Synergism of Cyclin-Dependent Kinase Inhibitors with Camptothecin Derivatives in Small Cell Lung Cancer Cell Lines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
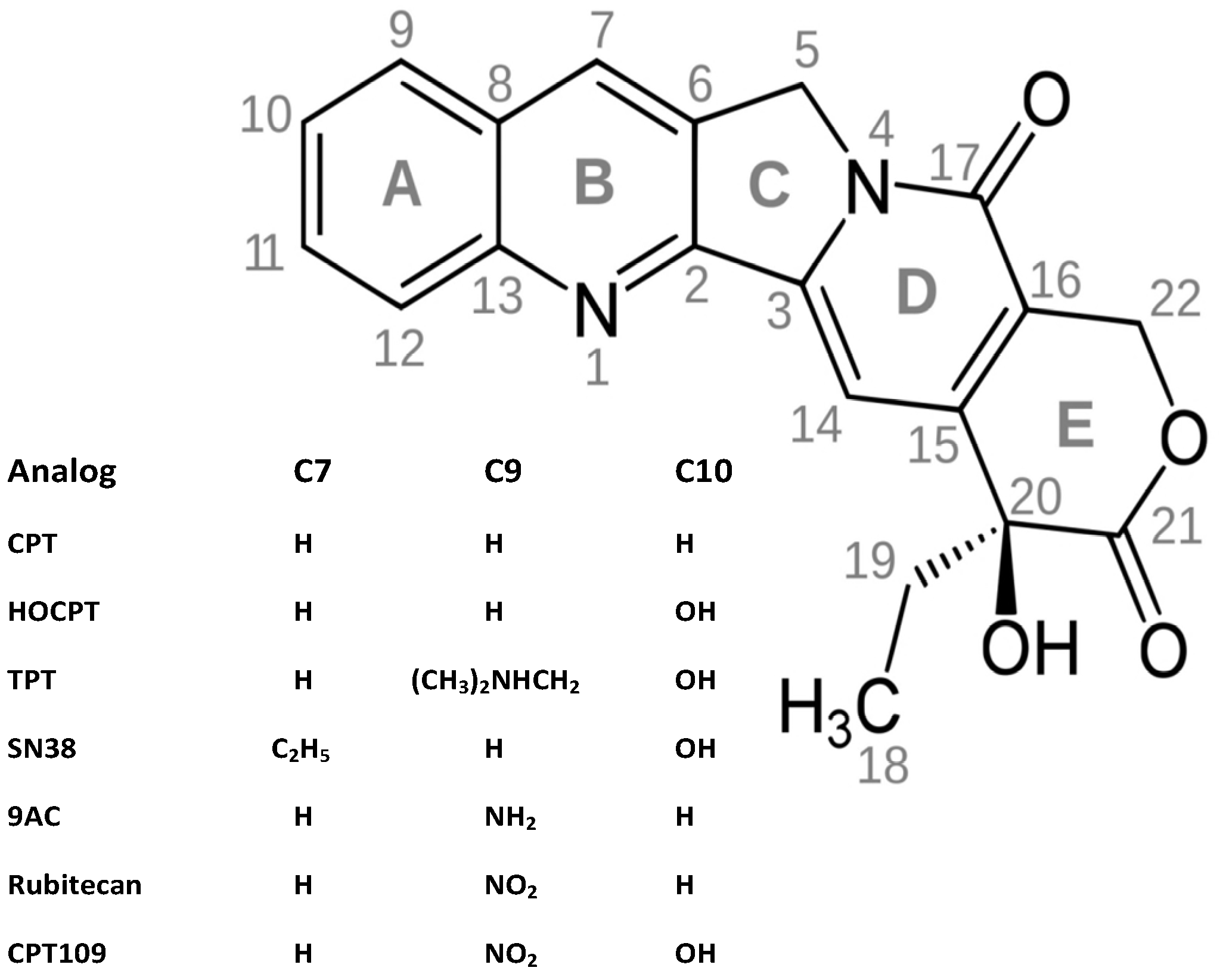
