1. Introduction
Acerola (
Malpighia emarginata DC.) is a shrub grown in tropical and subtropical areas. It has been introduced into many provinces of China including Guangxi, Guangdong, and Yunnan,
etc. Acerola fruits are mainly utilized by the supplement, pharmaceutical, and fruit-juices industries as a rich source of vitamin C [
1]. However, recent research showed that besides vitamin C, acerola fruits may be also a good source of phytochemicals such as anthocyanins [
2,
3], flavonoids and phenolic acids [
4], and polyphenols [
2,
5]. With respect to bioactivities, acerola showed antioxidant [
6], antimicrobial [
7], hepatoprotective [
8], and anti-hyperglycemic [
9] effects. Nevertheless, there is seldom report on the bioactivity constituents from aerial parts of acerola.
In our previous research, three norfriedelins with acetylcholinesterase inhibitory activity were found in acerola tree (
M. emarginata) [
10]. As continuation of this work, we have examined the lipophilic constituents of aerial parts of acerola collected in Nanning, Guangxi Province, China. Three new tetranorditerpenes (
1–
3) with a rare 2
H-benz[
e]inden-2-one substructure were obtained (
Figure 1). This report describes the isolation and structural determination of the compounds, as well as their cytotoxic activities.
Figure 1.
Structures of compounds 1–3 from the aerial parts of acerola.
Figure 1.
Structures of compounds 1–3 from the aerial parts of acerola.
2. Results and Discussion
Compound
1 was obtained as yellow power and had a molecular formula C
16H
14O
4 by HREI-MS ion at
m/z 270.0900 [M]
+ with 10 degrees of unsaturation.
13C-DEPT (
Table 1) revealed sixteen resonances consisting of two carbonyls, ten olefinic carbons, one quaternary carbon and three methyl groups. Seven out of ten degrees of unsaturation were occupied by two carbonyls and five double bonds and the remaining three indicated that compound
1 was tricyclic. The 1D NMR data of
1 was similar to those of substructure B in fimbricalyx A [
11]. In the HMBC spectrum (
Figure 2), cross-peaks between δ
H 1.44 (H
3-18 and H
3-19) and δ
C 209.4 (C-3), 46.2 (C-4), and 133.6 (C-5) suggested that the carbonyl was located at C-3. According to the HSQC spectrum, two hydroxyl proton signals were identified at δ
H 7.27 and 12.08. Correlations from δ
H 7.27 to 133.6 (C-5), 141.0 (C-6), and 184.6 (C-7), from δ
H 12.08 to 112.9 (C-8), 131.5 (C-13), and 160.9 (C-14) in the HMBC spectrum suggested that two hydroxyl groups were attached to C-6 and C-14, respectively. Two aromatic proton signals at δ
H 7.41 (1H, d,
J = 6.0 Hz) and δ
H 7.45 (1H, d,
J = 6.0 Hz) were attributed to H-11 and H-12, respectively, by their correlations with each other in the
1H-
1H COSY spectrum and by the HMBC correlations from δ
H 7.41 to C-13, C-8, and C-10, from δ
H 7.45 to C-14 and Me-15. The formation of an intramolecular hydrogen bond between 14-OH with 7-(C=O) shifted the proton signal of 14-OH to lower field at δ
H 12.08, which further confirmed the C-7 location of the carbonyl group. Thus, the structure of compound
1, named acerolanin A, was identified as shown in
Figure 1.
Table 1.
1H- and 13C-NMR data for compounds 1–3 (in CDCl3, 600 MHz for 1H and 150 MHz for 13C, δ in ppm).
Table 1.
