Pharmacophore Generation from a Drug-like Core Molecule Surrounded by a Library Peptide via the 10BASEd-T on Bacteriophage T7

Abstract
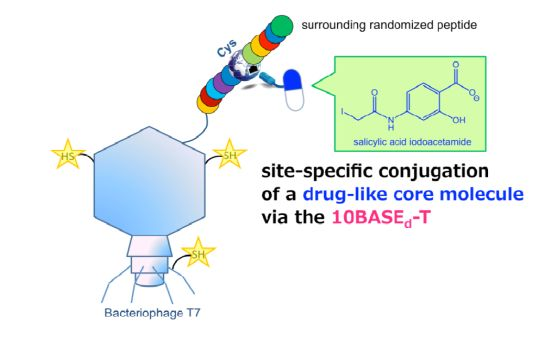
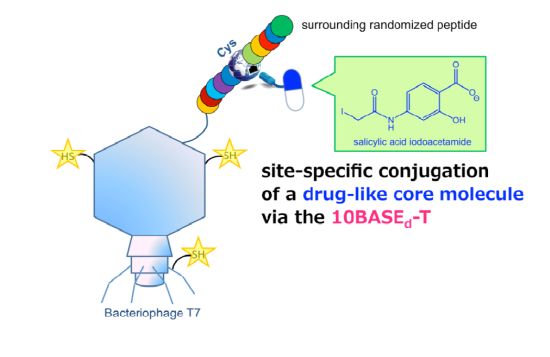
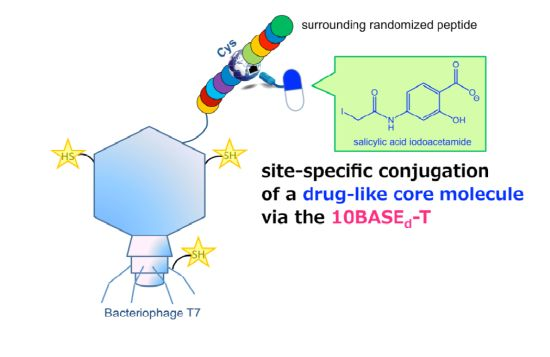
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

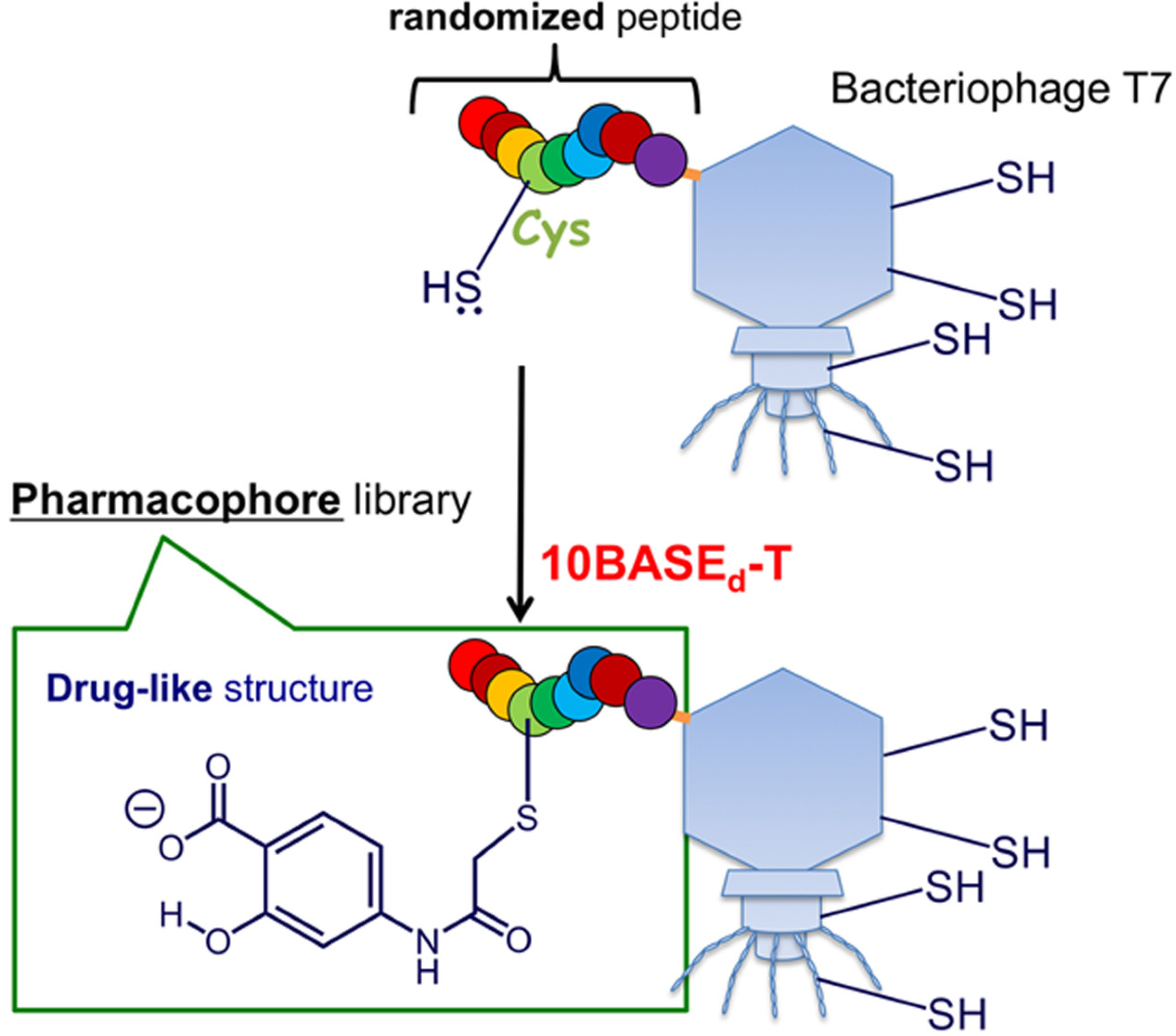
2. Results and Discussion
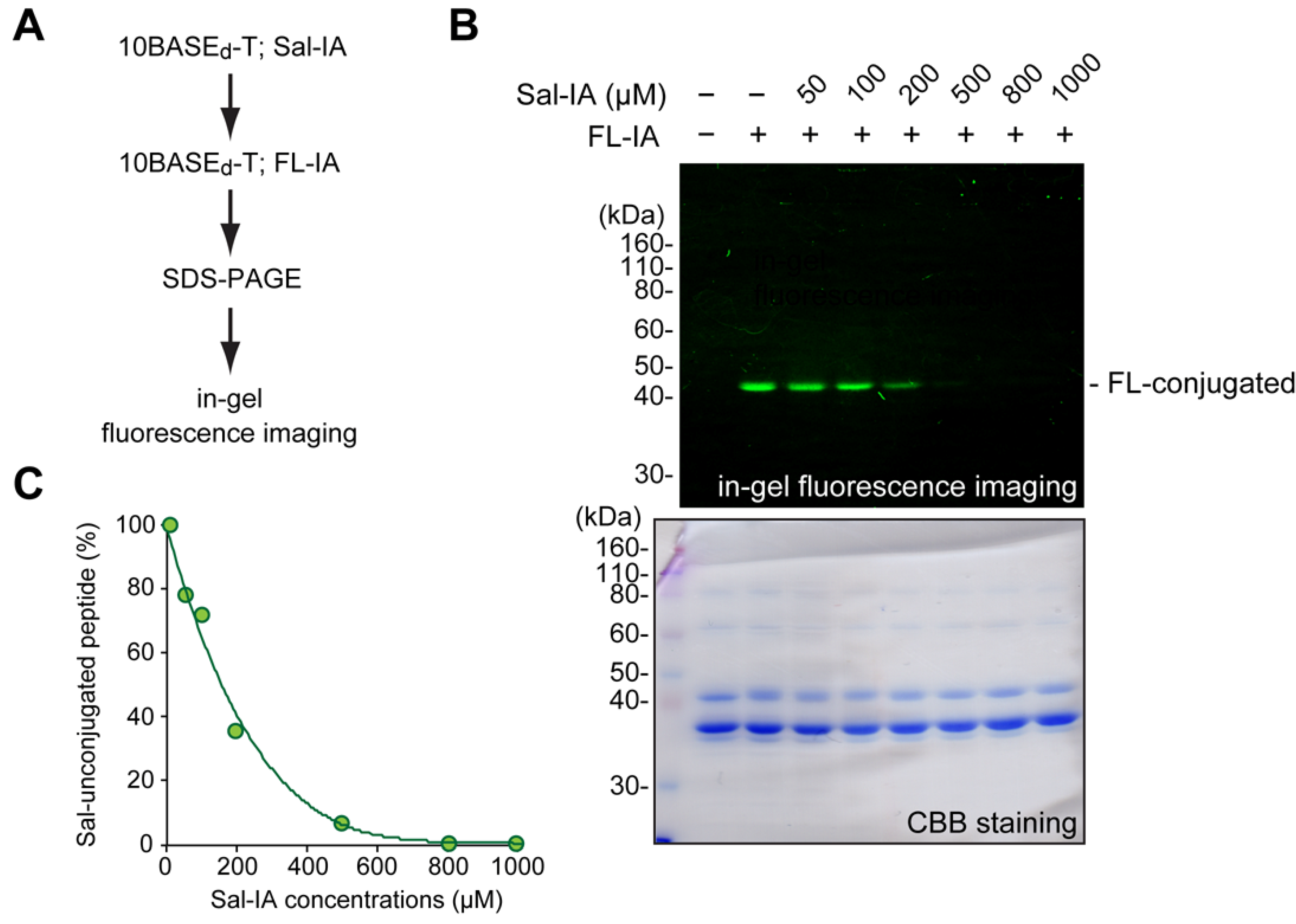
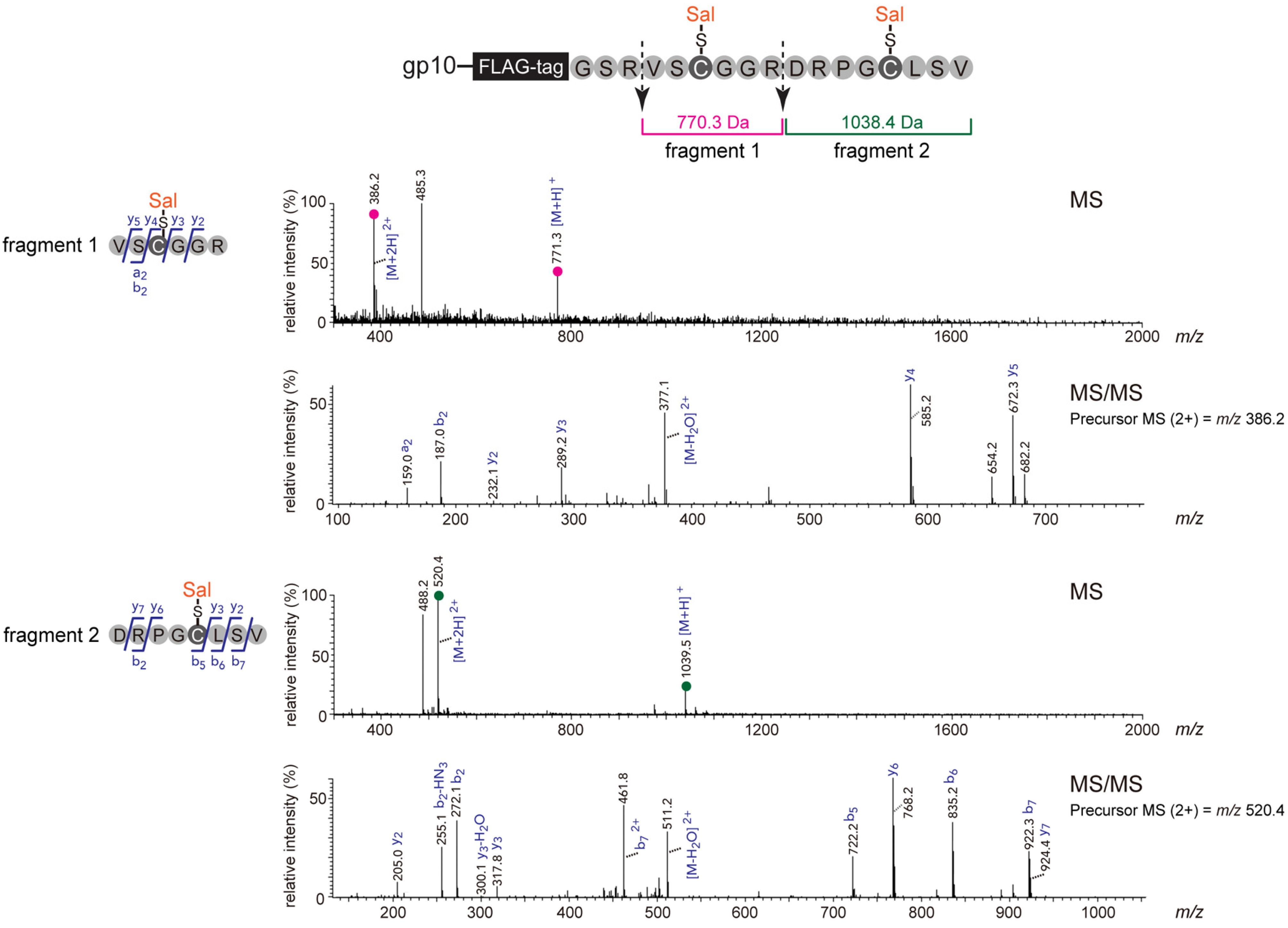
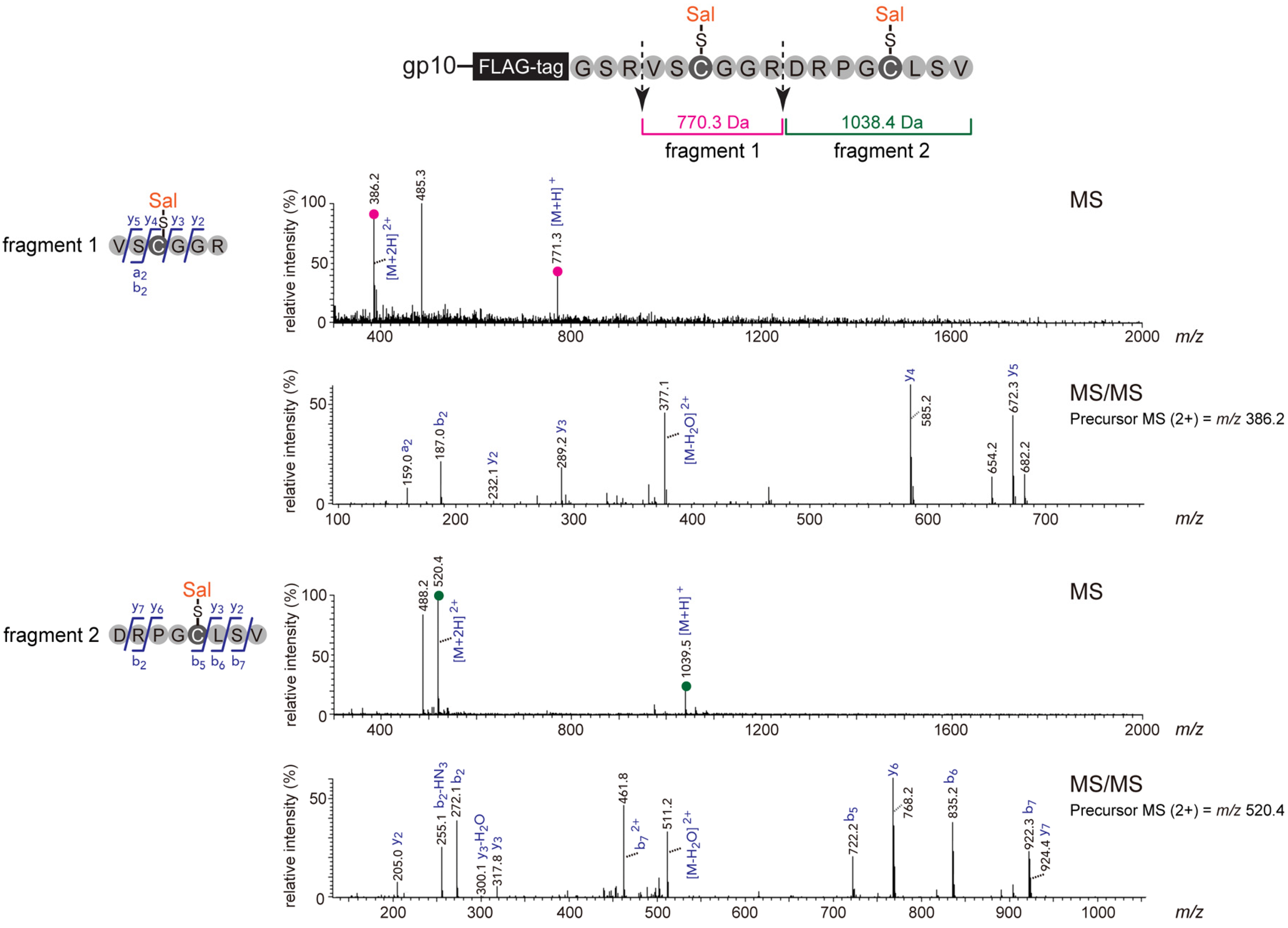
2.1. Optimization and Identification of Site-Specific Introduction of Acetamidosalicylic Acid via the 10BASEd-T


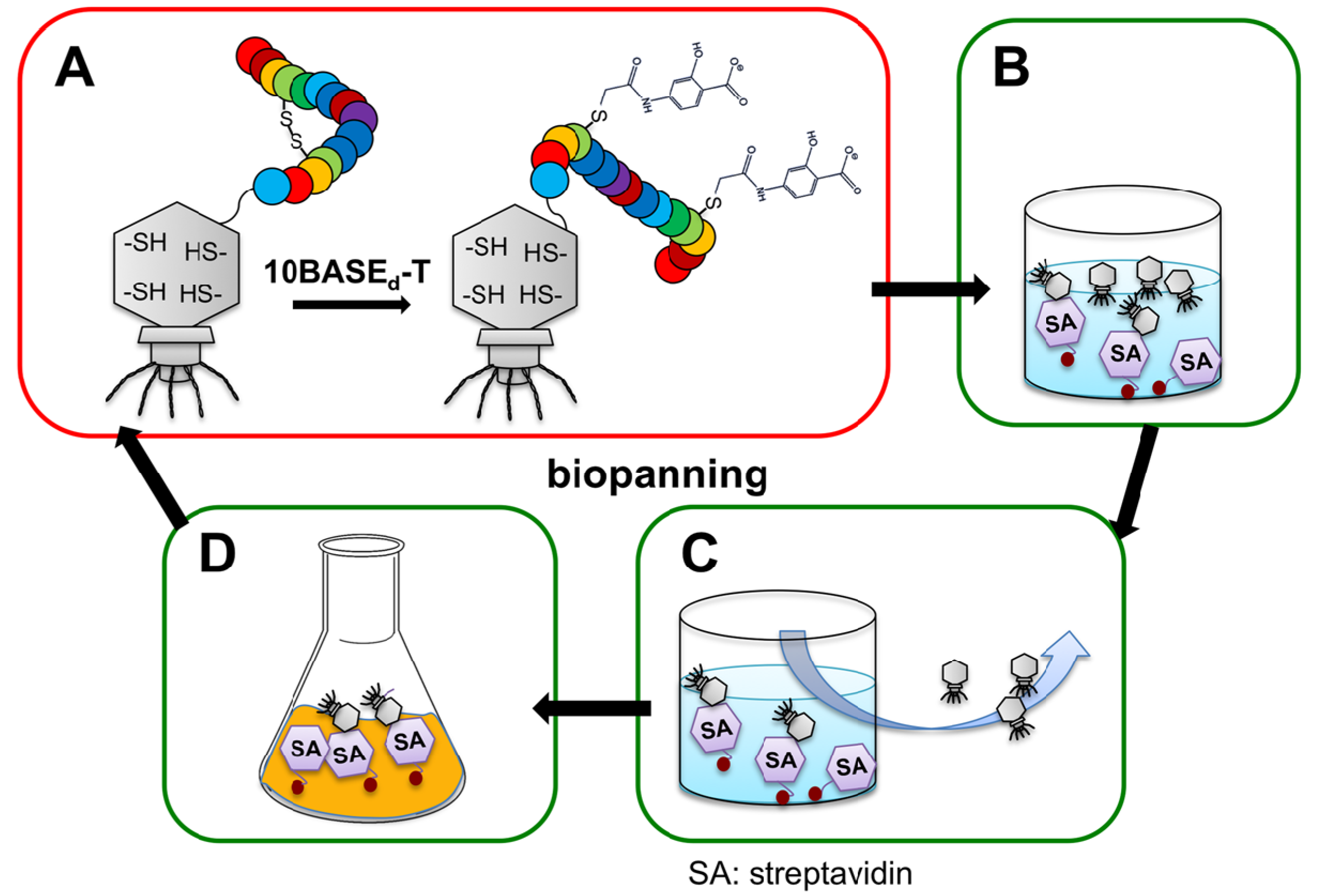
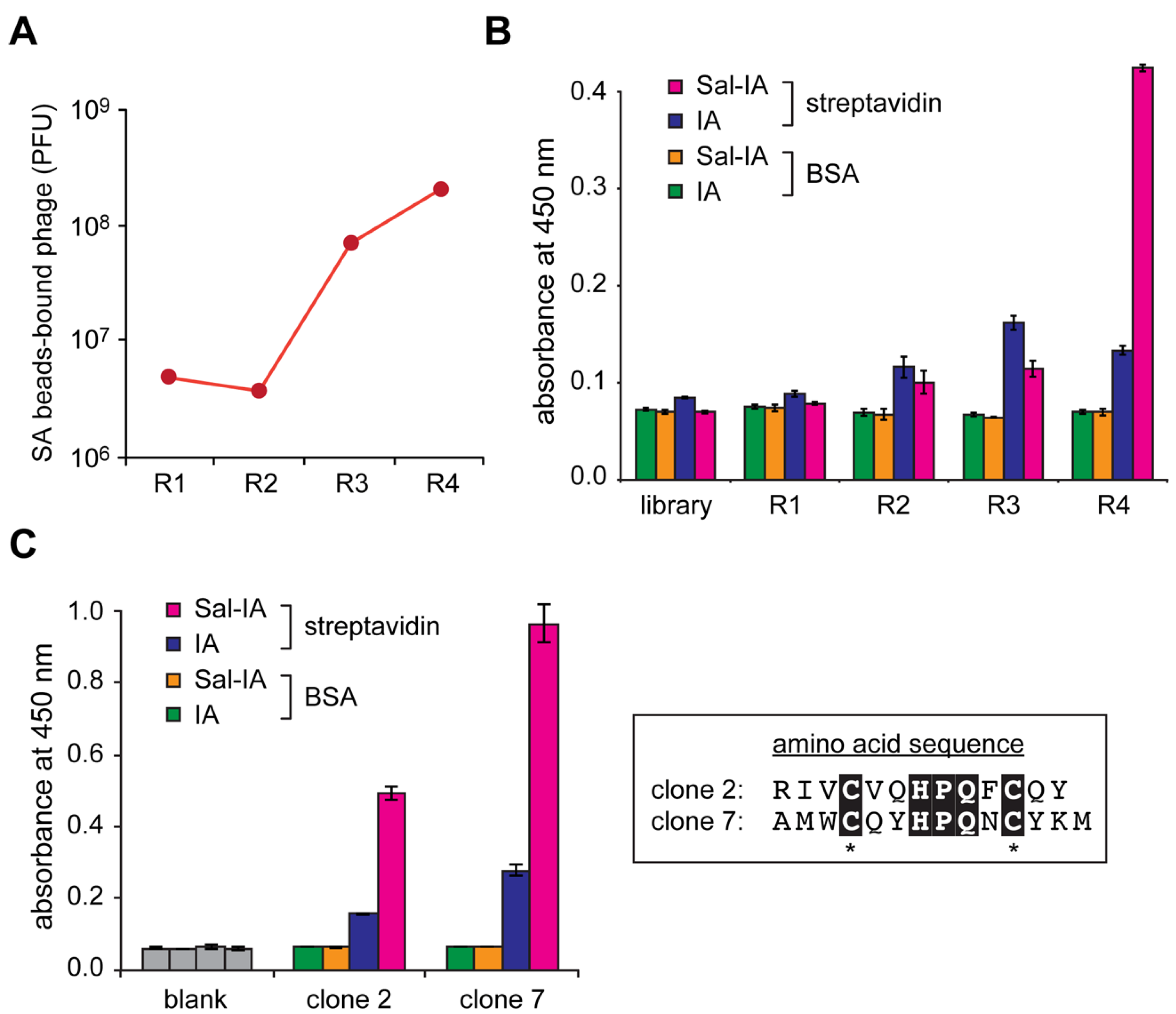
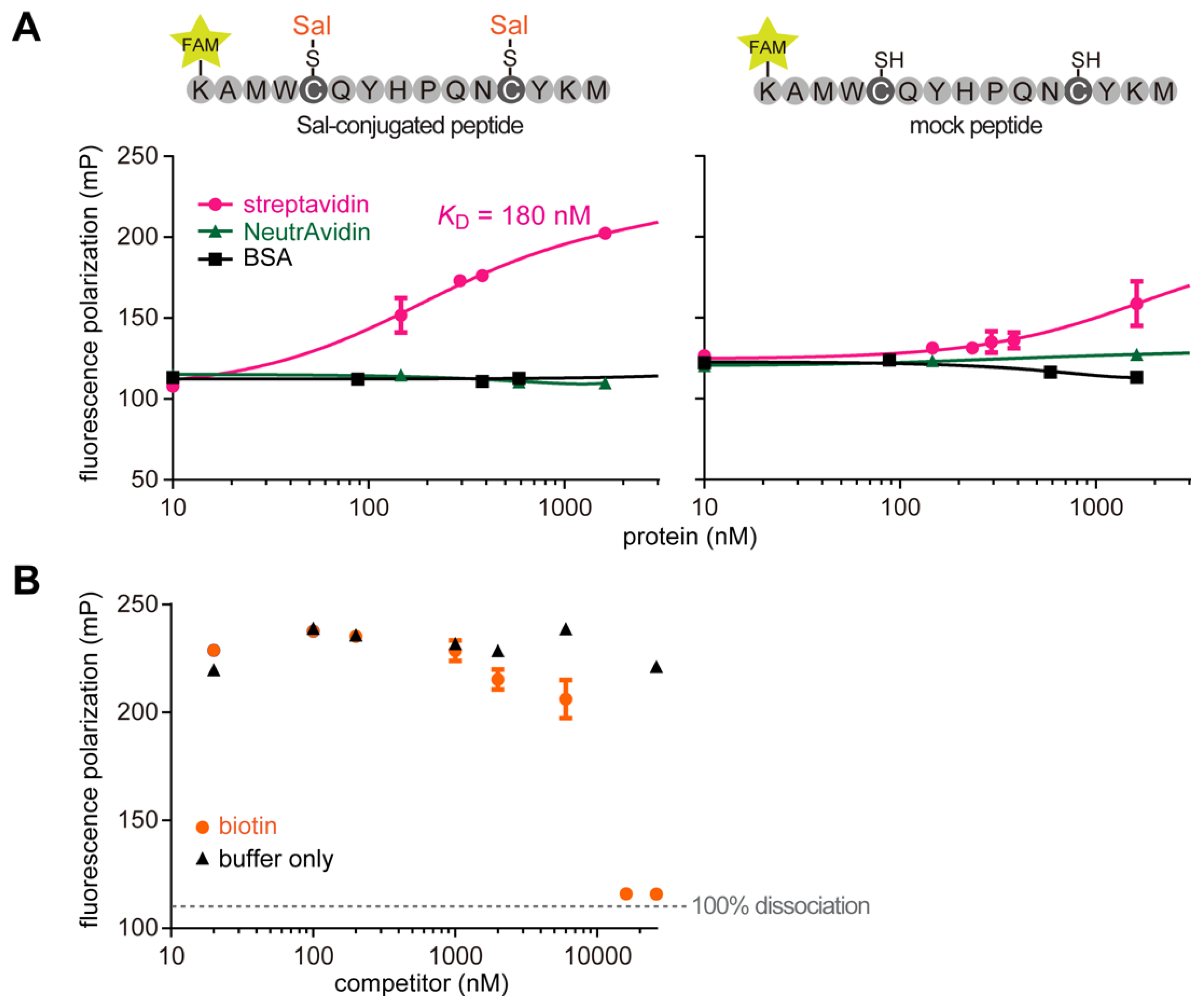
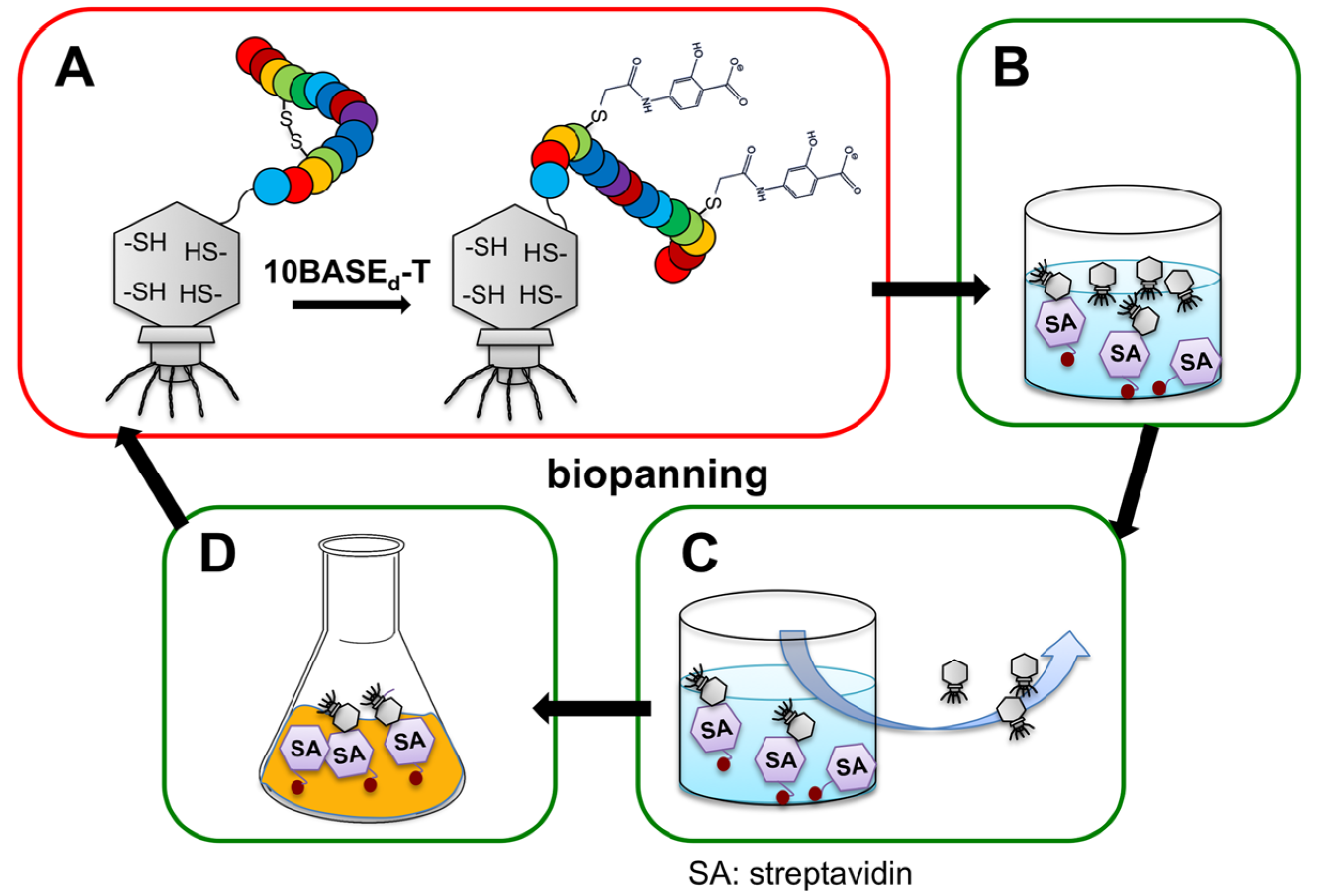
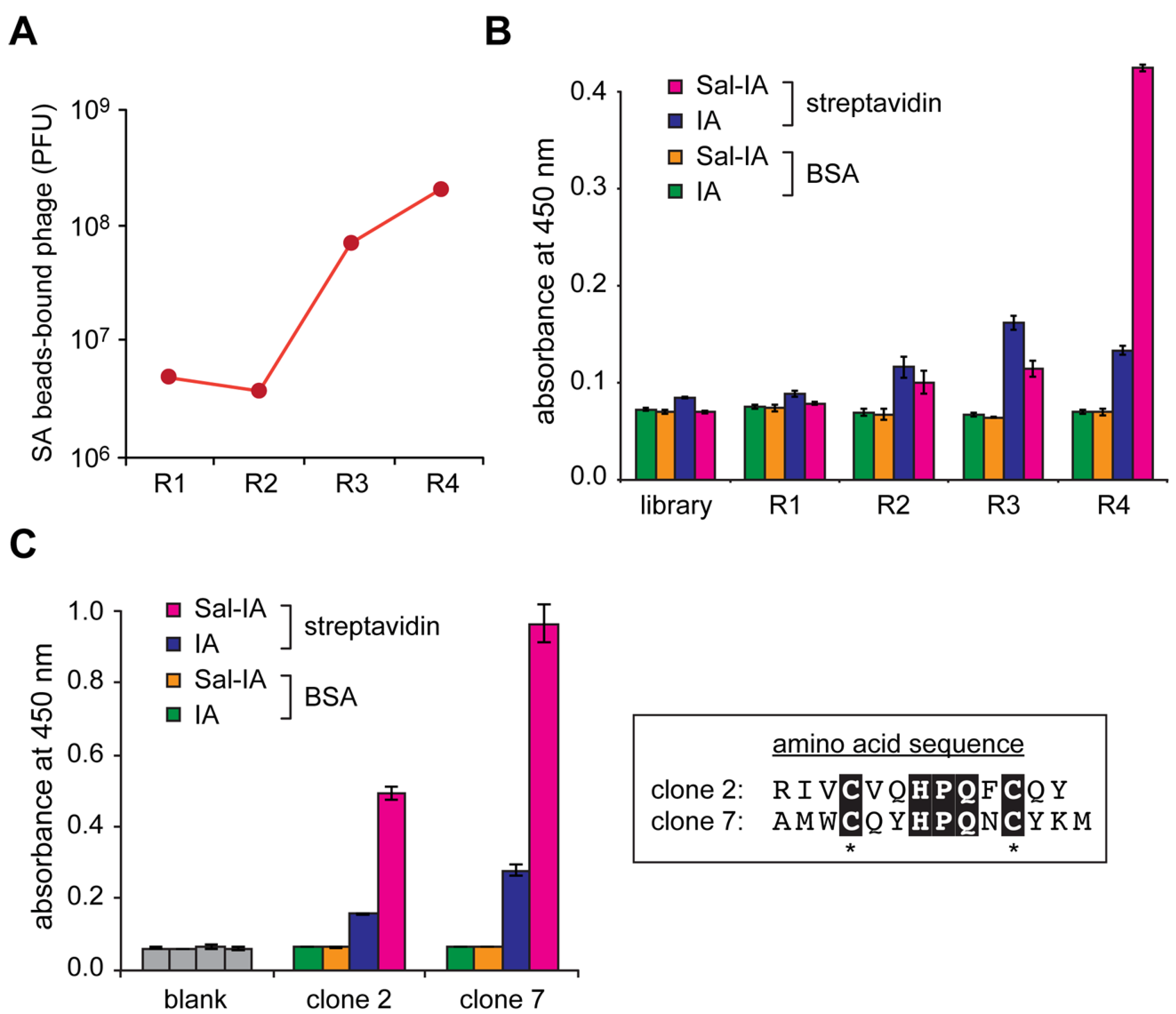
2.2. Construction of Peptide-Conjugated Acetamidosalicylic Acid Library and Selection of a Streptavidin-Specific Binder

3. Experimental
3.1. General
3.2. Construction of T7 Phage Display Libraries
3.3. Chemical Modification of T7 Phage-Displayed Peptide via the 10BASEd-T
3.4. In-Gel Fluorescence Imaging
3.5. Mass Spectrometric Analysis
3.6. Biopanning against Streptavidin
