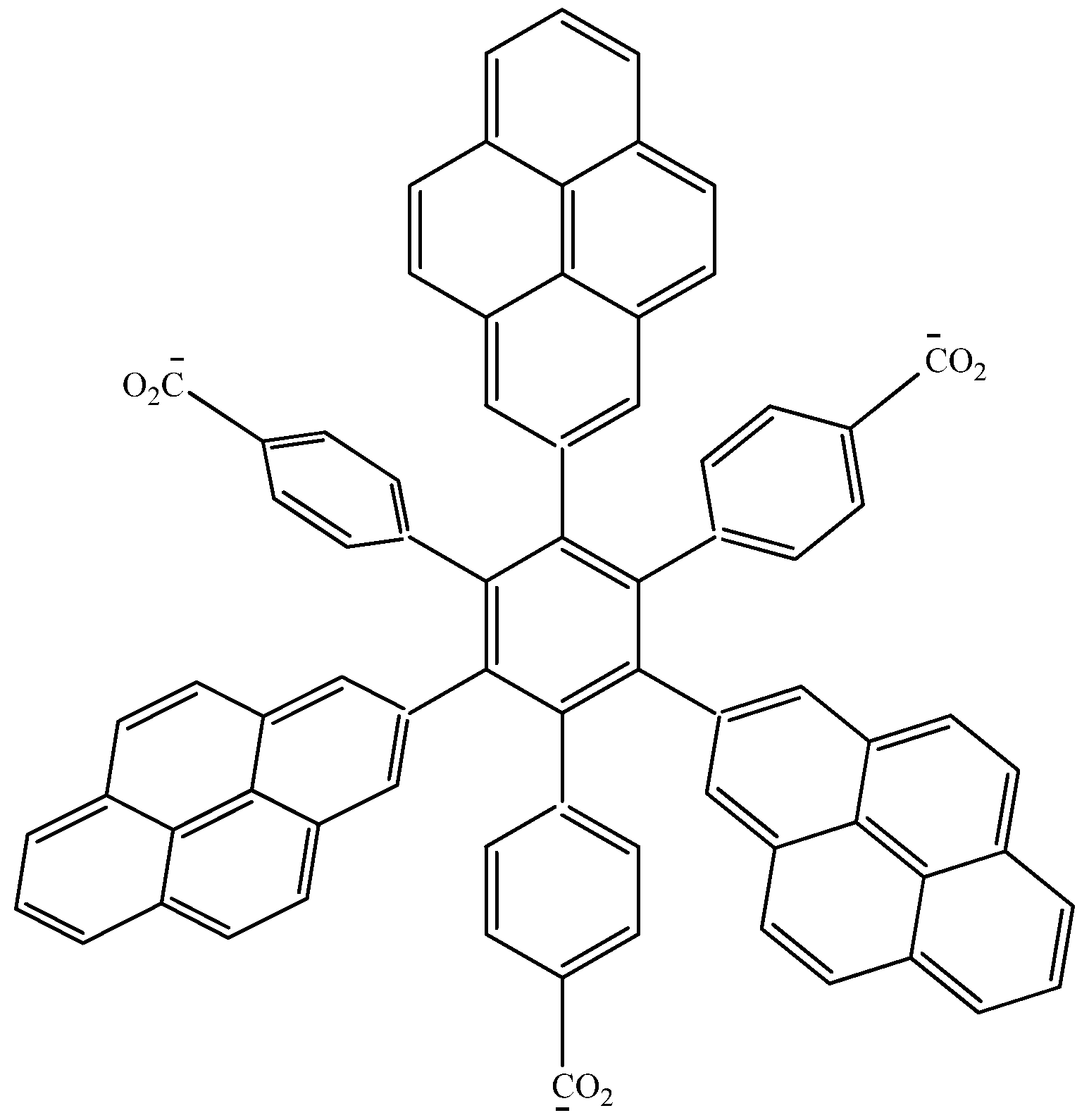
Therefore, the value of the energy for
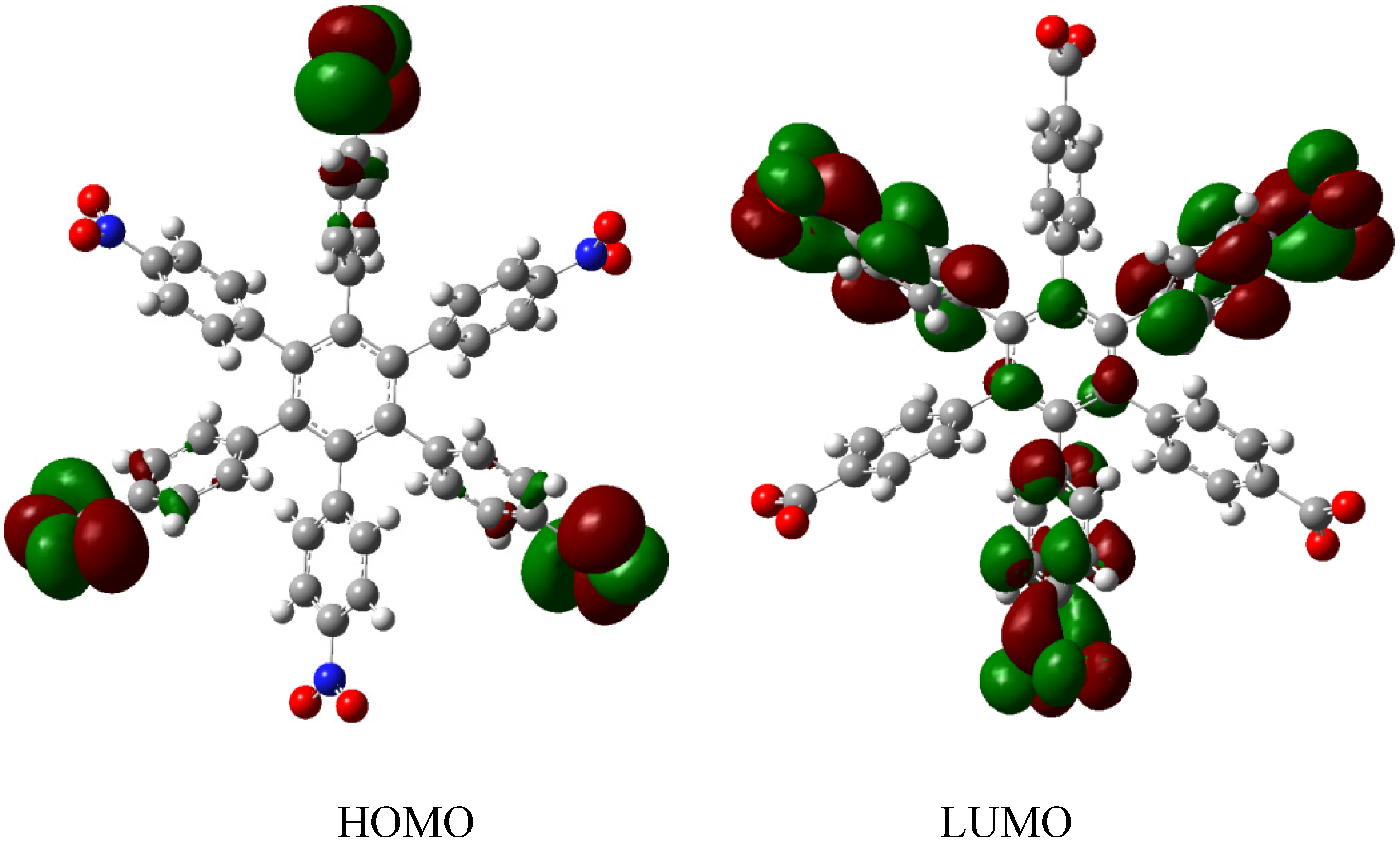
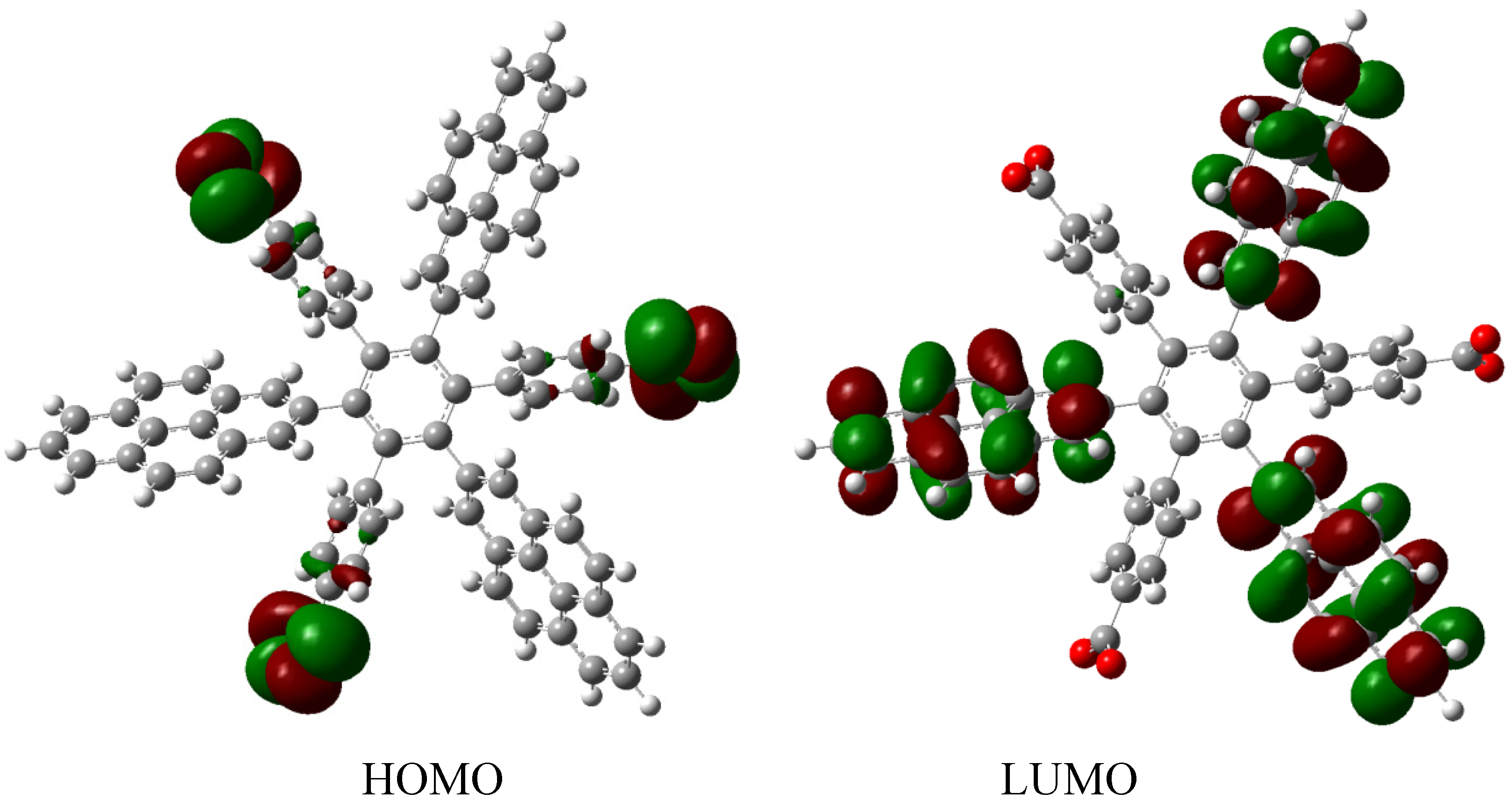
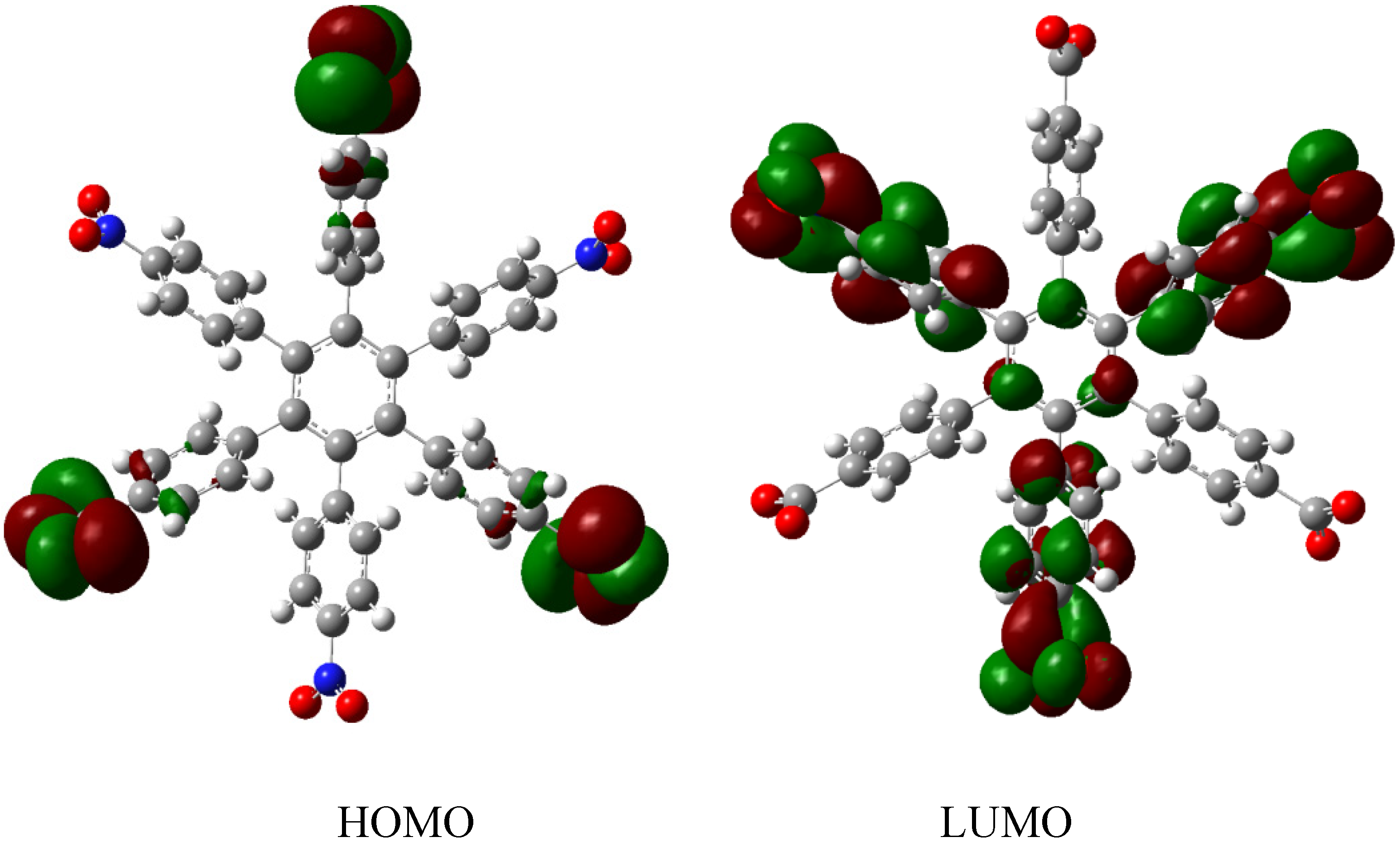
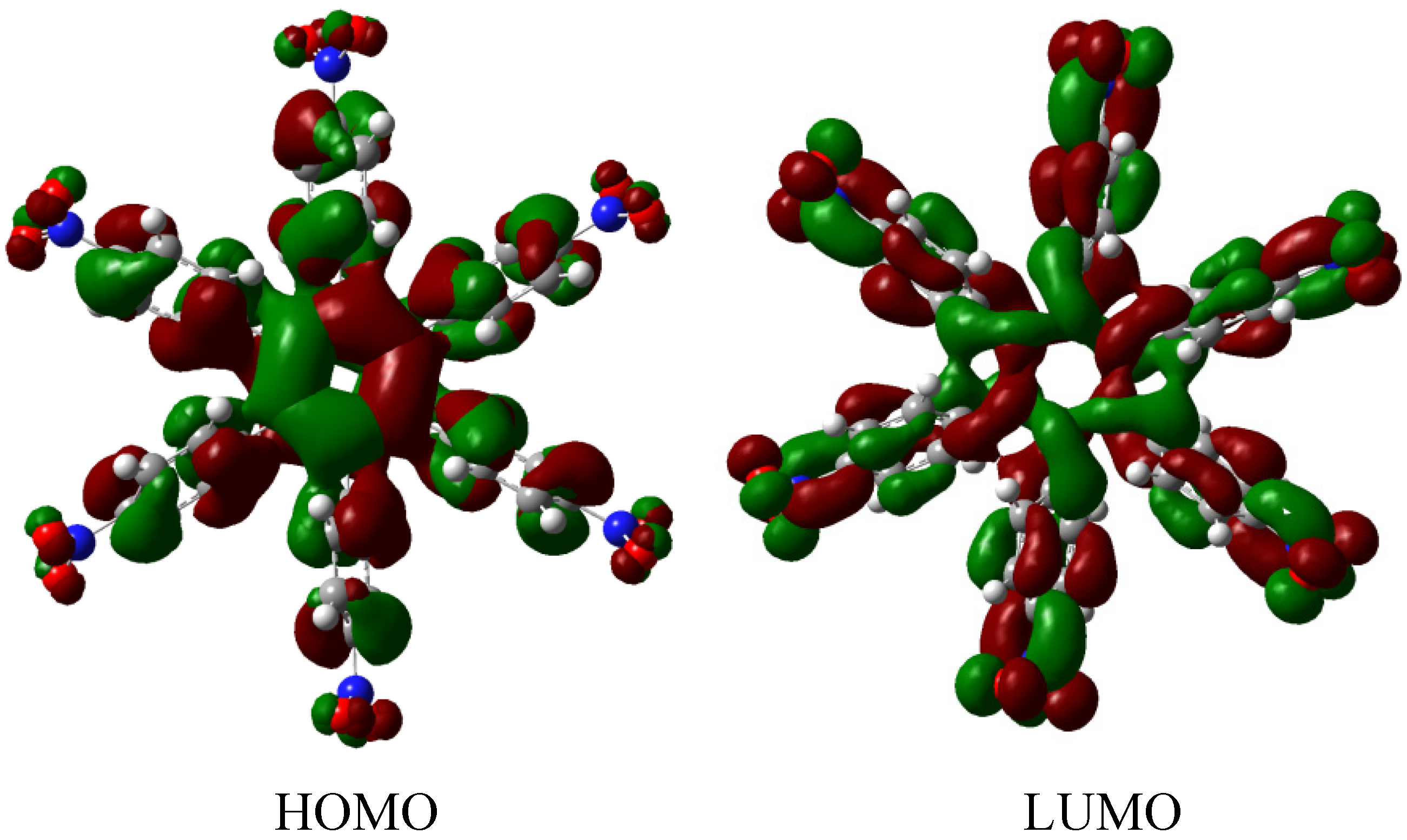
HAB1 is −139.62 kcal/mol; we made a subtraction of this result and the set of three (4-carboxylate) phenyl anions and three nitrobenzene molecules (plus 21.1 kcal/mol from the central ring) and the result is 69.72 kcal/mol. Notably this result was achieved, assuming three negative charges on the original molecule, as this factor would generate electron mobility. Similarly, it is evident that the energy gap between HOMO and LUMO is too short compared with organic semiconductor species [
20], with a value of 1.007 eV, although this result is derived from the fact that the molecular orbitals drastically change their position because of the presence of the negative charges. The shape of the molecular orbitals is presented in
Figure 3.
The nature of the molecule is reflected in these molecular orbitals, where the HOMO produces the entire negative charge coming from the electron pairs and correspondingly the LUMO operates as an acceptor region centered on the 4-nitrophenyl substituents. It appears that this represents a very good target