1H- and 13C-NMR data for compounds 1–3 (in CDCl3, 600 MHz for 1H and 150 MHz for 13C, δ in ppm).
| No. | 1 | 2 | 3 |
|---|
| | δC | δH (mult, J in Hz) | δC | δH (mult, J in Hz) | δC | δH (mult, J in Hz) |
|---|
| 1 | 125.9, d | 6.77 (s) | 125.2, d | 6.76 (s) | 184.3, s | |
| 3 | 209.4, s | | 209.9, s | | 205.6, s | |
| 4 | 46.2, s | | 46.3, s | | 43.2, s | |
| 5 | 133.6, s | | 131.0, s | | 163.4, s | |
| 6 | 141.0, s | | 141.3, s | | 98.7, d | 6.80 s |
| 7 | 184.6, s | | 180.1, s | | 164.6, s | |
| 8 | 112.9, s | | 130.7, s | | 133.2, s | |
| 9 | 128.5, s | | 131.9, s | | 128.8, s | |
| 10 | 118.4, s | | 156.3, s | | 125.2, s | |
| 11 | 155.6, d | 7.41 (d, 6.0) | 106.5, d | 7.25 (s) | 124.8, d | 9.05 (d, 8.4) |
| 12 | 136.7, d | 7.45 (d, 6.0) | 162.1, s | | 133.1, d | 7.62 (d, 8.4) |
| 13 | 131.5, s | | 122.8, s | | 137.9, s | |
| 14 | 160.9, s | | 129.8, s | 8.02 (s) | 122.4, d | 8.12 (s) |
| 15 | 16.1, q | 2.35 (s) | 16.8, q | 2.33 (s) | 22.1, q | 2.56 (s) |
| 18 | 21.6, q | 1.44 (s) | 21.7, q | 1.44 (s) | 25.0, q | 1.50 (s) |
| 19 | 21.6, q | 1.44 (s) | 21.7, q | 1.44 (s) | 25.0, q | 1.50 (s) |
| -OMe | | | 56.0, q | 4.00 (s) | 56.6, q | 4.20 (s) |
| 6-OH; | | 7.27 (s) | | 7.01 (s) | | |
| 14-OH | | 12.08 (s) | | | | |
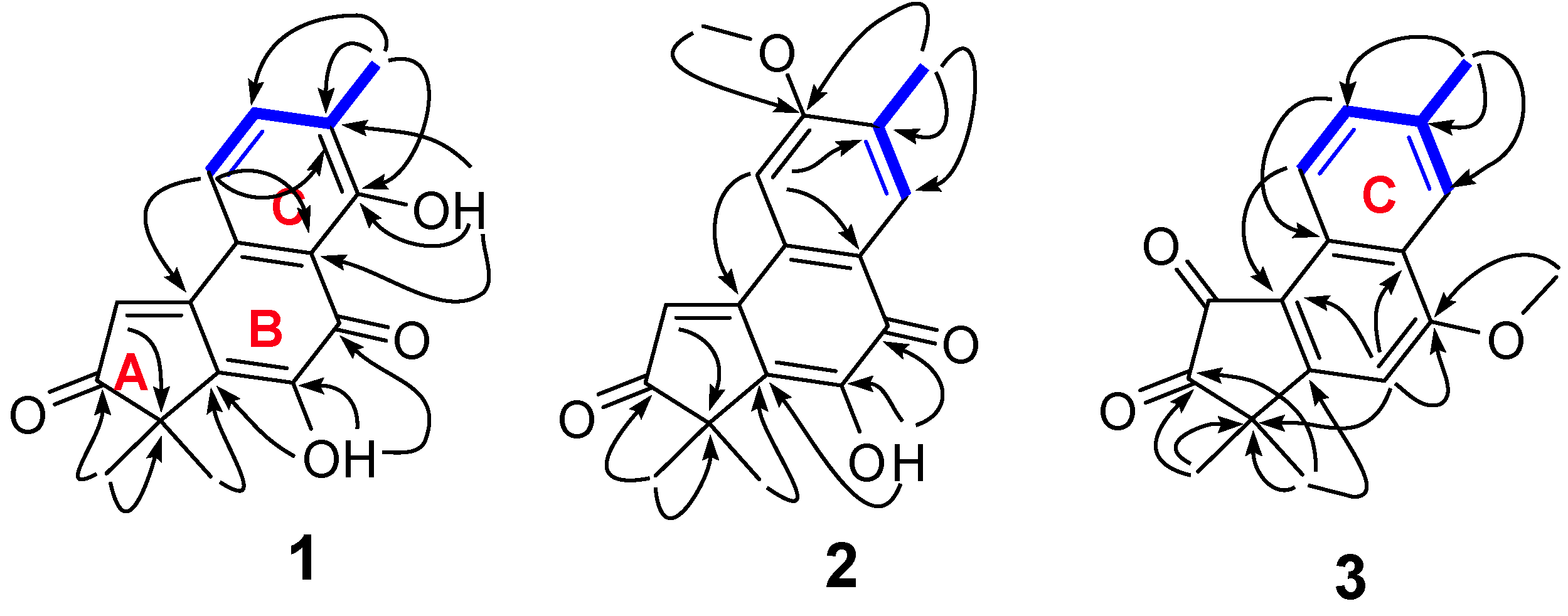
Figure 2.