3.7. Enzyme-Linked Immunosorbent Assay (ELISA)
3.8. Peptide Synthesis
3.9. Fluorescence Polarization Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Angelini, A.; Heinis, C. Post-translational modification of genetically encoded polypeptide libraries. Curr. Opin. Chem. Biol. 2011, 15, 355–361. [Google Scholar] [CrossRef]
- Ng, S.; Jafari, M.R.; Derda, R. Bacteriophages and viruses as a support for organic synthesis and combinatorial chemistry. ACS Chem. Biol. 2012, 7, 123–138. [Google Scholar] [CrossRef]
- Uzawa, T.; Tada, S.; Wang, W.; Ito, Y. Expansion of the aptamer library from a “natural soup” to an “unnatural soup”. Chem. Commun. 2013, 49, 1786–1795. [Google Scholar] [CrossRef]
- Jespers, L.; Bonnert, T.P.; Winter, G. Selection of optical biosensors from chemisynthetic antibody libraries. Protein Eng. Des. Sel. 2004, 17, 709–713. [Google Scholar] [CrossRef]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef]
- Ng, S.; Jafari, M.R.; Matochko, W.L.; Derda, R. Quantitative synthesis of genetically encoded glycopeptide libraries displayed on M13 phage. ACS Chem. Biol. 2012, 7, 1482–1487. [Google Scholar] [CrossRef]