for toroidal delocalization, because the frontier molecular orbitals are localized in different zones of the molecule and the energy gap is too short, therefore, it is expected that the electrons flow easily between them, even though this behavior obeys the ionic nature of the species, a feature that is altered in the next example.
Figure 3.
Frontier molecular orbitals of HAB1.
Figure 3.
Frontier molecular orbitals of HAB1.
The power to generate low energy toroidal dissipation was investigated in order to discover whether it might be caused by strong inductive effect or be totally independent of this. Thus a molecule was designed, hypothetically manifesting very strong electron withdrawal activity, in order to study the inductive effect where the electron-withdrawal characteristic is very dominant.
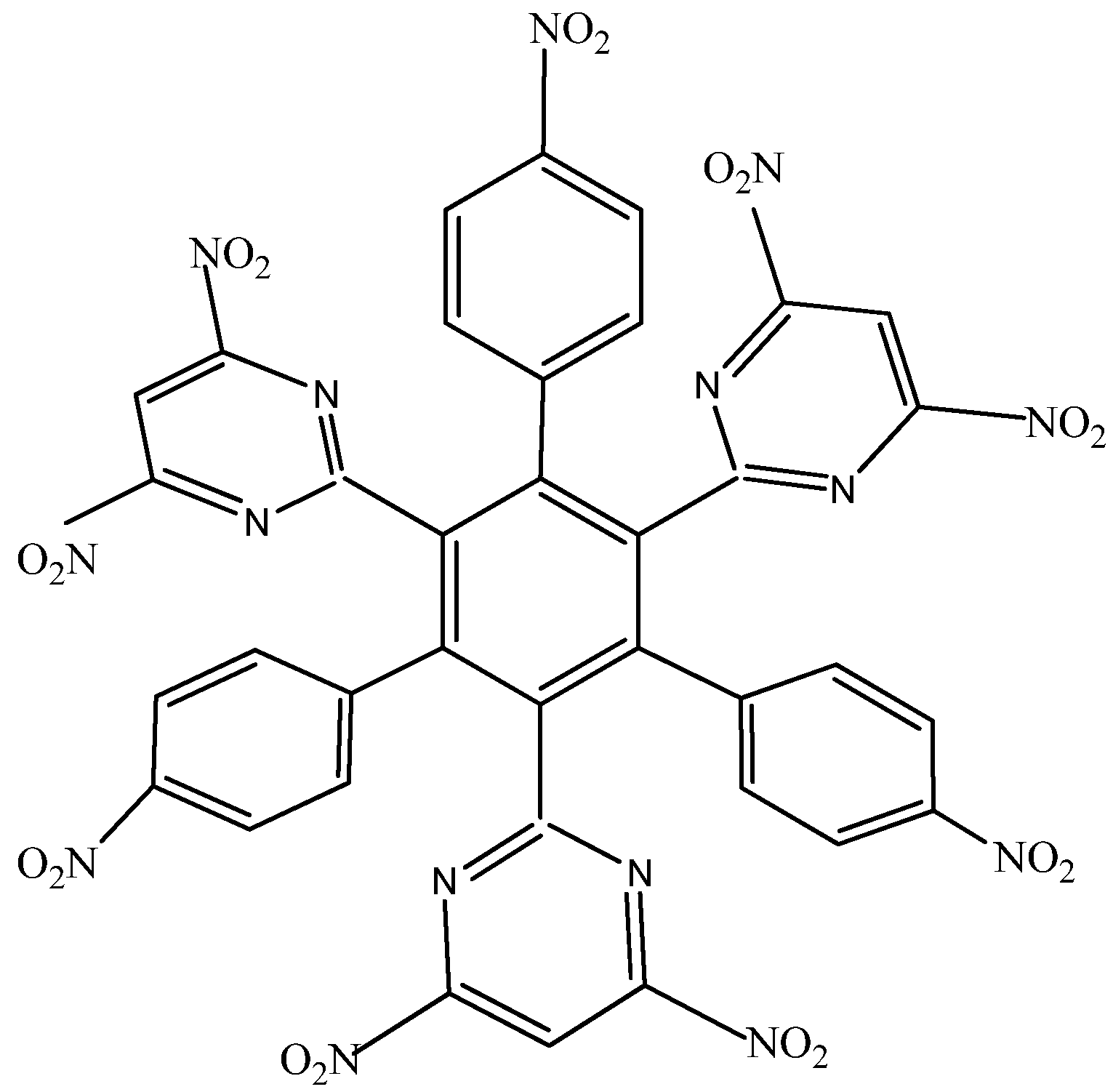
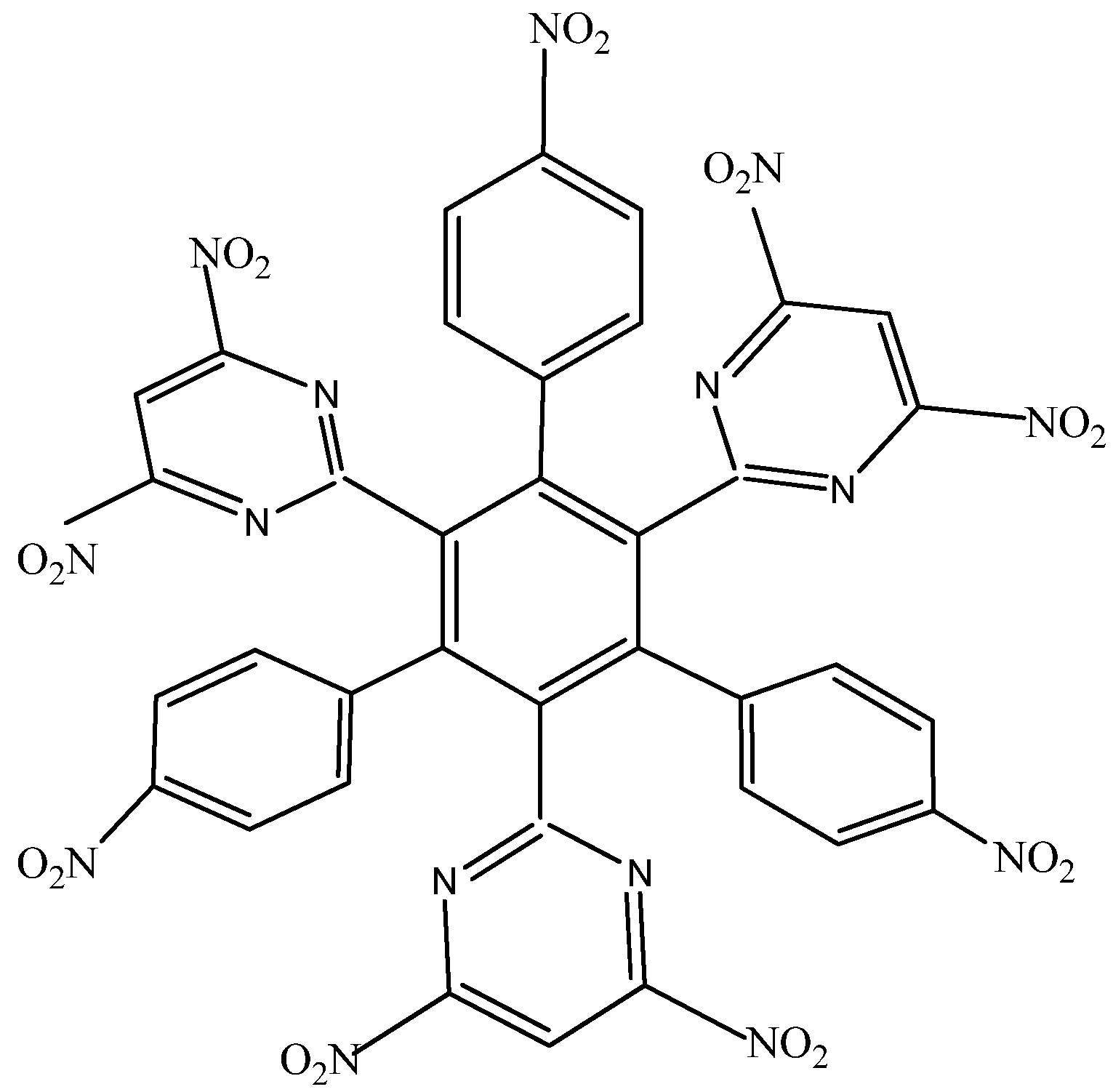
HAB2 consists of three 4-nitrophenyl and three 4,6-dinitro-2-pyrimidyl fragments and its shape is presented in
Figure 4. The azines are a species known for manifesting very strong electron-withdrawal effect [
21], so this kind of fragment is a good target for illustrating the influence that these phenomena have on electronic mobility.
Figure 4.
Structure of HAB2.
Figure 4.
Structure of HAB2.