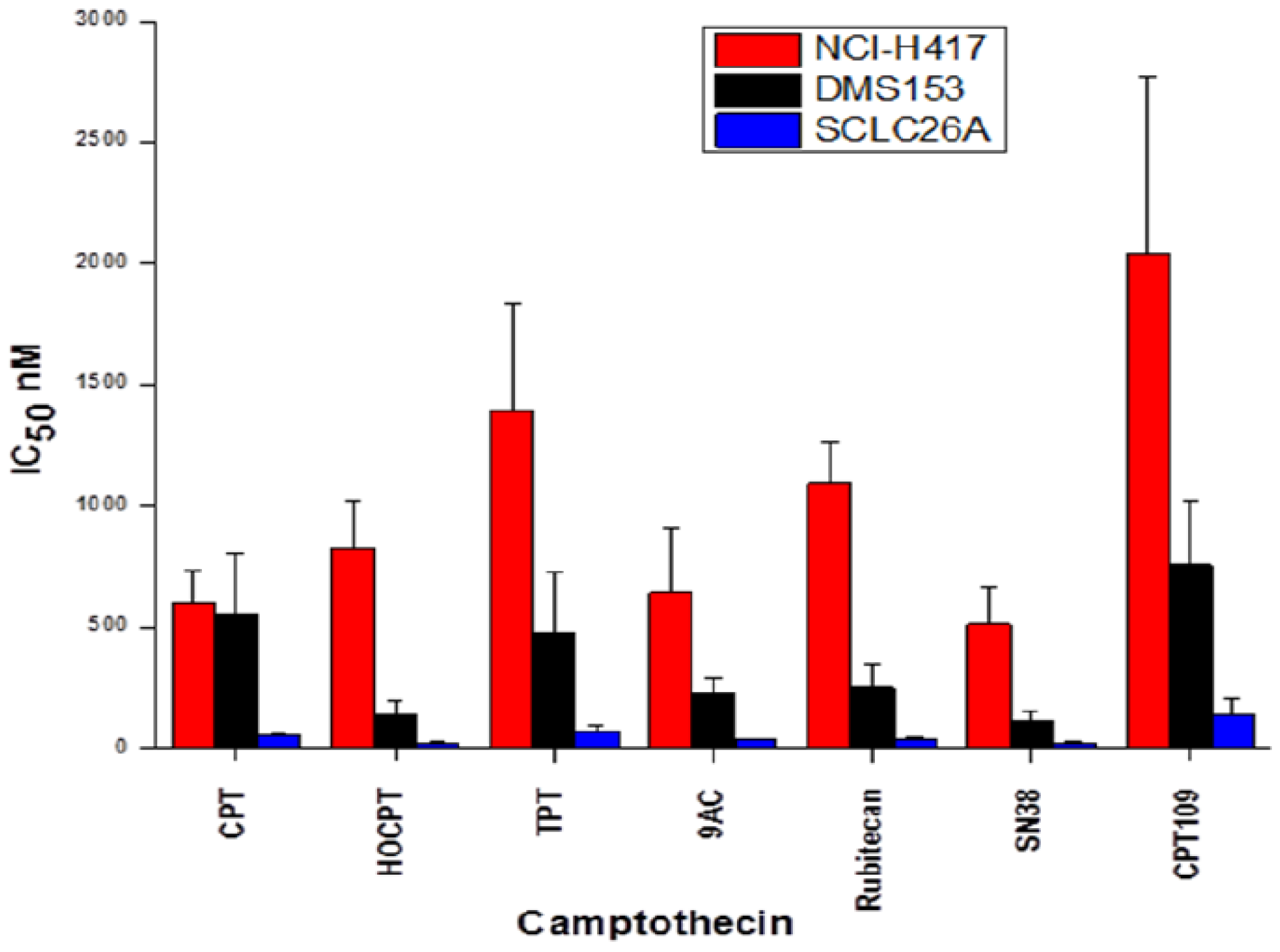
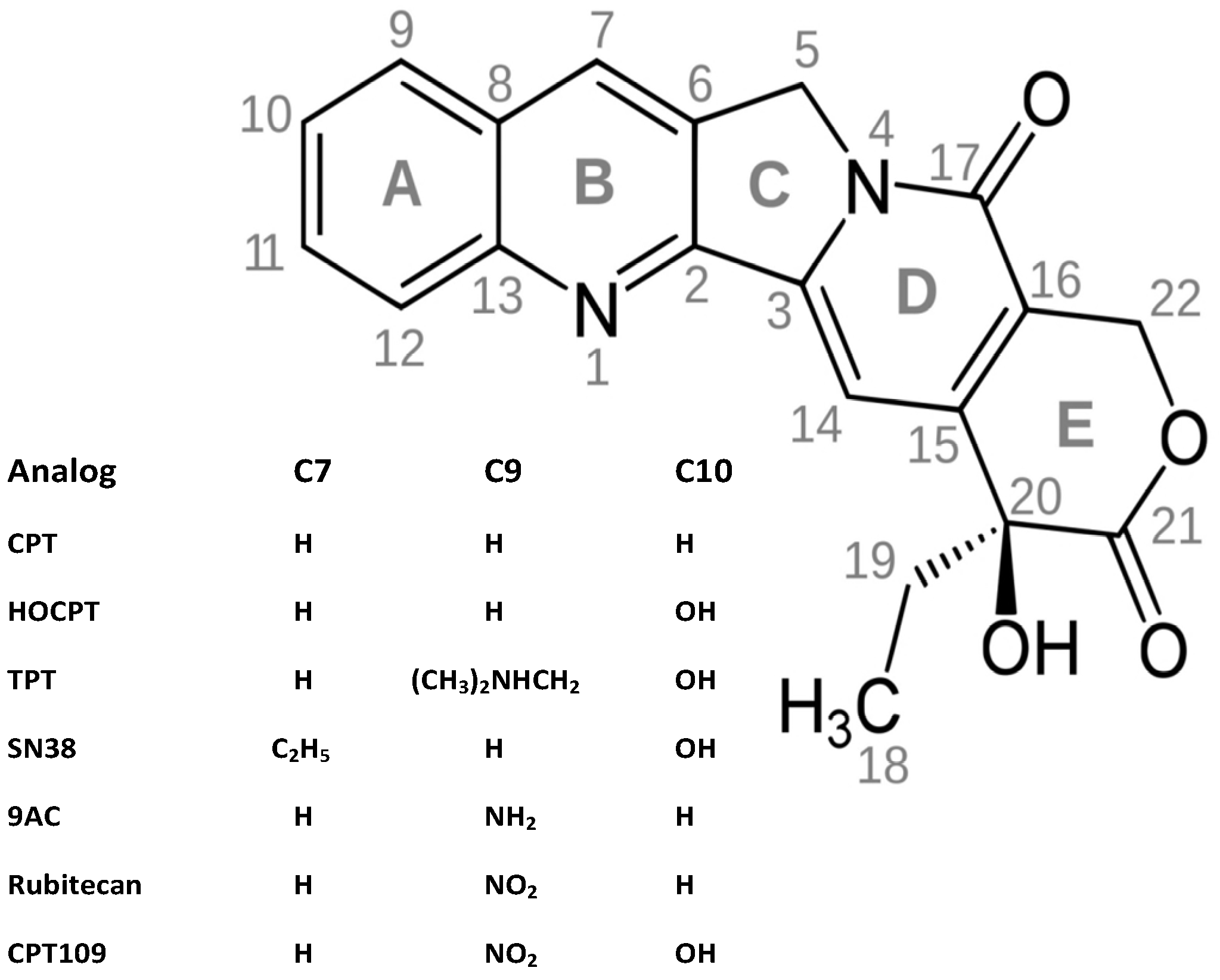