- Baeriswyl, V.; Calzavarini, S.; Gerschheimer, C.; Diderich, P.; Angelillo-Scherrer, A.; Heinis, C. Development of a selective peptide macrocycle inhibitor of coagulation factor XII toward the generation of a safe antithrombotic therapy. J. Med. Chem. 2013, 56, 3742–3746. [Google Scholar] [CrossRef]
- Arai, K.; Tsutsumi, H.; Mihara, H. A monosaccharide-modified peptide phage library for screening of ligands to carbohydrate-binding proteins. Bioorg. Med. Chem. Lett. 2013, 23, 4940–4943. [Google Scholar] [CrossRef]
- Santoso, B.; Lam, S.; Murray, B.W.; Chen, G. A simple and efficient maleimide-based approach for peptide extension with a cysteine-containing peptide phage library. Bioorg. Med. Chem. Lett. 2013, 23, 5680–5683. [Google Scholar] [CrossRef]
- Jafari, M.R.; Deng, L.; Kitov, P.I.; Ng, S.; Matochko, W.L.; Tjhung, K.F.; Zeberoff, A.; Elias, A.; Klassen, J.S.; Derda, R. Discovery of light-responsive ligands through screening of a light-responsive genetically encoded library. ACS Chem. Biol. 2013, in press. [Google Scholar] [CrossRef]
- Sakamoto, K.; Ito, Y.; Hatanaka, T.; Soni, P.B.; Mori, T.; Sugimura, K. Discovery and characterization of a peptide motif that specifically recognizes a non-native conformation of human IgG induced by acidic pH conditions. J. Biol. Chem. 2009, 284, 9986–9993. [Google Scholar]