The corresponding isodesmic reaction for
HAB2 is:
Whereas the isodesmic reaction for 4, 6-dinitropyrimidine is:
The behavior of this molecule is very similar to that found in the previous example; the homodesmotic reaction for the
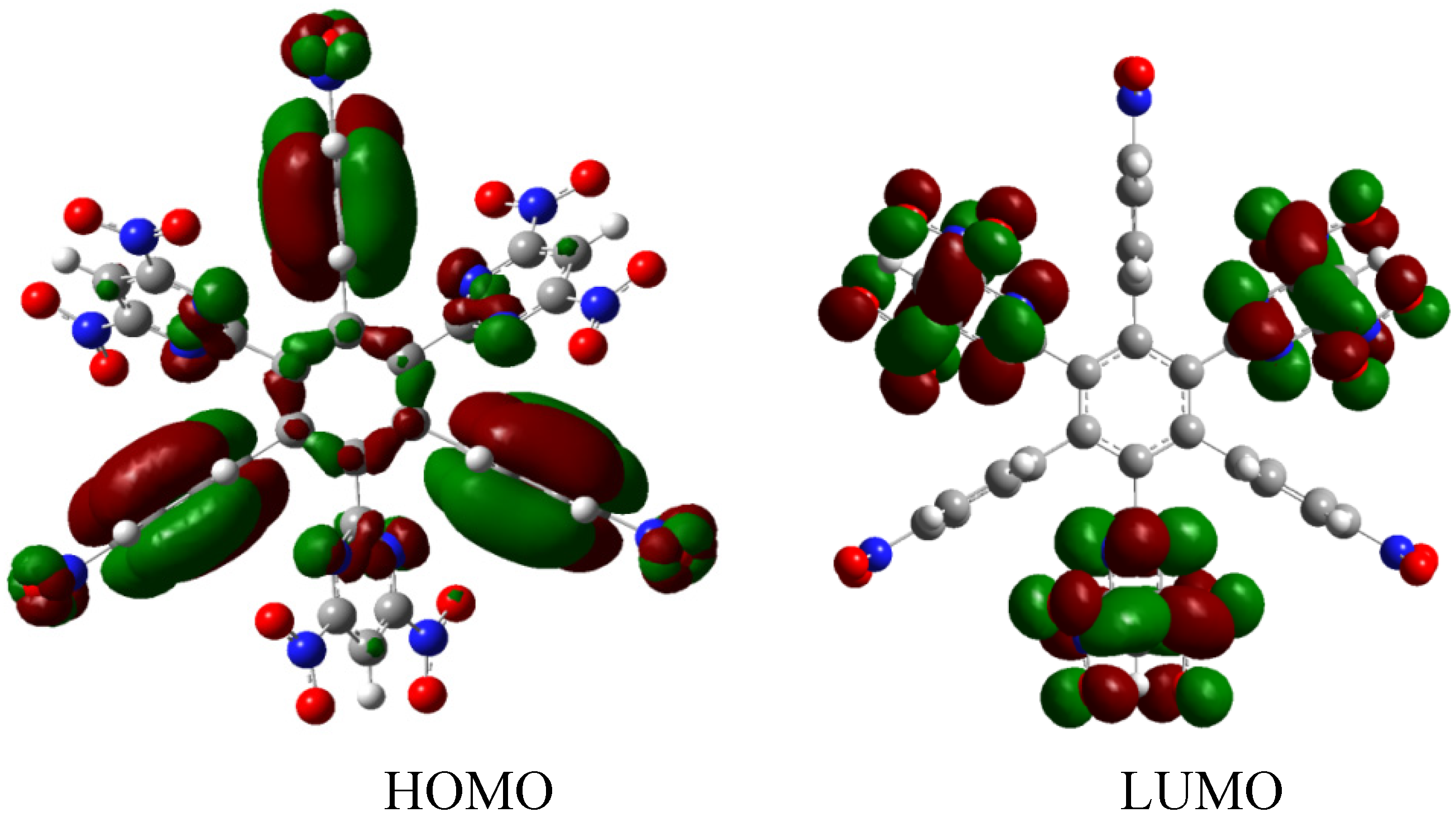
HAB2 is −92.118 kcal/mol, whereas the result for three diazine (−40.16 kcal/mol) plus three nitrobenzene molecules and the central ring is 118.16 kcal/mol with the same alternative frontier molecular orbitals (
Figure 5), the nature of these molecular orbitals depends on the presence of the diazine substituent because the strong electron withdrawal effect caused that the LUMO was totally localized on this fragment yielding an electron flow to this zone of the molecule. However, in this instance there are no negative charges, in contrast this molecule is totally attractive to electrons, a fact reflected in the energy gap between HOMO and LUMO, which is 3.61 eV, suggesting weak semiconductor behavior. However the similarities in behavior between both molecules are evident in the marked energy for toroidal delocalization, suggesting that the alternation of certain specific substituents may account for the improvement in electronic flow.
Figure 5.
Frontier molecular orbitals for HAB2.
Figure 5.
Frontier molecular orbitals for HAB2.
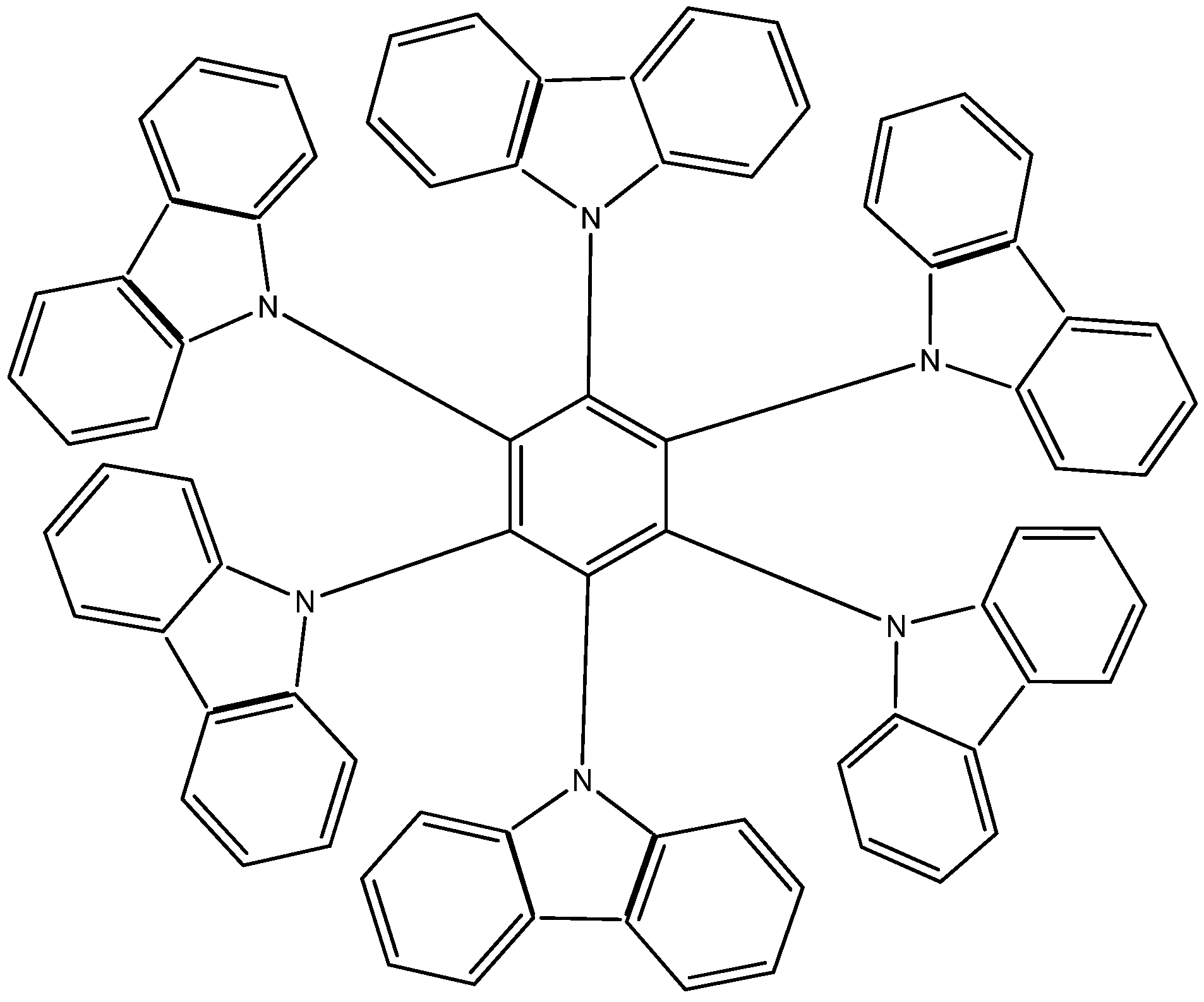
The third example
HAB3 corresponds to the molecule proposed by Lambert [
2,
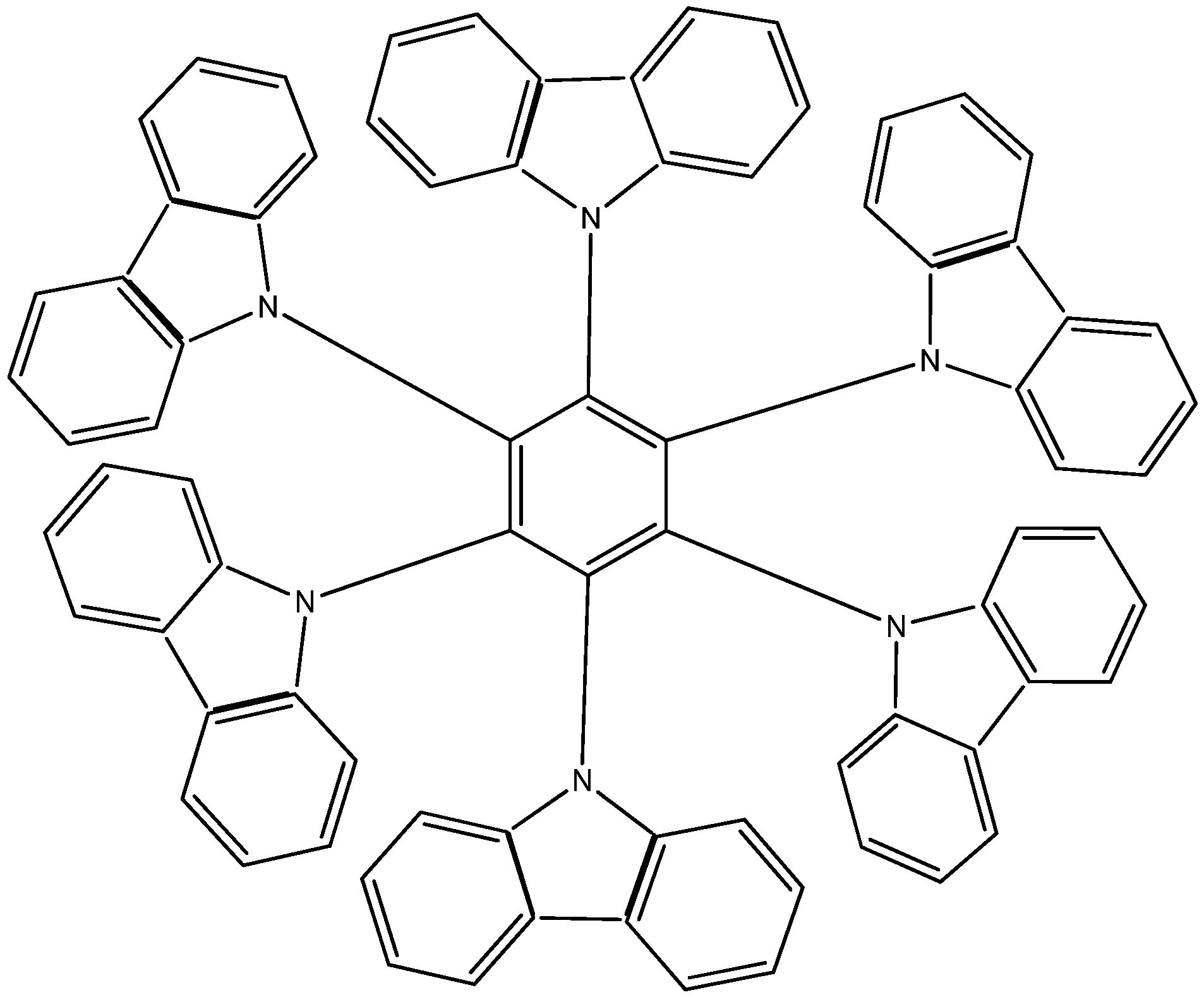
22], where six 9-carbazolyl substituents surround the central ring (see
Figure 6).
Figure 6.
Structure of HAB.
Figure 6.
Structure of HAB.
This is the isodesmic reaction for
HAB3, whereas the corresponding reaction for dibenzenepyrrole is:
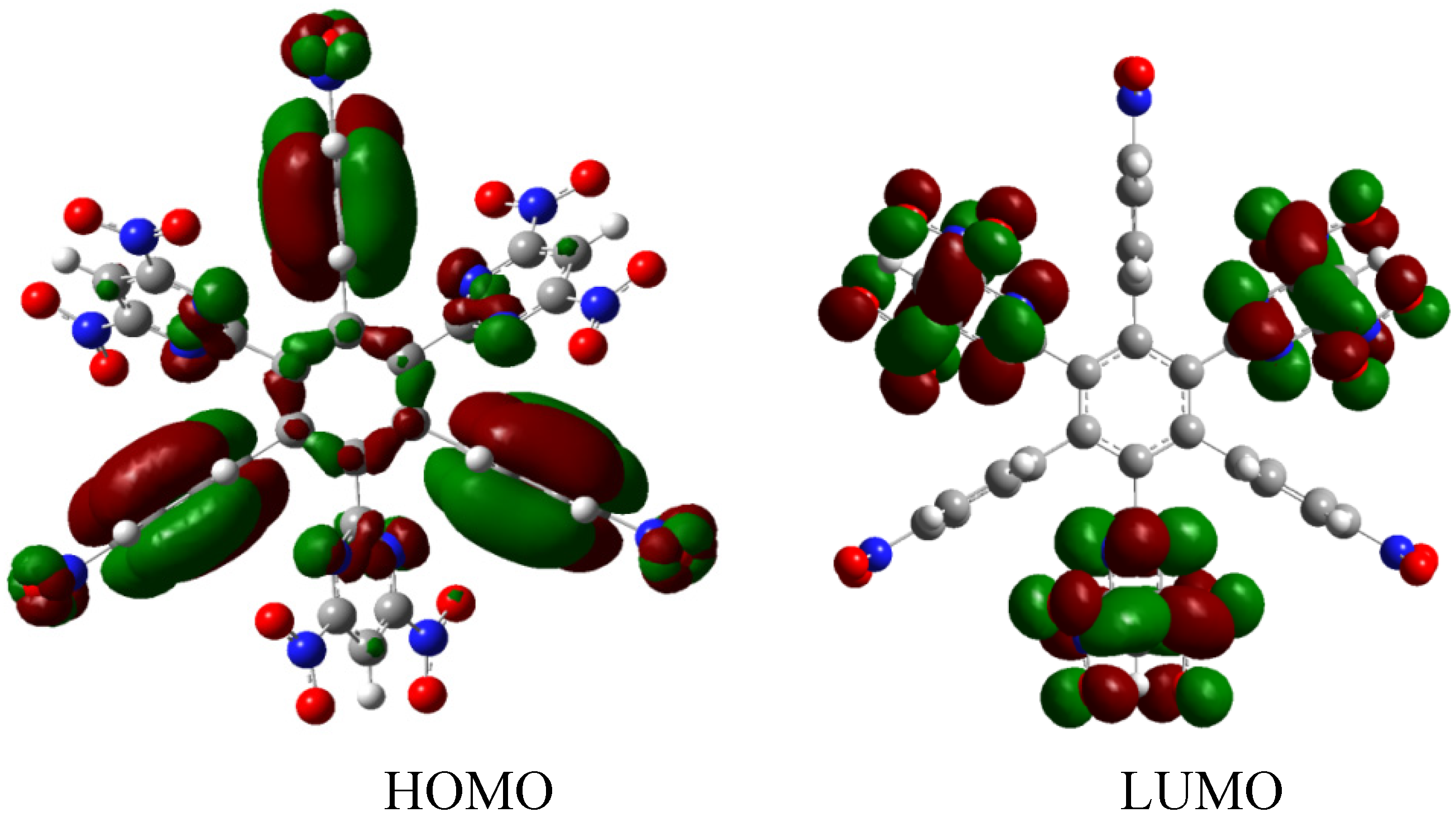
The results of the energy are −432.29 kcal/mol for HAB3, and −79.69 kcal/mol for dibenzenpyrrol. Therefore, applying the same mechanism, the result of the energy obtained for toroidal dissipation in this instance is 66.95 kcal/mol.