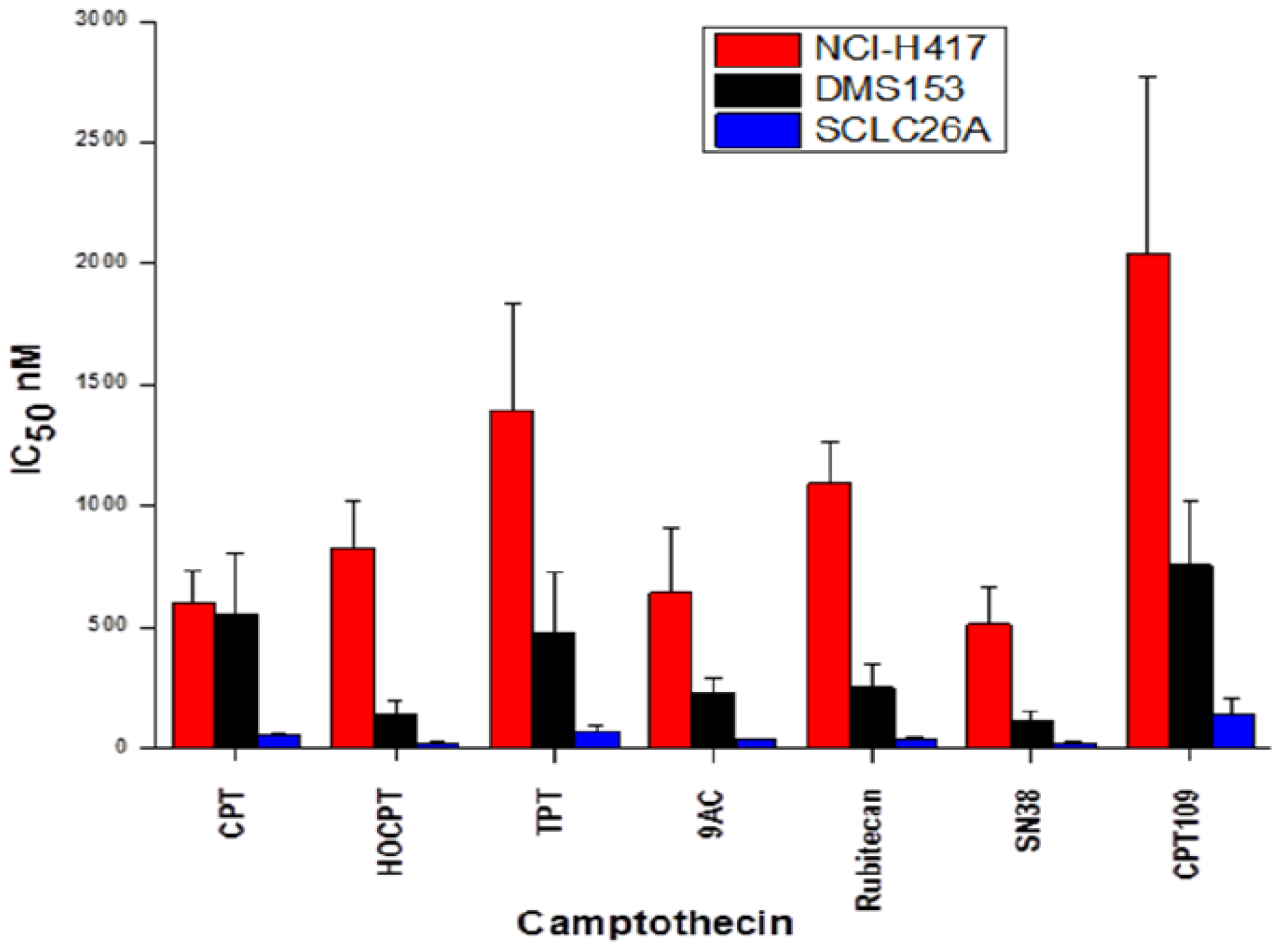
2.1. Cytotoxicity of CPT and Analogs against SCLC Cell Lines