- Hatanaka, T.; Ohzono, S.; Park, M.; Sakamoto, K.; Tsukamoto, S.; Sugita, R.; Ishitobi, H.; Mori, T.; Ito, O.; Sorajo, K.; et al. Human IgA-binding peptides selected from random peptide libraries: Affinity maturation and application in IgA purification. J. Biol. Chem. 2012, 287, 43126–43136. [Google Scholar] [CrossRef]
- Fukunaga, K.; Taki, M. Practical tips for construction of custom peptide libraries and affinity selection by using commercially available phage display cloning systems. J. Nucl. Acids 2012, 2012, 295719. [Google Scholar]
- Fukunaga, K.; Hatanaka, T.; Ito, Y.; Taki, M. Gp10 based-thioetherification (10BASEd-T) on a displaying library peptide of bacteriophage T7. Mol. BioSyst. 2013, 9, 2988–2991. [Google Scholar] [CrossRef]
- Forouhar, F.; Yang, Y.; Kumar, D.; Chen, Y.; Fridman, E.; Park, S.W.; Chiang, Y.; Acton, T.B.; Montelione, G.T.; Pichersky, E.; et al. Structural and biochemical studies identify tobacco SABP2 as a methyl salicylate esterase and implicate it in plant innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 1773–1778. [Google Scholar] [CrossRef]