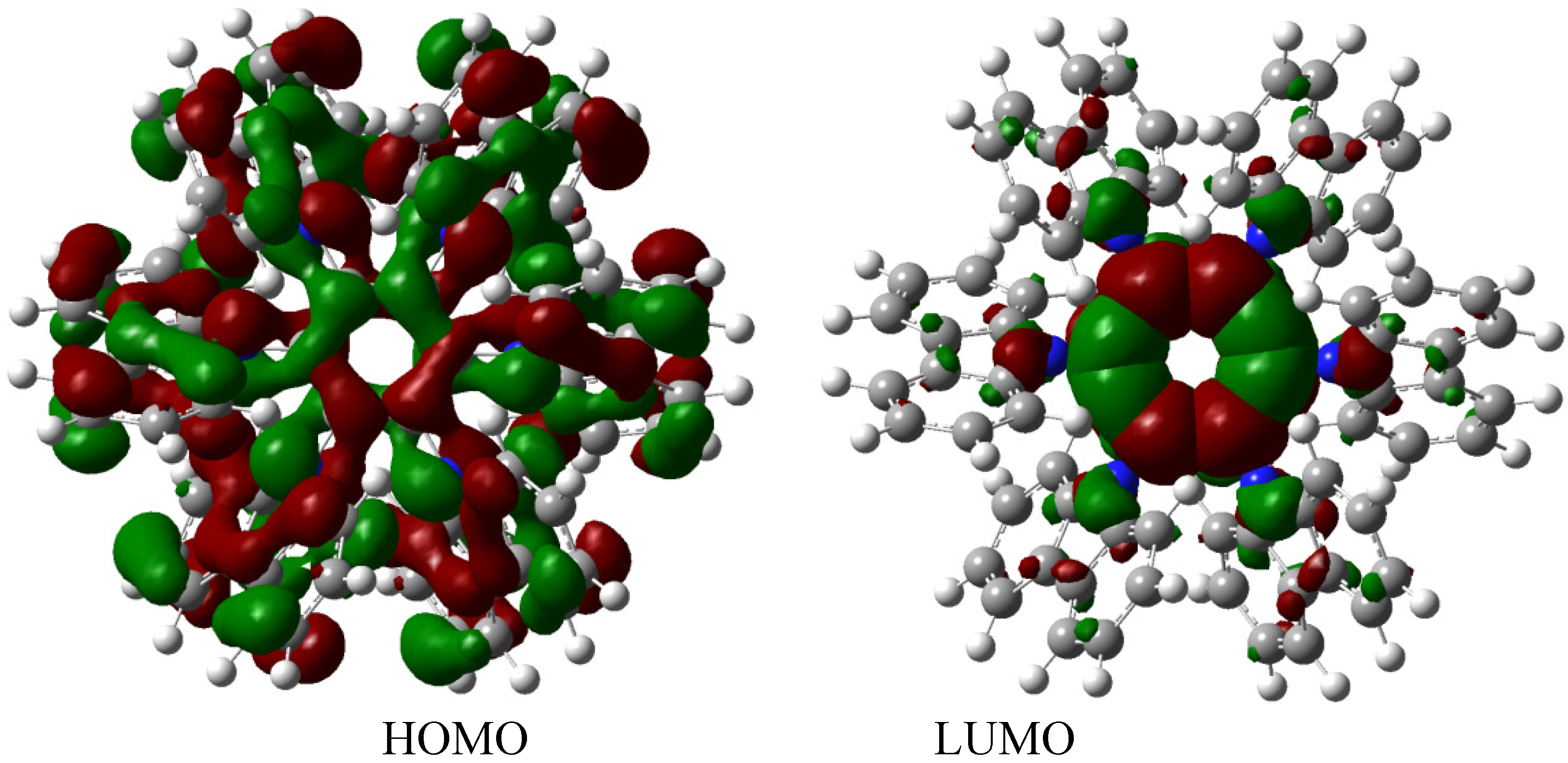
The description of this molecular orbital shares certain characteristics with previous examples because there are three molecular orbitals contributing to the set of the HOMO in an accidental degeneration process, this strong concentration, that is shown in
Figure 7, causes a massive electronic flux on this wave function. However, the LUMO has a very low value (−0.06eV) and the energy gap between frontier molecular orbitals is 3.87 eV; once again demonstrating weak semiconductor behavior. However, in this case, the nature of the LUMO is very different because the greatest concentration of the wave function is found near the central ring and perhaps for this reason the value for toroidal dissipation is so different than those manifested by the last molecule considering that the electrons have the tendency to remain in the central ring and not in the toroidal dispersion.
The behavior of the last three molecules is more or less similar, because all are notable for the substantial energy that they display, indicating toroidal delocalization and semiconductor behavior (or conductor behavior in the case of the ion). However, the situation is not clear in terms of inductive effect, as it must be important to consider the idea of alternation of fragments manifesting varied electronic behavior, if optimum situations for revealing interesting electronic features are to be designed.
Figure 7.
Frontier molecular orbitals for HAB3 derivative.
Figure 7.
Frontier molecular orbitals for HAB3 derivative.
3.2.2. Molecules Manifesting Limited Toroidal Delocalization
The proposal concerning the influence of inductive effect on the generation of extensive or limited toroidal delocalization effects was explored, presenting two molecules with totally contrasting behavior; the first one consisting of a benzene ring totally substituted by the 4-nitrophenyl groups, and the second one, by the 4-aminophenyl groups. The idea is to carry out a similar study to that conducted for the last cases, and then compare the results to a large electron withdrawal species (hexa(4-nitrophenyl)benzene) and an example of total electron release (hexa(4-aminophenyl)benzene). However, there appears to be no clear relationship between the inductive effect and toroidal dissipation, as contrarily both molecules manifest almost the same behavior with the respect to the electronic dissipation, it seems that the presence of a substituent with a known inductive effect can affect the direction of the electronic flux considering electron release or withdraw, but do not affect the quality of this flux The results are as follows:
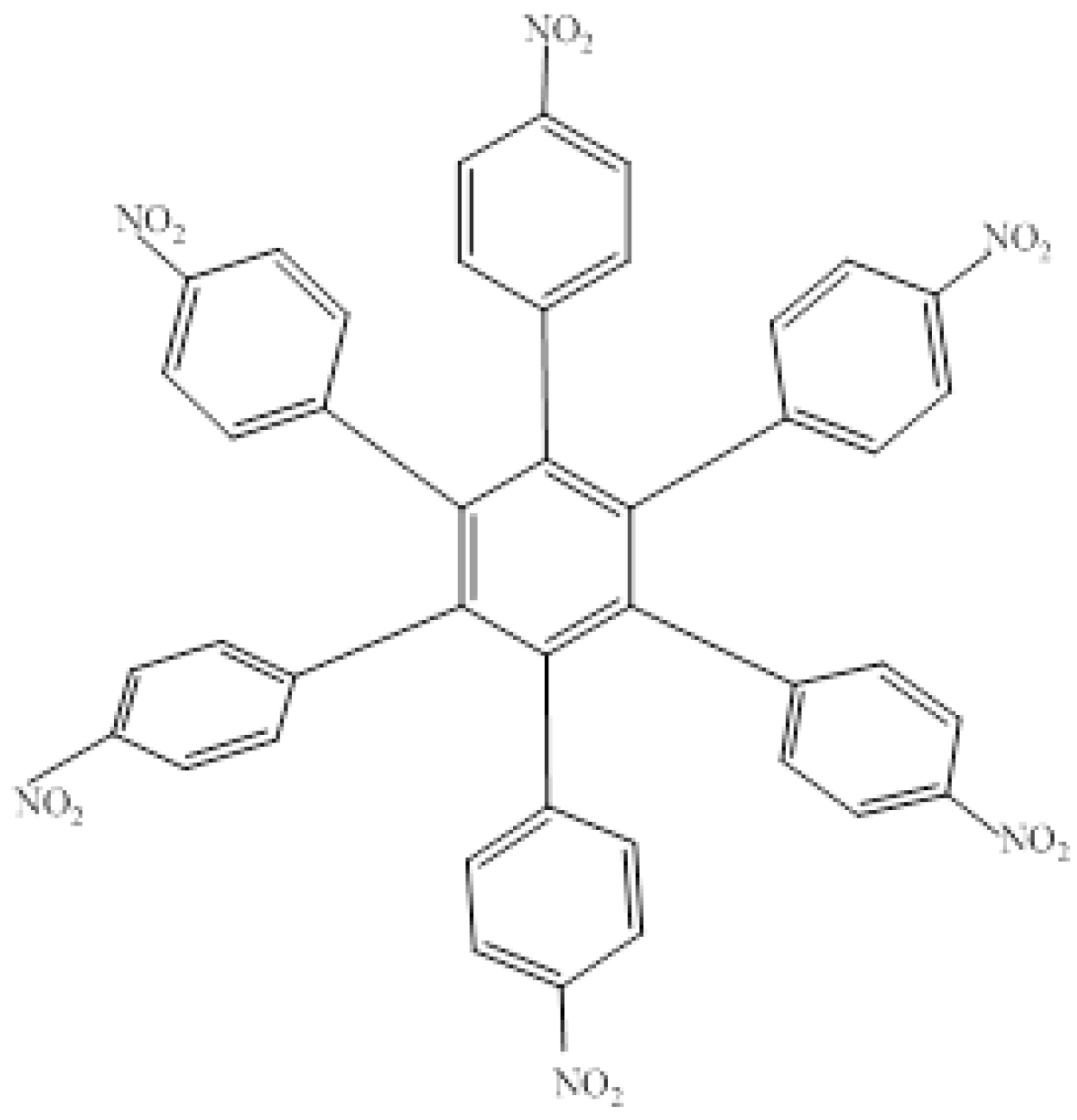
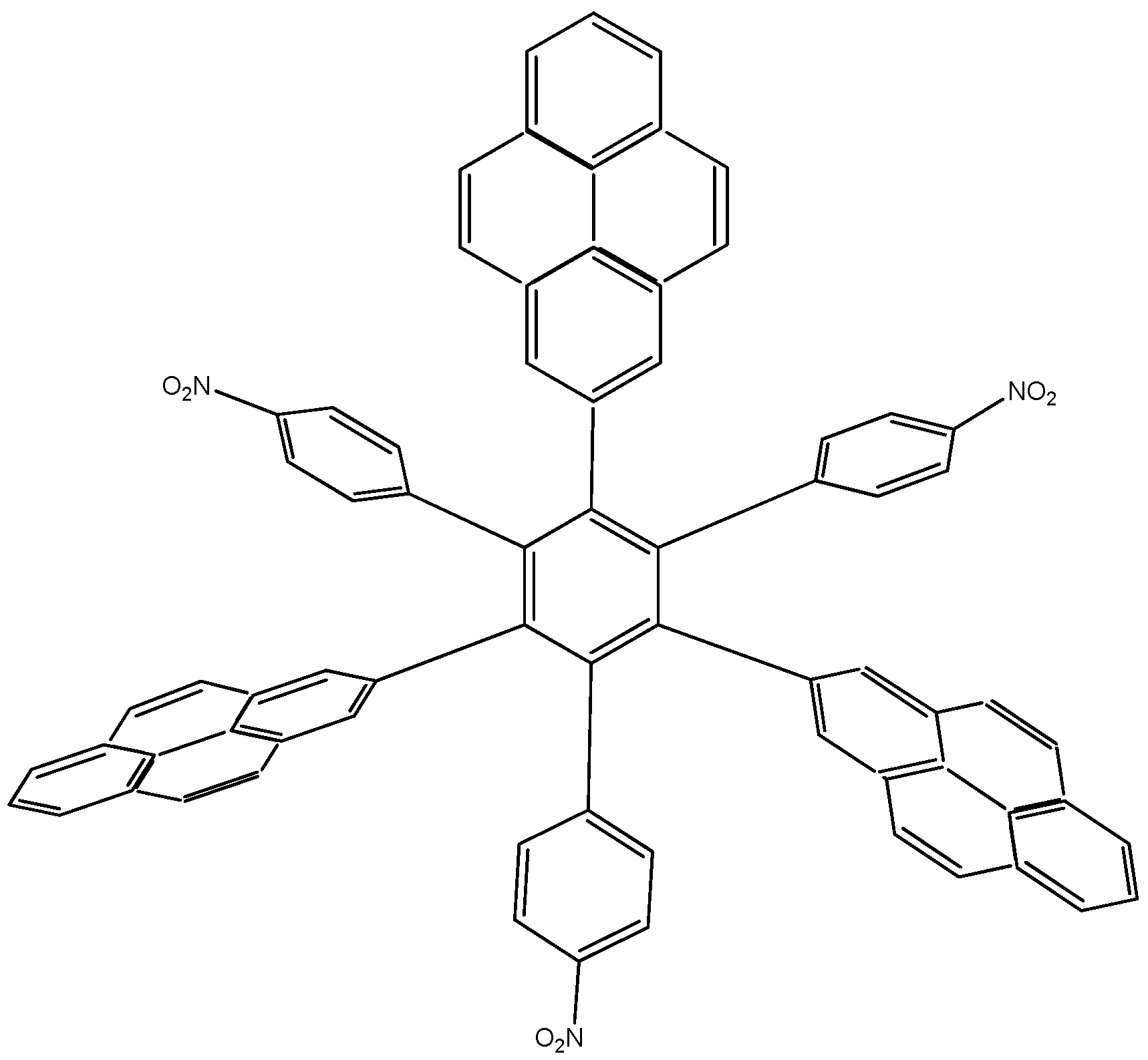
The first molecule to be studied from this set was
HAB4, containing six 4-nitrophenyl groups (see
Figure 8), and the corresponding isodesmic reaction is presented in Equation (8).