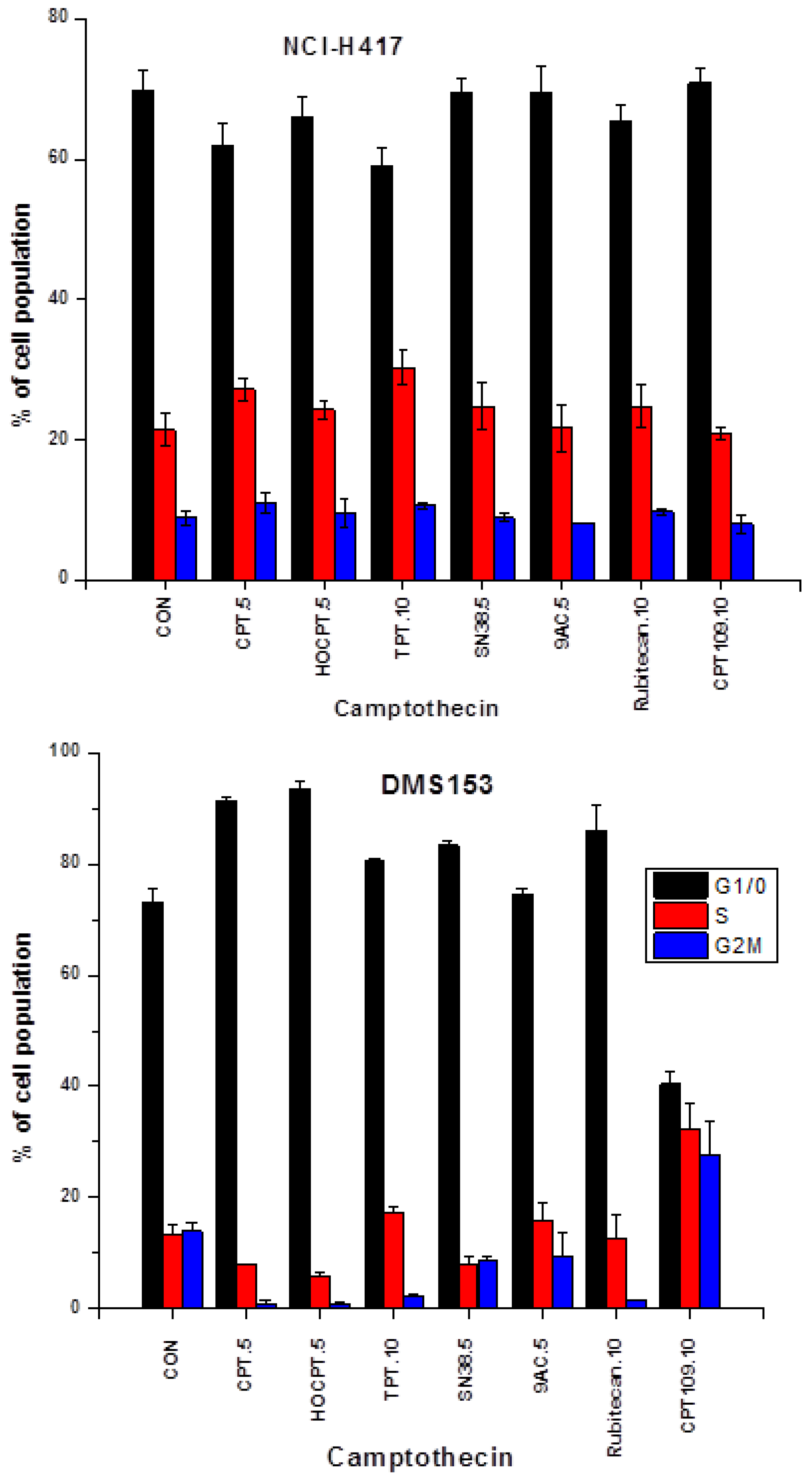
2.2. Cell Cycle Effects of CPT and Analogs on NCI-H417 