- Seidler, J.; McGovern, S.L.; Doman, T.N.; Shoichet, B.K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003, 46, 4477–4486. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Rosenberg, A.; Griffin, K.; Studier, F.W.; McCormick, M.; Berg, J.; Novy, R.; Mierendorf, R. T7 select phage display system: A powerful new protein display system based on bacteriophage T7. inNovations 1996, 6, 1–6. [Google Scholar]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Mapping of vascular Zip Codes by Phage Display. Methods Enzymol. 2012, 503, 35–56. [Google Scholar] [CrossRef]
- Carrico, Z.M.; Farkas, M.E.; Zhou, Y.; Hsiao, S.C.; Marks, J.D.; Chokhawala, H.; Clark, D.S.; Francis, M.B. N-terminal labeling of filamentous phage to create cancer marker imaging agents. ACS Nano 2012, 6, 6675–6680. [Google Scholar] [CrossRef]
- Chen, S.; Touati, J.; Heinis, C. Tracking chemical reactions on the surface of filamentous phage using mass spectrometry. Chem. Commun. 2014, in press. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Fukunaga, K.; Hatanaka, T.; Ito, Y.; Taki, M. Selection of Streptavidin-Binding Artificial Peptide Possessing Salicylic Acid Moiety via the 10BASEd-T on the Bacteriophage T7. Pept. Sci. 2014, in press. [Google Scholar]