Figure 8.
Structure of HAB4.
Figure 8.
Structure of HAB4.
Isodesmic Reaction for HAB4
The corresponding isodesmic reaction for the nitrobenzene molecule is presented above (Equation (3)). It is a known fact that the 4-nitrophenyl group is one of the most aromatic electron withdrawing species [
23], therefore this case is expected to represent an outstanding example of electronic transportation. The result for toroidal delocalization makes a very interesting contrast, when compared with electron-donor examples. The energy value for
HAB4 is −128.95 kcal/mol, and this result subtracted from six nitrobenzene molecules and the central ring (−158.5 kcal/mol) yields 29.57 kcal/mol for the toroidal dissipation. This result is very low when compared with the molecules analyzed above, suggesting that the best targets for designing electronic active molecules must be electron release substituents, a possibility that will be analyzed later. However, the results relating to this molecule deserve careful analysis in order to establish the real nature of these derivatives and their potential application.
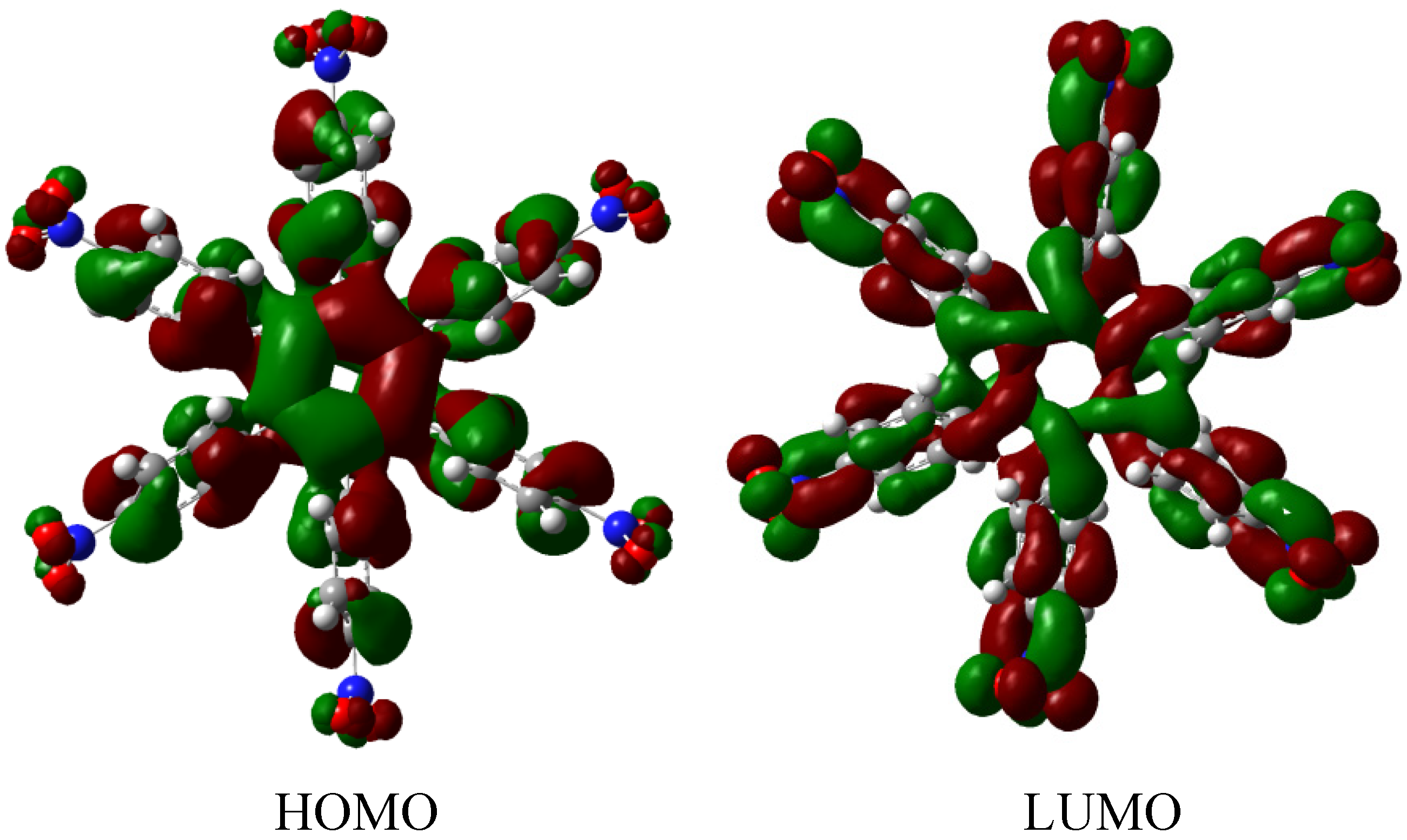
The molecular orbital scheme indicates a very interesting situation because both orbitals are in the same molecular zone, causing that the electronic flow exists between the frontier molecular orbitals; the shape of which are depicted in
Figure 9.
Figure 9.
Frontier molecular orbitals for HAB4.
Figure 9.
Frontier molecular orbitals for HAB4.
However, the behavior will not be as good as this scheme predicts because the energy gap between HOMO and LUMO is 4.32 eV, suggesting insulator behavior and as both frontier molecular orbitals occupy the same probabilistic regions; this predicts serious difficulties in terms of electronic transportation, leading to the conclusion that this does not represent a good example of an active electronic molecule.
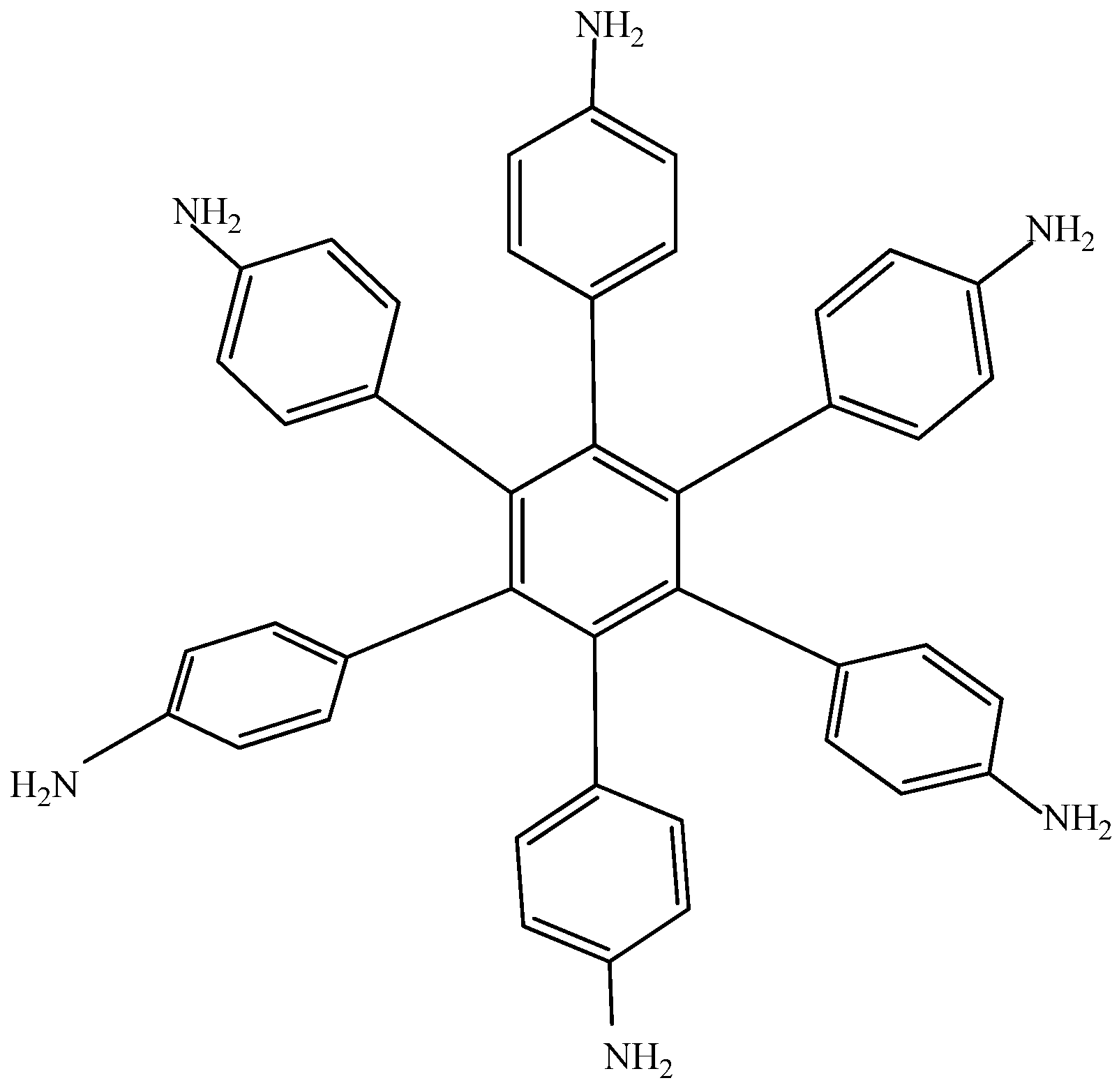
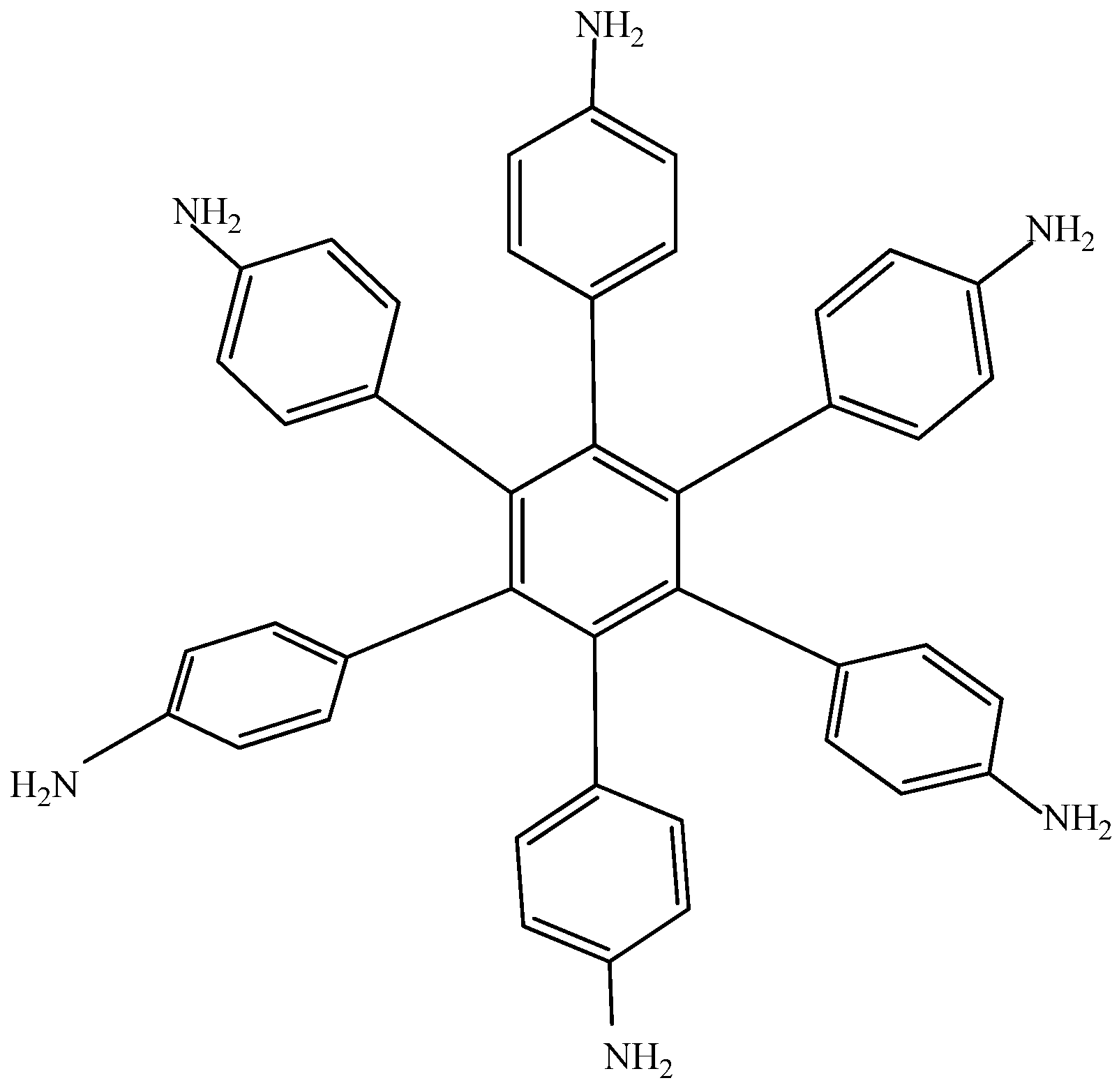
Electronic-withdrawal substituents represent predictably bad targets in contrast to molecules surrounded by electron release substituents, which comprise very good targets. For this reason,
HAB5 was designed. This species contains six 4-aminophenyl substituents and shows very interesting results. The shape of
HAB5 is presented in
Figure 10.
Figure 10.
Structure of HAB5.
Figure 10.
Structure of HAB5.
The isodesmic reaction for
HAB5 is as follows:
Whereas the isodesmic reaction for evaluating aniline is presented in equation 10.