and DMS153 Cell Lines
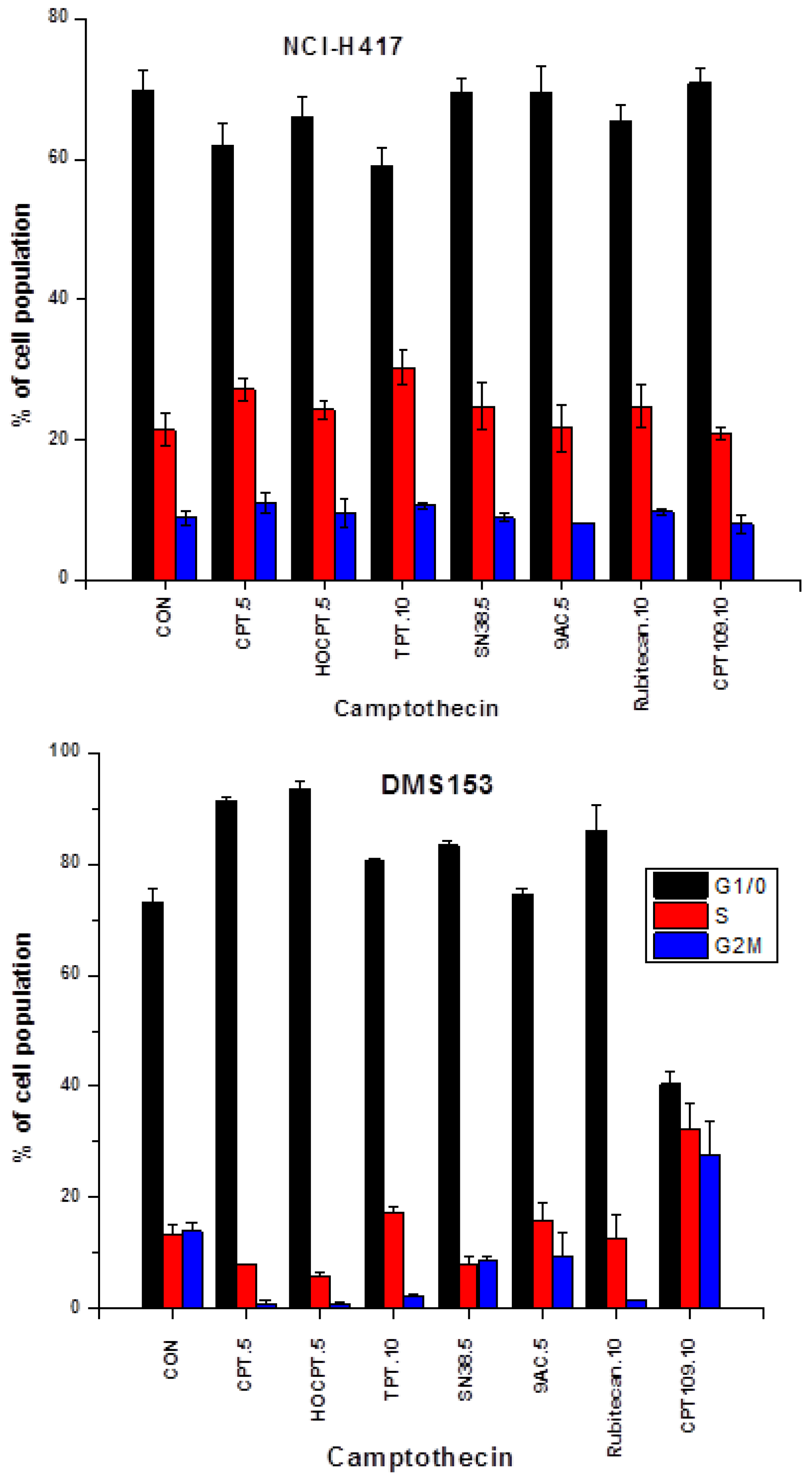
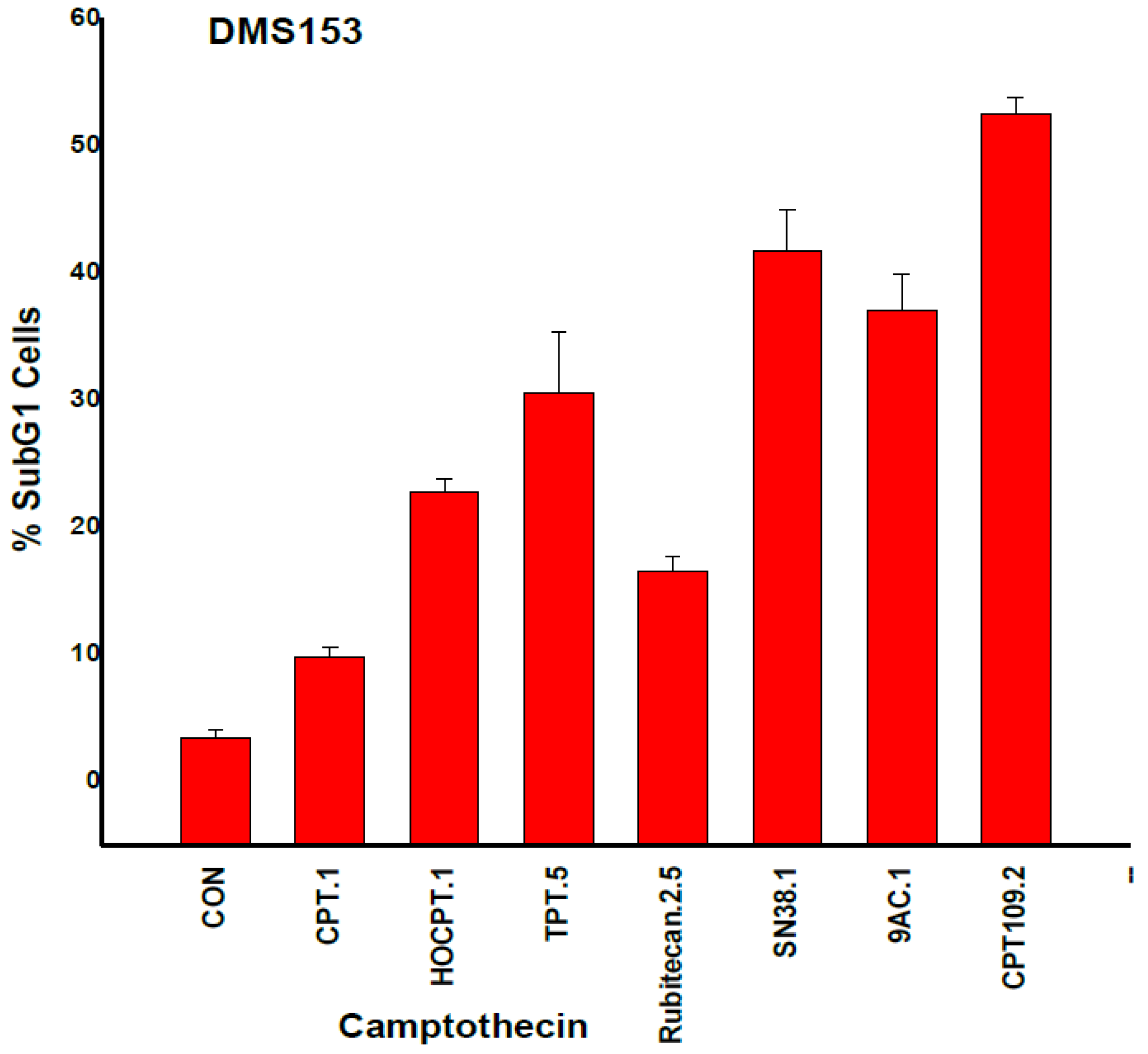