- Vodnik, M.; Zager, U.; Strukelj, B.; Lunder, M. Phage display: Selecting straws instead of a needle from a haystack. Molecules 2011, 16, 790–817. [Google Scholar] [CrossRef]
- Krumpe, L.R.; Atkinson, A.J.; Smythers, G.W.; Kandel, A.; Schumacher, K.M.; McMahon, J.B.; Makowski, L.; Mori, T. T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics 2006, 6, 4210–4222. [Google Scholar] [CrossRef]
- Kay, B.K.; Adey, N.B.; He, Y.S.; Manfredi, J.P.; Mataragnon, A.H.; Fowlkes, D.M. An M13 phage library displaying random 38-amino-acid peptides as a source of novel sequences with affinity to selected targets. Gene 1993, 128, 59–65. [Google Scholar] [CrossRef]
- Frost, J.R.; Smith, J.M.; Fasan, R. Design, synthesis, and diversification of ribosomally derived peptide macrocycles. Curr. Opin. Struct. Biol. 2013, 23, 571–580. [Google Scholar] [CrossRef]
- Katz, B.A.; Cass, R.T. In crystals of complexes of streptavidin with peptide ligands containing the HPQ sequence the pKa of the peptide histidine is less than 3.0. J. Biol. Chem. 1997, 272, 13220–13228. [Google Scholar] [CrossRef]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucl. Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef]