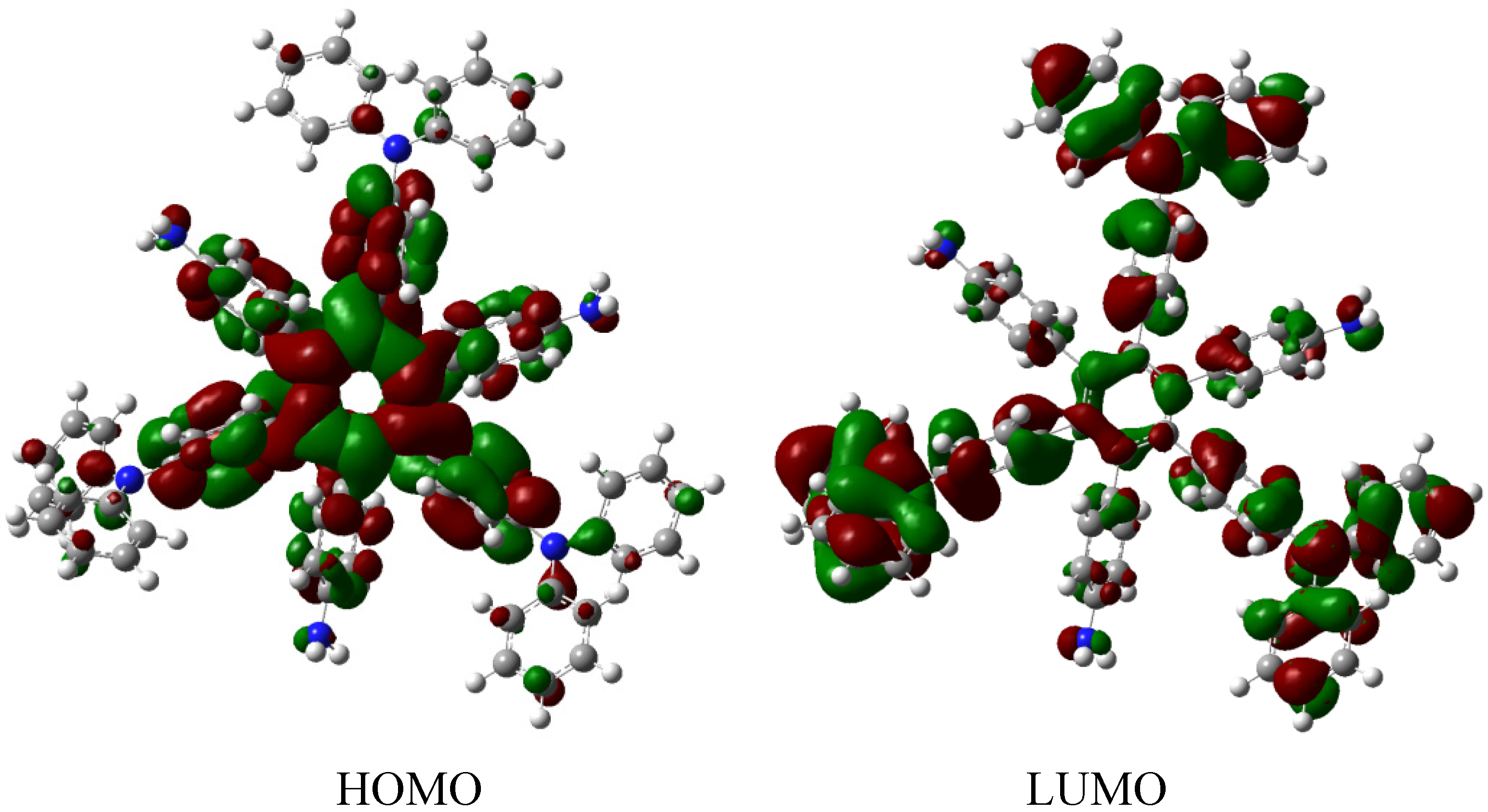
The result of the energy is −264.49 kcal/mol for HAB5, and −44.23 kcal/mol for the aniline molecule, therefore considering the central ring and six 4-nitrobenzene molecules, the toroidal dissipation for this molecule is 21.99 kcal/mol, indicating a very similar situation to that found in the previous example. However, the nature of the substituents is completely different because in this case the amino substituent causes the electron release effect. Likewise, the different origin and the values for the inductive effect are not sufficient to make a significant difference, thus it seems to be imperative to have regions with very similar energy i.e. short energy gap between HOMO and LUMO, where the flux of the electrons is able to take place and where electrons are very mobile.
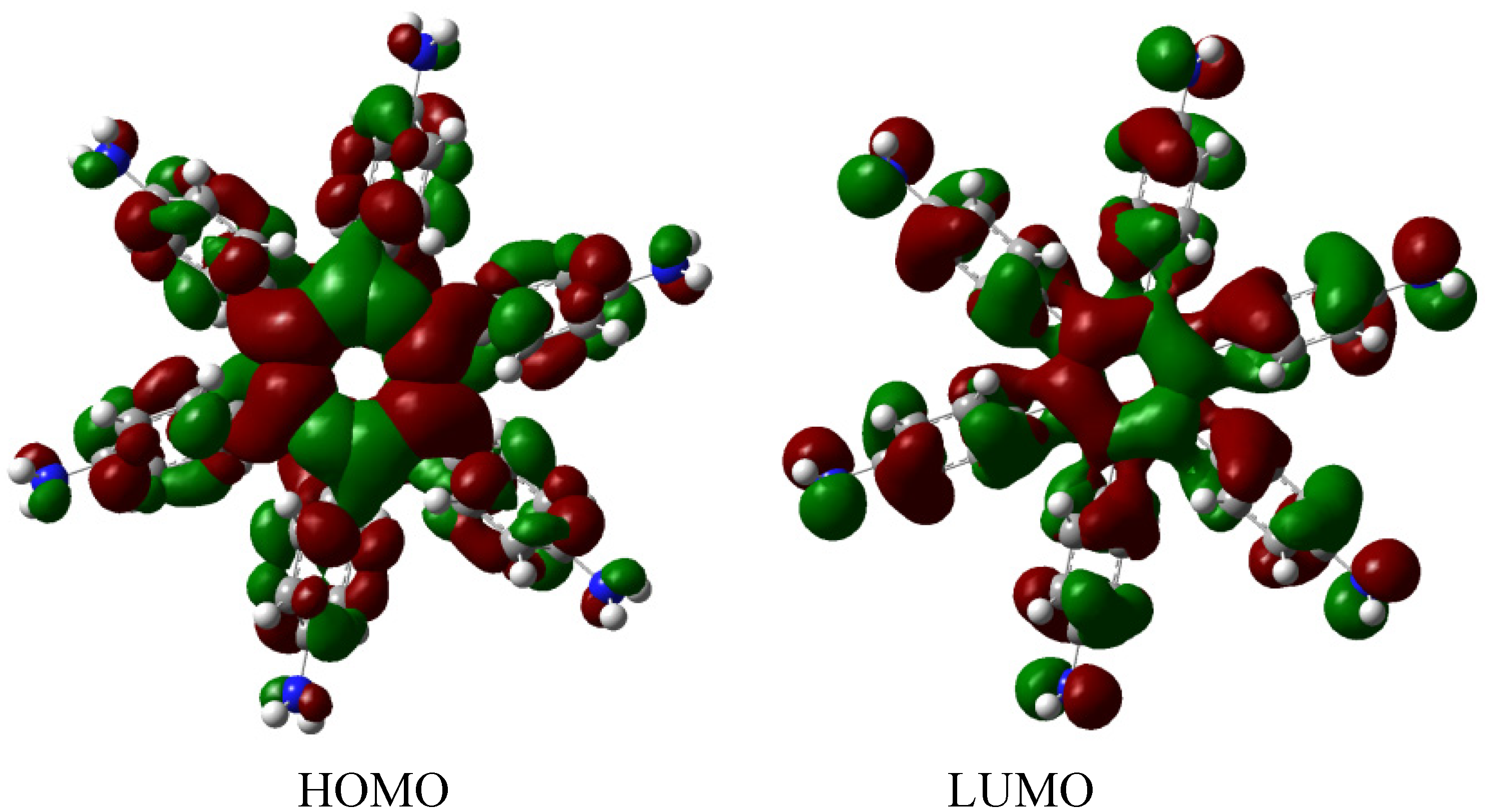
The molecular orbital distribution indicates an unusual situation because the molecule in the frozen structure belongs to the C
6 point group, a group that has no triple degenerated irreducible representations; however the distribution in the HOMO, HOMO-1 and HOMO-2 manifests such minor differences that there is a suggestion of triple accidental degeneration. LUMO is a positive molecular orbital and the energy gap between HOMO and LUMO is 4.51 eV, once again indicating insulator behavior, therefore, the energy gap seems to be fundamental in order to explain the electronic dissipation because large gaps lead to the lack of relationship between the frontier molecular orbitals. Therefore, an important point is the nature of the shape of these molecular orbitals, which are presented in
Figure 11.
Figure 11.
Frontier molecular orbitals for HAB5.
Figure 11.
Frontier molecular orbitals for HAB5.
These distributions suggest that the electronic flux should be guaranteed because there are important concentrations in both frontier molecular orbitals, however, the large energy gap gives place to a lack of relationship between the mentioned orbitals as in HAB4; thus it would appear that an electronic flow between these two functions is improbable.
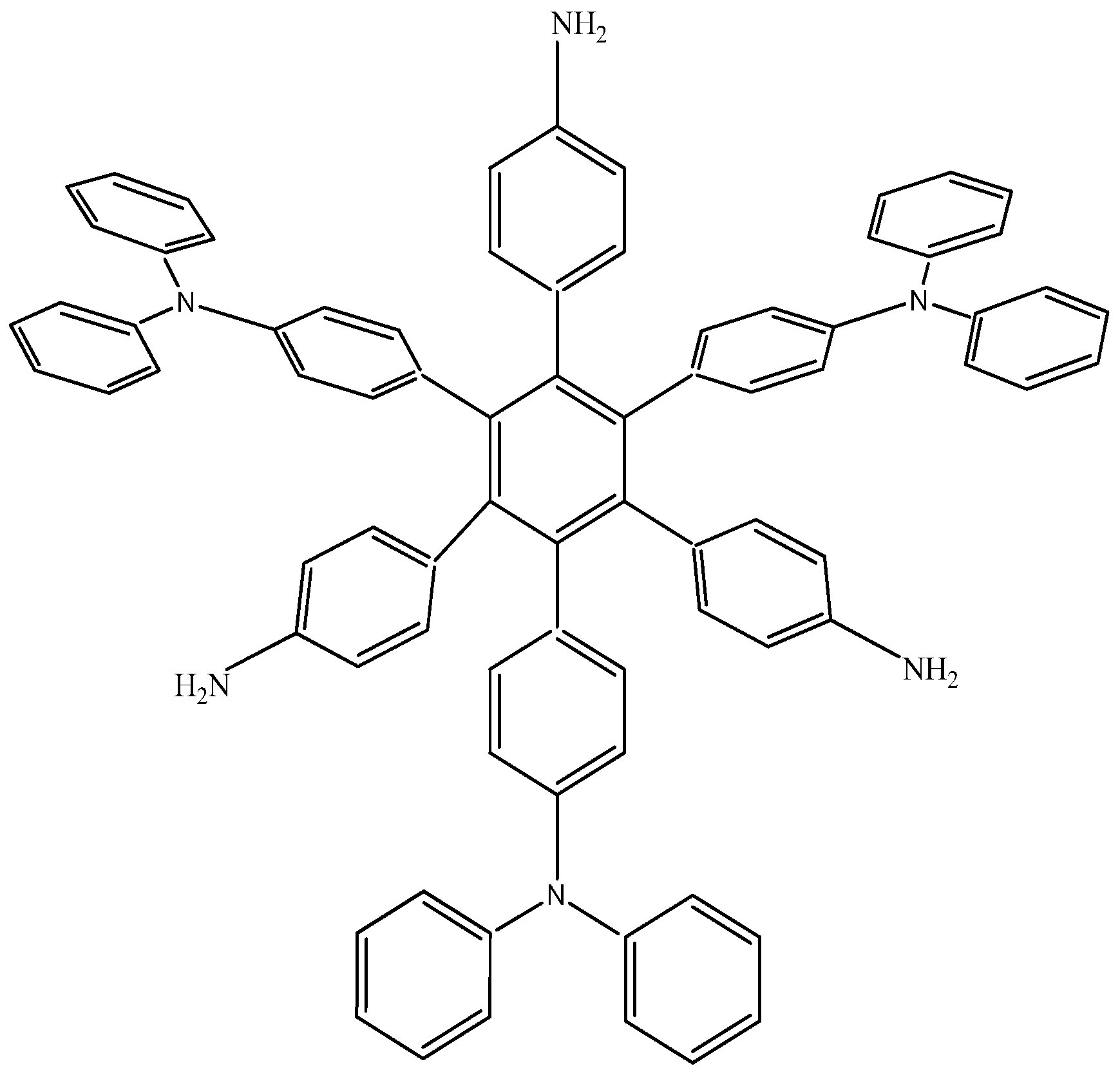
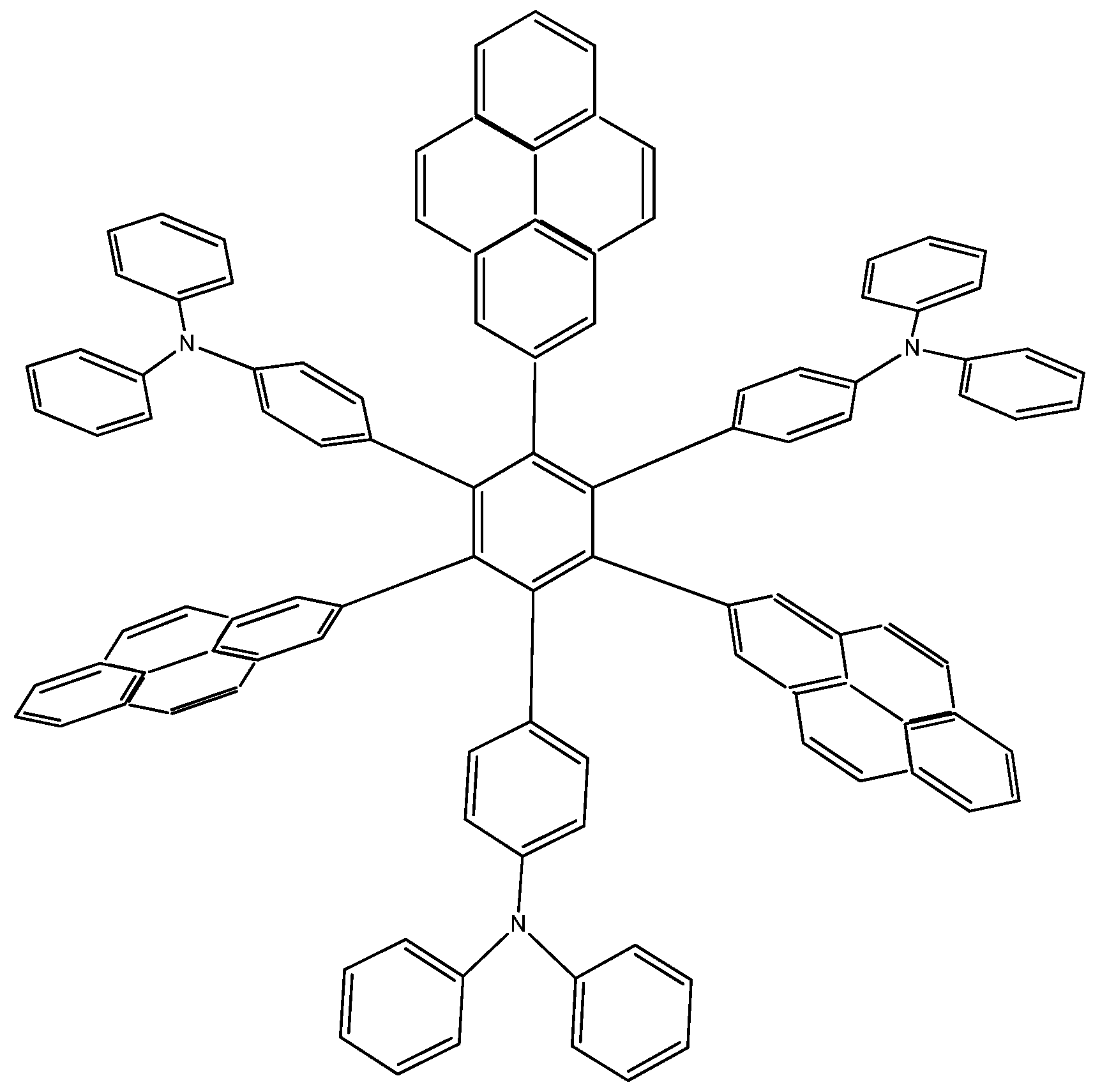
The last example in this section shows a combination of electron-donor substituents in an attempt to confirm the last hypothesis; it has three 4-aminophenyl groups and three 4-(diphenylamino)phenyl groups and was denominated
HAB6 (see
Figure 12).