The key HMBC (H→C) and
1H-
1H COSY correlations (
![Molecules 19 02629 i001]()
) of
1–
3.
Figure 2.
The key HMBC (H→C) and
1H-
1H COSY correlations (
![Molecules 19 02629 i001]()
) of
1–
3.
Compound
2 was obtained as a light yellow power and had a molecular formula C
17H
16O
4 as evidenced by HREI-MS at
m/
z 284.1056 [M]
+. The 1D NMR data (
Table 1) of
2 showed high similarities to those of
1 except that there was one methoxyl group (δ
C 56.0 and
δH 4.00) in
2 instead of one hydroxyl group in
1. The methoxyl group was determined to be attached to C-12 by the following evidence: HMBC (
Figure 2) correlations from
δH 2.33 (15-CH
3) to δ
C 129.8 (C-14), 162.1 (C-12), and 122.8 (C-13), from δ
H 4.00 (MeO-12) to δ
C 162.1 (C-12); Furthermore, the fact that δ
H 7.25 (H-11, 1H) and 8.02 (H-14, 1H) were singlets in the
1H-NMR demonstrated they were not in the
ortho-position as in compound
1. Further detailed study of the HMBC and
1H-
1H COSY data (
Figure 2) determined the other parts of the structure of
2. Therefore, the structure of compound
2 was elucidated as shown in
Figure 1. The compound was named acerolanin B.
Acerolanin C (
3) was obtained as colorless monoclinic crystals from CHCl
3/MeOH (1:3). The molecular formula C
17H
16O
3 was established by the positive HREI-MS (found [M]
+ at
m/
z 268.1097, calcd for C
17H
16O
3 at
m/
z 268.1099), corresponding to 10 degrees of unsaturation. The skeleton of
3 was the same as
2 according to its 1D NMR data (
Table 1). The difference between
3 and
2 was the absence of one hydroxyl group in
3 according to comparison of their formula and
13C-DEPT data (four =CH in
3 and three =CH in
2). As supported by the HMBC correlations from Me-15 to C-12, C-13, and C-14 and by the
1H-
1H COSY between H-11 (9.05, d,
J = 8.4 Hz) with H-12 (7.62, d,
J = 8.4 Hz), there was no substituent at C-11, C-12, and C-14 Thus, the only methoxyl group could be located at C-7 by the HMBC correlations from H-6 to C-4, C-8, and C-10, from MeO-7 to C-7. In the HMBC spectrum, two methyl-proton signals at δ
H 1.50 correlated with a carbonyl-carbon signal at δ
C 205.6 suggested one carbonyl at C-3. Therefore, the other carbonyl should be located at C-1. In order to confirm its structure, the X-ray crystallography of
3 was completed and the result (
Figure 3) allowed unambiguous assignment of its planar structure.
Figure 3.
X-Ray crystal structure of 3.
Figure 3.
X-Ray crystal structure of 3.
Acerolanins A–C are a class of tetranorditerpenoids possessing a rare 2
H-benz[
e]inden-2-one substructure. Analogues have been isolated previously from four species of the Euphorbiaceae, namely,
Neoboutonia glabrescens [
12],
Neoboutonia mannii [
13],
Trigonostemon howii [
14], and
Strophioblachia fimbricaly [
11]. Some compounds of this class were reported to have cytotoxic and antimicrobial activities, so compounds
1–
3 were evaluated for cytotoxicity against HL-60, SMMC-7721, A-549, MCF-7, and SW480 human tumor cell lines using the MTS method. Cisplatin was used as a positive control. Results are summarized in
Table 2. As can be observed, compounds
1–
3 showed moderate cytotoxicity against above five cell lines with IC
50 values from 10 to 40 μM.