- Sandhu, A.; Handa, H.; Abe, M. Synthesis and applications of magnetic nanoparticles for biorecognition and point of care medical diagnostics. Nanotechnology 2010, 21, 442001. [Google Scholar] [CrossRef]
- Hamamoto, T.; Sisido, M.; Ohtsuki, T.; Taki, M. Synthesis of a cyclic peptide/protein using the NEXT-A reaction followed by cyclization. Chem. Commun. 2011, 47, 9116–9118. [Google Scholar] [CrossRef]
- Li, S.; Roberts, R.W. A novel strategy for in vitro selection of peptide-drug conjugates. Chem. Biol. 2003, 10, 233–239. [Google Scholar] [CrossRef]
- Lloyd, D.G.; Buenemann, C.L.; Todorov, N.P.; Manallack, D.T.; Dean, P.M. Scaffold hopping in de novo design. Ligand generation in the absence of receptor information. J. Med. Chem. 2004, 47, 493–496. [Google Scholar] [CrossRef]
- Reardon, S. Project ranks billions of drug interactions. Nature 2013, 503, 449–450. [Google Scholar] [CrossRef]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Sample Availability: All of the chemicals used in this study are commercially available. The model T7 phage monoclone is available from corresponding author.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tokunaga, Y.; Azetsu, Y.; Fukunaga, K.; Hatanaka, T.; Ito, Y.; Taki, M. Pharmacophore Generation from a Drug-like Core Molecule Surrounded by a Library Peptide via the 10BASEd-T on Bacteriophage T7. Molecules 2014, 19, 2481-2496. https://doi.org/10.3390/molecules19022481
Tokunaga Y, Azetsu Y, Fukunaga K, Hatanaka T, Ito Y, Taki M. Pharmacophore Generation from a Drug-like Core Molecule Surrounded by a Library Peptide via the 10BASEd-T on Bacteriophage T7. Molecules. 2014; 19(2):2481-2496. https://doi.org/10.3390/molecules19022481
Chicago/Turabian StyleTokunaga, Yuuki, Yuuki Azetsu, Keisuke Fukunaga, Takaaki Hatanaka, Yuji Ito, and Masumi Taki. 2014. "Pharmacophore Generation from a Drug-like Core Molecule Surrounded by a Library Peptide via the 10BASEd-T on Bacteriophage T7" Molecules 19, no. 2: 2481-2496. https://doi.org/10.3390/molecules19022481
APA StyleTokunaga, Y., Azetsu, Y., Fukunaga, K., Hatanaka, T., Ito, Y., & Taki, M. (2014). Pharmacophore Generation from a Drug-like Core Molecule Surrounded by a Library Peptide via the 10BASEd-T on Bacteriophage T7. Molecules, 19(2), 2481-2496. https://doi.org/10.3390/molecules19022481