Figure 12.
Structure of HAB6.
Figure 12.
Structure of HAB6.
The isodesmic reaction for
HAB6 follows:
Whereas the isodesmic reaction for triphenylamine is:
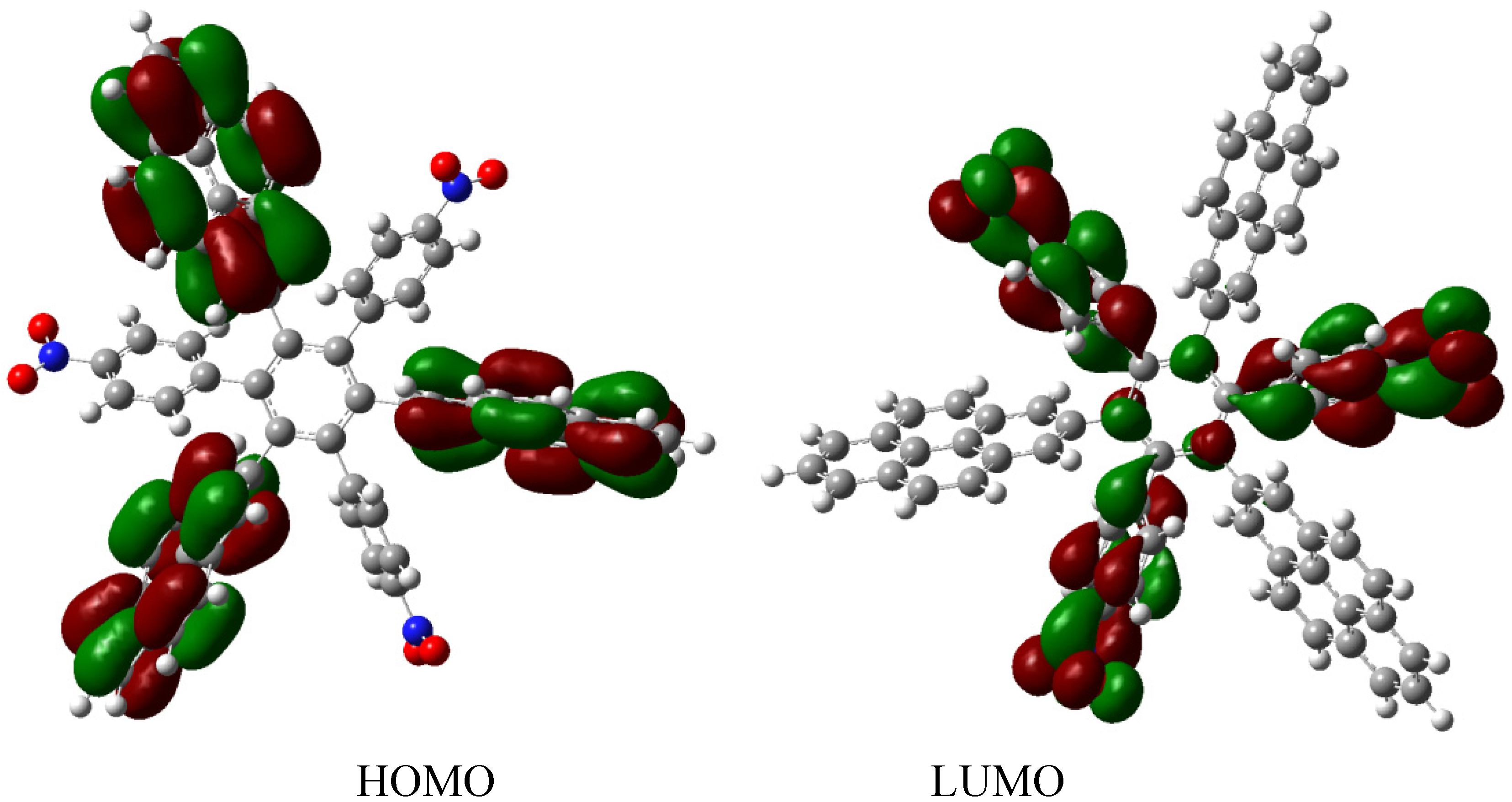
The energy result for this reaction is −97.21 kcal/mol. Therefore, considering this and the other results, the last values are −423.54 kcal/mol for the
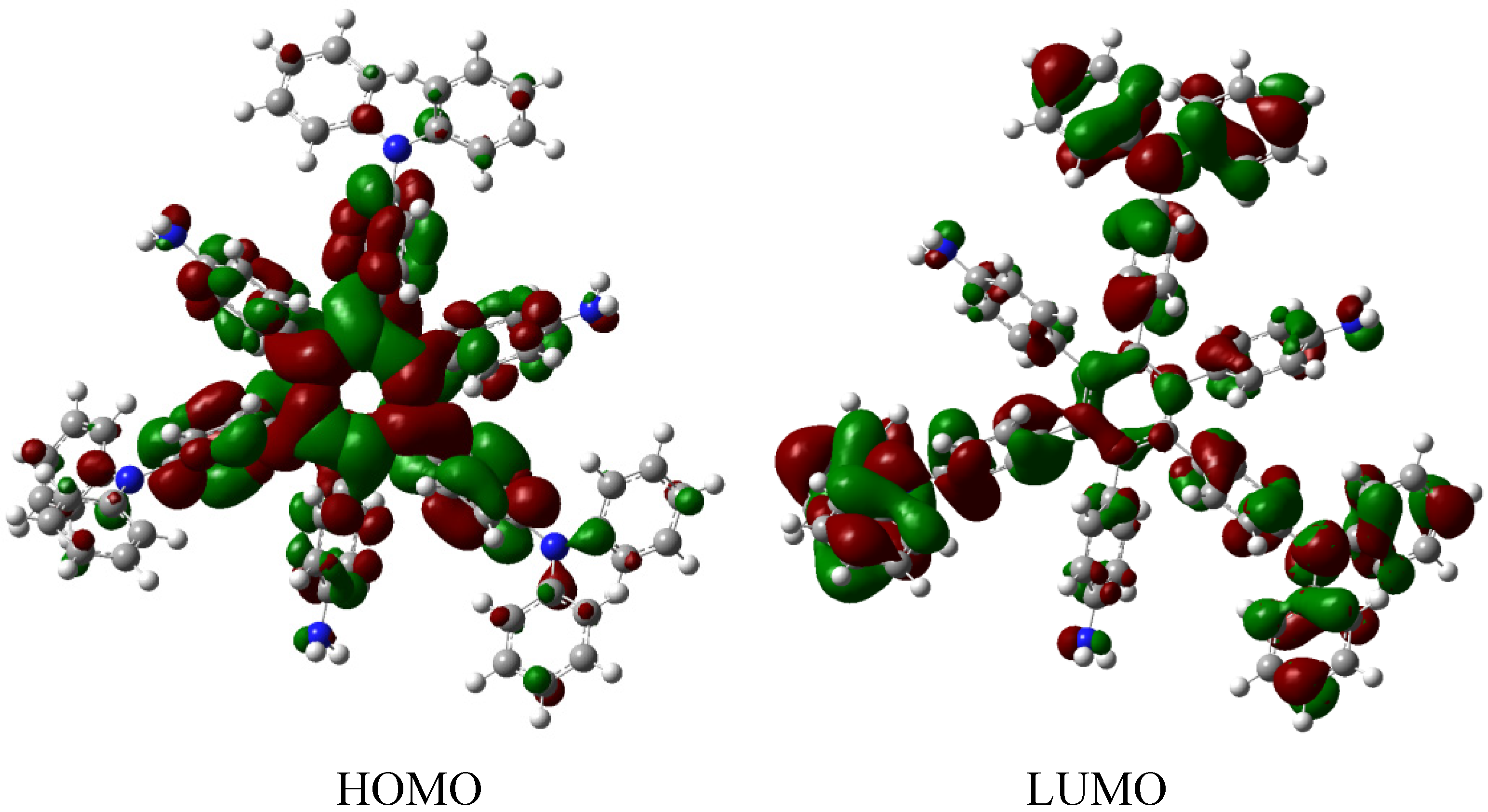
HAB6, −443.53 kcal/mol for the three aniline (see equation 10), and three triphenylamine plus −21.1 from the central ring with a resulting toroidal dissipation value of 19.76 kcal/mol. Considering these results of this molecule it seems that the combination is not appropriated for the electronic dispersion and this feature can be explained with reference to the molecular orbital analysis and the shape of HOMO and LUMO that is shown in
Figure 13.
Figure 13.
Frontier molecular orbitals for HAB6.
Figure 13.
Frontier molecular orbitals for HAB6.
The situation of the molecular orbitals for HAB6 is somewhat different to that indicated in the other examples. Similar behavior to that of hexa(4-aminophenyl) benzene (HAB5) is predicted because it only contains electron donor substituents. However, the HOMO shows that (4-diphenylamino) phenyl substituents are likely to be present, mainly with the aromatic ring joined to the central ring whereas the 4-aminophenyl orbitals indicate medium participation. In contrast, the LUMO indicates participation on the part of all substituents, including the central ring. Therefore the description suggests that electronic flow goes from an electron donor (i.e., the (4-diphenylamino) phenyl fragments) towards the periphery and also towards the central regions. This phenomenon is the result of combining two types of electron donor species; however, it seems that the mixing of two donors does not result in a very good generator of toroidal delocalization, as the energy gap in this case is 4.08 eV, once again suggesting insulator behavior. Thus it appears that electronic flow is inhibited when fragments are of the same nature.
3.2.3. Molecules Containing the Pyrenyl Groups
Pyrene is one of the most peculiar polyaromatic hydrocarbons [
24,
25]. It would appear to have different aromatic regions within its perimeter and to be able to work either as an electron donor or as an electron acceptor depending on the environment. It has particular electronic characteristics and is also known to be a very strong carcinogenic agent. This chameleonic fragment is likely to be useful for designing OLED’s [
26,
27] and is thus likely to be apt for designing good toroidal delocalization derivatives; indeed this idea is proposed by Lambert [
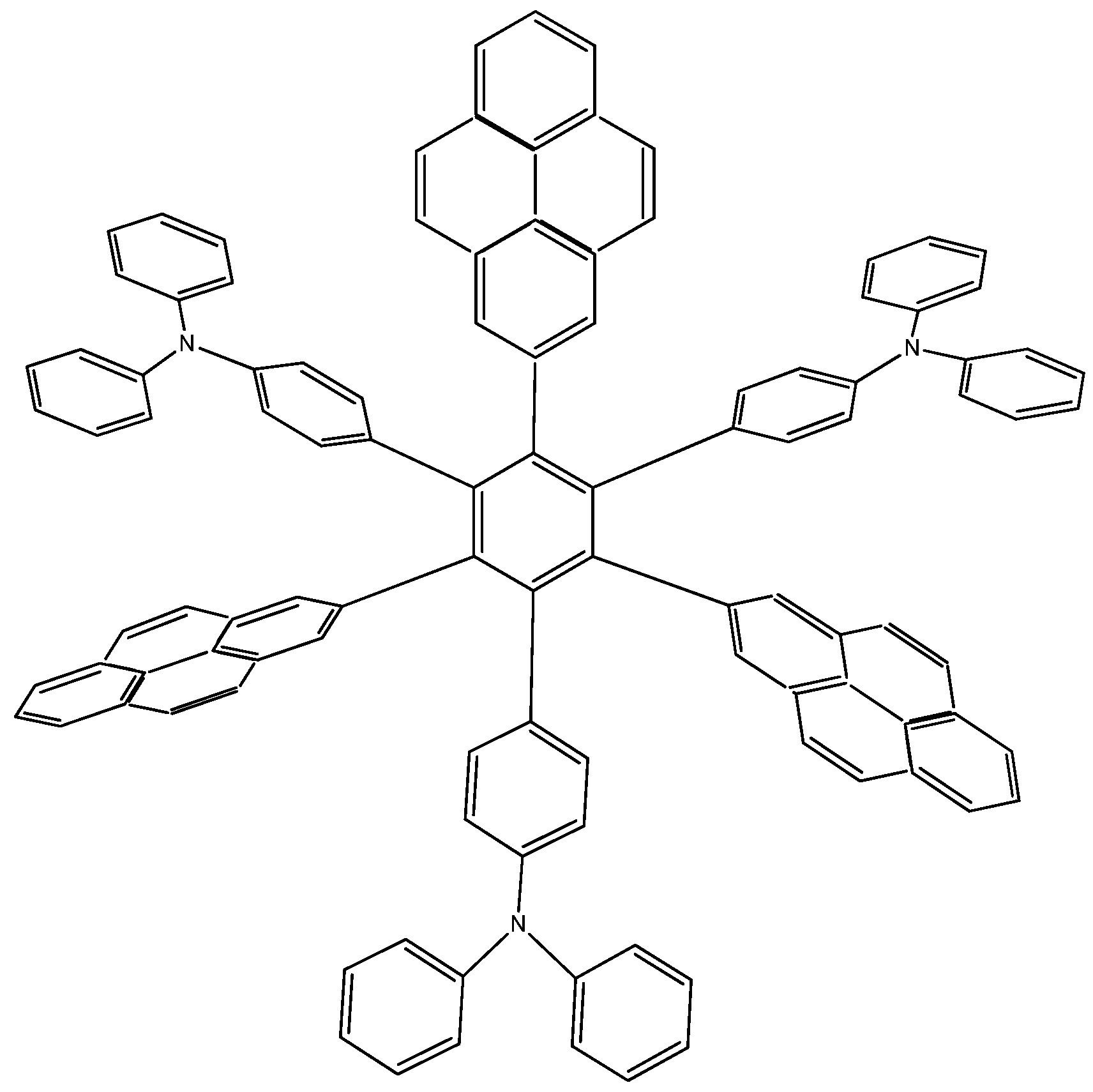
9]. Due to the ambiguous nature of this molecule, it can act either as an electron acceptor or donor, so that the molecule proposed by Lambert, as well as two more locally designed species were studied here. Results contrast with those presented above; the first example
HAB7, that contains three 2-pyrenyl and three (4-diphenylamino)phenyl substituents (
Figure 14). This molecule is precisely that provided by Lambert.