Table 2.
Cytotoxicity data of compounds 1−3 with IC50 values (μM) a.
Table 2.
Cytotoxicity data of compounds 1−3 with IC50 values (μM) a.
| Compounds | HL-60 | SD | SMMC-7721 | SD | A-549 | SD | MCF-7 | SD | SW480 | SD |
|---|
| 1 | 10.23 | 0.32 | 12.20 | 0.42 | 12.32 | 0.45 | 16.22 | 0.72 | 18.12 | 0.86 |
| 2 | 14.11 | 0.61 | 16.54 | 0.63 | 18.27 | 0.72 | 22.08 | 1.12 | 24.32 | 1.21 |
| 3 | 22.17 | 1.80 | 20.10 | 1.07 | 31.65 | 1.22 | 28.04 | 1.47 | >40 | - |
| Cisplatin | 1.86 | 0.10 | 6.13 | 0.34 | 7.27 | 0.42 | 15.27 | 0.65 | 16.23 | 0.76 |
3. Experimental
3.1. General Procedures
1H- and 13C-NMR spectra were measured on Bruker AVANCE III-600 instruments (Bruker, BioSpin International AG, Karlsruhe, Germany) with trimethylsilane (TMS) as the internal standard. ESIMS were recorded on a VG Auto Spec-3000 mass spectrometer (VG, Manchester, UK), while HREIMS was measured on an AutoSpec Premier P776 mass spectrometer (Water Corporation, Billerica, MA, USA). IR spectra were obtained on a Bio-Rad FTS-135 spectrometer (Bio-Rad Laboratories Inc., Richmond, CA, USA).
TLC was performed on precoated TLC plates (200–250 μM thickness, F254 Si gel 60, Qingdao Marine Chemical, Inc., Qingdao, China) with compounds visualized by spraying the dried plates with 10% aqueous H2SO4 followed by heating until dryness. Silical gel (200–300 mesh, Qingdao Marine Chemical, Inc.) and Lichroprep RP-18 (40–63 μm, Merck, Darmstadt, Germany) were used for column chromatography. Methanol, trichloromethane, n-hexane, and acetone were purchased from Tianjing Chemical Reagents Co. (Tianjing, China).
3.2. Plant Material
The aerial parts of acerola (Malpighia emarginata) were collected in May 2012 from Nanning, Guangxi province, China. The samples were identified by Prof. Zongyu Wang, Kunming Institute of Botany, Chinese Academy of Science. A voucher specimen (No. KIB 2012-04-20) has been deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3. Extraction and Isolation
The powder and dried aerial parts of M. emarginata (10 kg) were extracted with acetone at room temperature (20 L × 3, 3 days each time) and concentrated in vacuo to give a crude extract (400 g), which was then partitioned in succession between H2O and CHCl3. The CHCl3 fraction (85 g) was chromatographed on silica gel with CHCl3/MeOH gradient elution (100:0→20:1) to afford four fractions: Fr.1 (35 g, 100:0), Fr. 2 (12 g, 100:1), Fr.3 (10 g, 50:1), and Fr.4 (25 g, 10:1). By reverse-phase silica gel (MeOH/H2O, step gradients), Fr.2 was divided into three parts (MeOH/H2O: 40%, 60%, 80%), and 60% part was on chromatography over silica gel with n-hexane/CHCl3 (2:1) to yield acerolanin C (3, 7 mg). Using reverse-phase silica gel (MeOH/H2O, step gradient), Fr.3 was also divided into three fractions (MeOH/H2O: 40%, 60%, 80%). 60% fraction was further chromatographed on a silica gel column gradient eluting with CHCl3/acetone (15:1) to obtain acerolanin B (2, 5 mg), while 40% fraction was subjected to silica gel column chromatography (CHCl3/MeOH, 30:1) to give acerolanin A (1, 8 mg).
3.4. Characteristic Data of Compounds 1–3
Acerolanin A (
1). Yellow powder; UV in CHCl
3 λ
max (log ε): 444 (4.86), 326 (4.98), 239 (4.88). IR (KBr)
vmax 3450, 1680, 1601, 1311, 1236 cm
−1;
1H- and
13C-NMR data: see
Table 1; positive ESI-MS:
m/z 293 [M + Na]
+; positive HREIMS:
m/z 270.0900 [M]
+ (calcd for C
16H
14O
4, 270.0892).