Figure 14.
Structure of HAB7.
Figure 14.
Structure of HAB7.
The corresponding isodesmic reaction for
HAB7 is:
Whereas the isodesmic reaction for pyrene is:
The energy result for
HAB7 derivative is −599.58 kcal/mol, is subtracted from −612.213 kcal/mol, representing three pyrene and three triphenylamine molecules plus the central ring, and yielding 12.63 kcal/mol for the toroidal delocalization result. This value is not as good as that obtained for
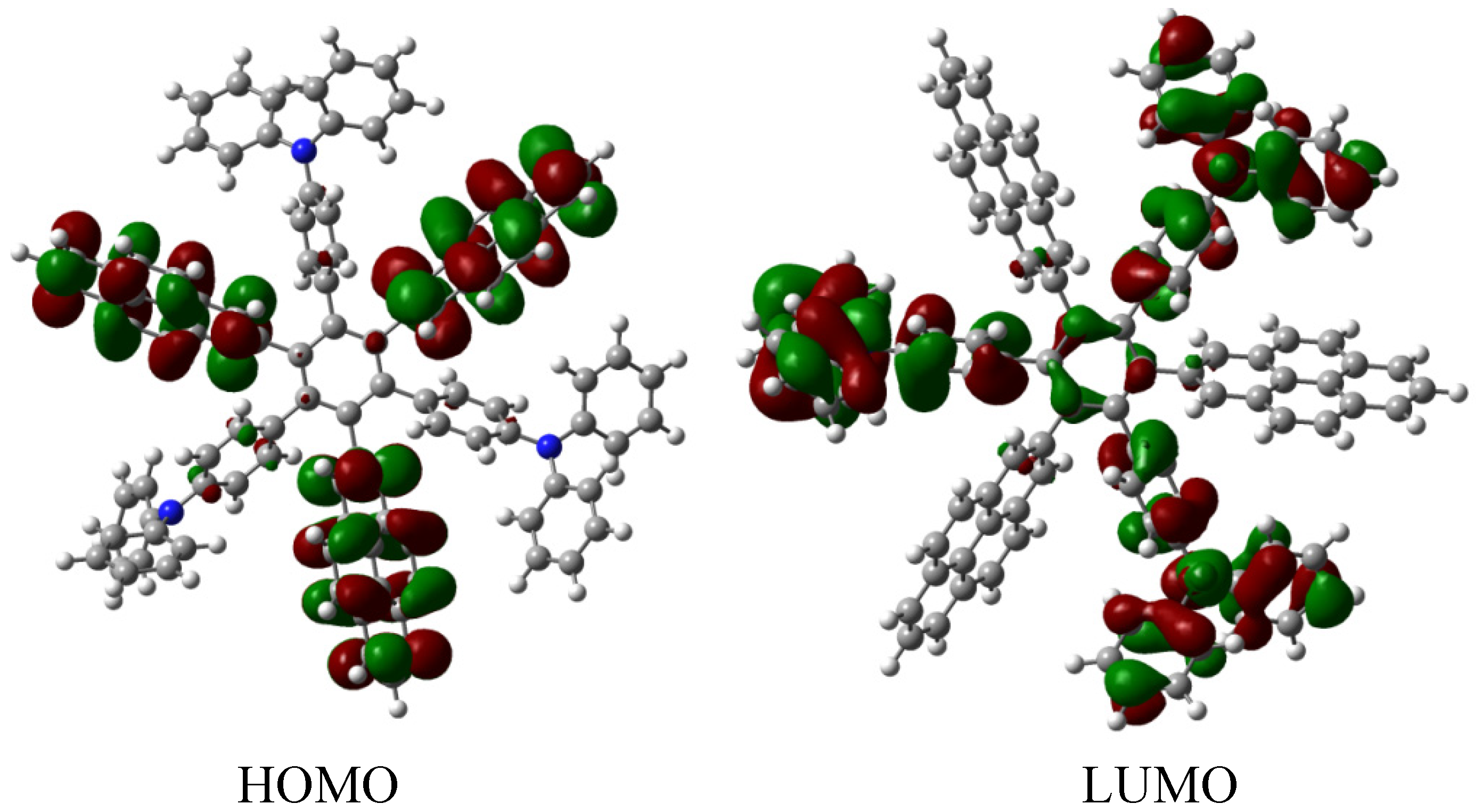
HAB5; however, it deserves to be analyzed as an example of frontier molecular orbitals seeking for a good orbital combination that promotes electronic transfer. The frontier molecular orbitals are presented in
Figure 15.
Figure 15.
Frontier molecular orbitals for HAB7.
Figure 15.
Frontier molecular orbitals for HAB7.
The frontier molecular orbitals manifest very different distributions; the only possibility in the case of the HOMO relates to the (4-diphenylamino) phenyl substituents with no other participation, whereas the LUMO is formed entirely from the wave functions derived from the 2-pyrenyl fragments. This behavior is very similar to the case of the electron withdrawal substituents studied above. The curious factor lies in the fact that the energy gap for this molecule is 3.2, eV, representing the best value obtained in these calculations for a set of neutral molecules, although it has the smallest value for toroidal dissipation, concurring with Lambert’s results, thus indicating that the molecule has good characteristics in terms of an active electronic species [
9]. This result establishes hope for the other cases because a small value for toroidal dissipation will not have great impact, if the molecular orbitals help to generate good communication for electronic transfer.
Another example was designed with the 2-pyrenyl fragment in the molecules, with the intention of including an electron-withdrawal fragment. This feature was used to evaluate possible influence in terms of electron mobility in this type of group, and thus pyrene was combined with nitrobenzene (
HAB8 derivative), once again giving an intriguing result. The shape of the molecule is depicted in
Figure 16.
Figure 16.
Structure of HAB8.
Figure 16.
Structure of HAB8.
The corresponding isodesmic reaction for
HAB8 is:
The energy results are similar to the previous example, −365.77 kcal/mol for this molecule that is subtracted from −390.06 kcal/mol, derived from the substituents and the central ring, so that the result for toroidal dissipation is 24.29 kcal/mol; a value that is very similar to that for HAB4.
The molecular orbitals scheme is notorious in the case of
HAB8 (see
Figure 17), because of the particular chemistry of pyrene [
24]. In LUMO, the only probabilities are derived from the 4-nitrophenyl fragments together with the central ring, whereas the HOMO only has the probability of the 2-pyrenyl fragments themselves, this behavior is the opposite to that mentioned above in
Figure 15 for the case of
HAB7 with the (4-diphenylamino) phenyl and 2-pyrenyl groups. This is predictable due to the contrary nature of these ligands, one manifesting electron-release activity and the other electron-withdrawal activity. This description can also be useful because electronic flow is directed entirely from the HOMO which contains the 2-pyrenyl fragments towards the LUMO with the 4-nitrophenyl substituents; the important point being that the energy gap between HOMO and LUMO for this case is 2.84 eV, a value which reveals strong semiconductor character and guarantees the electronic flow between all the lateral substituents. Thus this instance is very similar to that of other molecules containing pyrene,
i.e., a low energy gap, alternation between frontier molecular orbitals and moderate electron toroidal delocalization.
Figure 17.
Frontier molecular orbitals for HAB8.
Figure 17.
Frontier molecular orbitals for HAB8.
In order to assess these last assertions, a new molecule (
HAB9), containing pyrene was designed, in this case, the 2-pyrenyl fragment was accompanied by the (4-carboxylate) phenyl one, and the shape of the resulting molecule is presented in
Figure 18.
Figure 18.
Structure of HAB9.
Figure 18.
Structure of HAB9.
The corresponding isodesmic reaction for
HAB9 is:
The isodesmic reaction yields the energy value of −389.21 kcal/mol, whereas the value of three pyrene molecules, plus three (4-carboxylate) phenyl anions, plus the central ring is −440.902 kcal/mol, which again is subtracted from the value cited above; therefore the toroidal electronic delocalization energy is 51.692 kcal/mol, a greater value than that for the last two examples.
The frontier molecular orbitals for
HAB9 (see
Figure 19), manifest the same behavior as the other pyrene derivatives, a HOMO that supports the negative charge on the carboxylate surface and a LUMO that is completely localized on the 2-pyrenyl fragments, however the energy gap for this case in 1.74 eV, a predictably narrow value, owing to the three negative charges.
Figure 19.
Frontier molecular orbitals for HAB9.
Figure 19.
Frontier molecular orbitals for HAB9.
The behavior of the molecule is very similar to that of its counterparts; conductor species, isolated frontier molecular orbitals and narrow energy gap. However, it is important to note the role played by the 2-pyrenyl fragment, in the present case this represents a very pure probability function localized on the LUMO, but in the last description where it is complemented by the 4-nitrophenyl group, it works as a HOMO, besides the fact that the carboxylate group bears a negative charge causes that the HOMO is practically completely localized on this radical. However, in the other example where the pair is (4-diphenylamino) phenyl substituent, the 2-pyrenyl fragment is once again localized in the LUMO. This indicates the chameleonic behavior cited above, pyrene adapts easily to the environment and works as a donor when faced with an acceptor, or as an acceptor when faced with a donor. In conclusion, it seems to be a good idea to include this fragment or something similar when dealing with this kind of molecule, because it guarantees some kind of electron mobility. An interesting report exists describing how a
HAB derivative containing the 2-pyrenyl fragment was prepared [
28], where the compound shows very interesting spectroscopic properties and indeed manifests a strong and red-shifted fluorescence, a fact suggesting interesting behavior. However, in the present study this was not included because in this particular species there are no other fragments that can serve as electronic complements.
It is also important to note that a very interesting report on hexa(4-aminophenyl)benzene has been written describing its interesting electronic and spectroscopic characteristics [
1]. This fact is important in terms of the type of study presented here, because the electronic characteristics of this molecule were shown to constitute one of the most discrete in the entire context, therefore it is probable that the behavior manifesting stronger electronic manifestations can be addressed in experimental manner in the near future.
A curious detail worthy of comment is that the HOMO-LUMO energy gap for all cases was achieved and compared in
Table 1, where notably all the electron donor substituent species are shown to be insulators, furthermore the compound totally substituted by 4-nitrophenyl fragments indicates the same behavior, even though the NO
2 derivative is a strong electron acceptor. Moreover, all the species containing the 2-pyrenyl fragment manifest the behavior of semiconductors, whereas the one containing NO
2 and diazine appears to be a borderline species. All charged molecules manifest marked conductor behavior. This feature should be taken into account because the capacity for electron transfer (low energy gap) is also important for designing a good material, in spite of the magnitude of the energy corresponding to toroidal dissipation.
Table 1.
Comparison among HOMO-LUMO energy gap of the HAB derivatives under study.
Table 1.
Comparison among HOMO-LUMO energy gap of the HAB derivatives under study.
| Substituents | HOMO-LUMO Gap
(in eV) |
|---|
| 4-Aminophenyl (HAB5) | 4.38 |
| 4-Nitrophenyl (HAB1) | 4.307 |
| 4-Aminophenyl, (4-diphenylamino) phenyl (HAB6) | 4.084 |
| 9-Carbazolyl (HAB3) | 3.841 |
| 4-Nitrophenyl, (4,6-dinitro-2-pyrimidyl) (HAB2) | 3.62 |
| 2-Pyrenyl, (4-diphenylamino) phenyl (HAB7) | 3.205 |
| 4-Nitrophenyl, 2-pyrenyl (HAB8) | 2.836 |
| 2-Pyrenyl, (4-carboxylate-phenyl)(HAB9) | 1.747 |
| 4-Nitrophenyl, (4-carboxylate-phenyl) (HAB4) | 1.007 |
The global Mulliken charges values (from selected fragments) can be useful for assessing the donation of electrons from the substituents (and even from the central ring) to the toroidal delocalization. These values were estimated in a way similar to those for ASE,
i.e. the sum of local charges of the aromatic rings on the
HAB’s substituted species were subtracted from the same values derived from the free molecules, the results are known as relative charge and are presented in
Table 2.
Table 2.
Comparison among relative charges of the HAB derivatives under study.
Table 2.
Comparison among relative charges of the HAB derivatives under study.
| Substituents | Relative Charge |
|---|
| 4-Aminophenyl (HAB5) | 2.71 |
| 4-Nitrophenyl, (4,6-dinitro-2-pyrimidyl) (HAB2) | 2.26 |
| 9-Carbazolyl (HAB3) | 1.96 |
| 4-Nitrophenyl, (4-carboxylate-phenyl) (HAB4) | 1.64 |
| 4-Aminophenyl, (4-diphenylamino) phenyl (HAB6) | 1.37 |
| 2-Pyrenyl, (4-diphenylamino) phenyl (HAB7) | 0.98 |
| 4-Nitrophenyl (HAB1) | 0.96 |
| 2-Pyrenyl, (4-carboxylate-phenyl) (HAB9) | 0.89 |
| 4-Nitrophenyl, 2-pyrenyl (HAB8) | 0.68 |
From a summary of the phenomena, it is possible to suggest the best targets for compounds with potential electronic device application: in first place those compounds bearing a negative charge, obviously show a narrow energy gap, good energy dissipation and reasonable charge values; these can therefore be considered for this classification. The other cases present different situations; the species containing diazine or carbazole seem to be the best neutral targets, whereas those containing pyrene are also good in terms of gaps and charges and finally the more symmetrical cases with NO2 and NH2 are not as good as the other cases, possibly because of their marked symmetry and also taking into account that they fall into the category of extreme inductive effect. Thus this collection of phenomena is likely to indicate a more symmetric distribution of electrons, resulting in lack of mobility.
Indeed it would appear that a good target must constitute a molecule that fulfills several requirements; firstly manifesting toroidal dissipation, even though the energy magnitude of this should not constitute an important factor. However, this characteristic is very important for enabling electronic transit. Secondly, it should have a narrow HOMO-LUMO gap, although this may be larger than that of the classical representation of conductor, semiconductor and insulator species. Thirdly, it should manifest effective alternation in terms of the frontier molecular orbitals, as well as availability of electrons and/or holes to facilitate flow.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}