Acerolanin B (
2). Light yellow powder; UV in CHCl
3 λ
max (log ε): 345 (4.52), 275 (4.51), 230 (4.23). IR (KBr)
vmax 3461, 1683, 1637, 1597, 1383, 1270, 1247 cm
−1;
1H- and
13C-NMR data: see
Table 1; positive ESI-MS:
m/z 307 [M + Na]
+; positive HREIMS:
m/z 284.1056 [M]
+ (calcd for C
17H
16O
4, 284.1049).
Acerolanin C (
3).
Colorless Monoclinic crystals from CHCl
3/MeOH (1:3); UV in CHCl
3 λ
max (log ε): 389 (3.98), 274 (4.11), 239 (4.06). IR (KBr)
vmax 1755, 1682, 1570, 1463, 1231 cm
−1;
1H- and
13C-NMR data: see
Table 1; positive ESI-MS:
m/z 291 [M + Na]
+; positive HREIMS:
m/z 268.1097 [M]
+ (calcd for C
17H
16O
3, 268.1099).
Crystal data for 3: C
17H
16O
3,
M = 268.30, monoclinic, a = 8.8643(9) Å, b = 13.7477(14) Å,
c = 11.3957(12) Å, α = 90.00°, β = 104.141(2)°, γ = 90.00°, V = 1346.6(2) Å
3, T = 100(2) K, space group
P21
/c,
Z = 4,
μ(MoKα) = 0.090 mm
−1, 14026 reflections measured, 3824 independent reflections (
Rint = 0.0282). The final
R1 values were 0.0423 (
I > 2
σ(
I)). The final
wR(
F2) values were 0.1020 (
I > 2
σ(
I)). The final
R1 values were 0.0620 (all data). The final
wR(
F2) values were 0.1157 (all data). The goodness of fit on
F2 was 1.026. The crystal structure of
3 was solved by direct method SHELXS-97 (Sheldrich, G.M. University of Gottingen: Gottingen, Germany, 1997) and the full-matrix least-squares calculations. Crystallographic data for the structure of
2 have been deposited with the Cambridge Crystallographic Data Centre (deposition number: CCDC 939851). CCDC 939851 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via the Internet at
www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail:
deposit@ccdc.cam.ac.uk.
3.5. Cytotoxicity Assay
The cytotoxicity of compounds 1–3 was tested against human breast cancer (MCF-7), hepatocellular carcinoma (SMMC-7721), myeloid leukemia (HL-60), lung cancer (A-549) and colon cancer (SW480) cell lines using an MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt) assay, with cisplatin (Sigma-Aldrich, St. Louis, MO, USA) as the positive control. All the cell lines were obtained from Shanghai cell bank in China and were cultured in RPMI-1640 or DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA) at 37 °C in a humidified atmosphere containing 5% CO2. The viability of cells was determined by performing colorimetric measurements of soluble formazan formed through the reduction of MTS in living cells. In brief, 100 μL medium containing 5,000 cells were plated in each wells in 96 well plates and allowed to adhere for 24 h before drug treatment, while suspension cells were seeded just before drug addition at a concentration of 1 × 105 cells/mL. Cells were exposed to the test compound dissolved in dimethyl sulfoxide (DMSO) at different concentrations in triplicates at 37 °C for 48 h. At the end of the incubation, the medium were replaced with MTS medium (317 μg/mL), and then the incubation was continued for 4 h at 37 °C. The optical densities of the cell lysates were measured at 490 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). The cell viability was calculated by the following formula: cell viability (%) = (ODsample/ODcontrol) × 100%. The IC50 value of each compound was calculated by Reed and Muench’s method based on the corresponding dose response curve, and data were obtained from triplicate experiments. Statistical analysis was performed using the commercially available statistical software (SPSS 11.5 for Windows, SPPS Incorporation, Chicago, IL, USA).




{kind=link}
{kind=link}
{kind=link}
 ) of 1–3.
) of 1–3.