2.3. CPT Analogs-induced Apoptotic Cell Death in DMS153 Cells

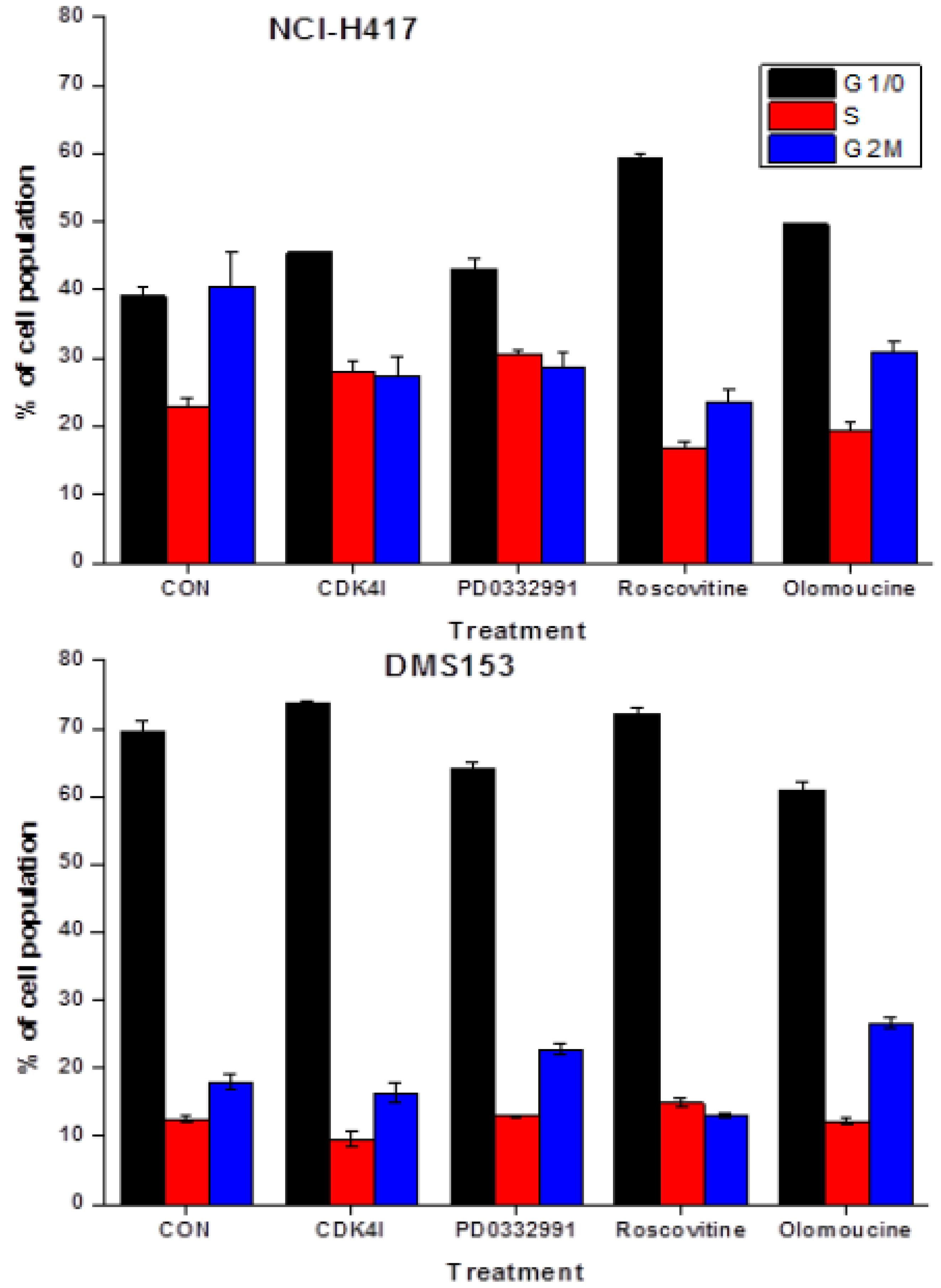
2.4. Effects of CDK Inhibitors on Cell Cycle Distribution of NCI-H417 and DMS153 Cells
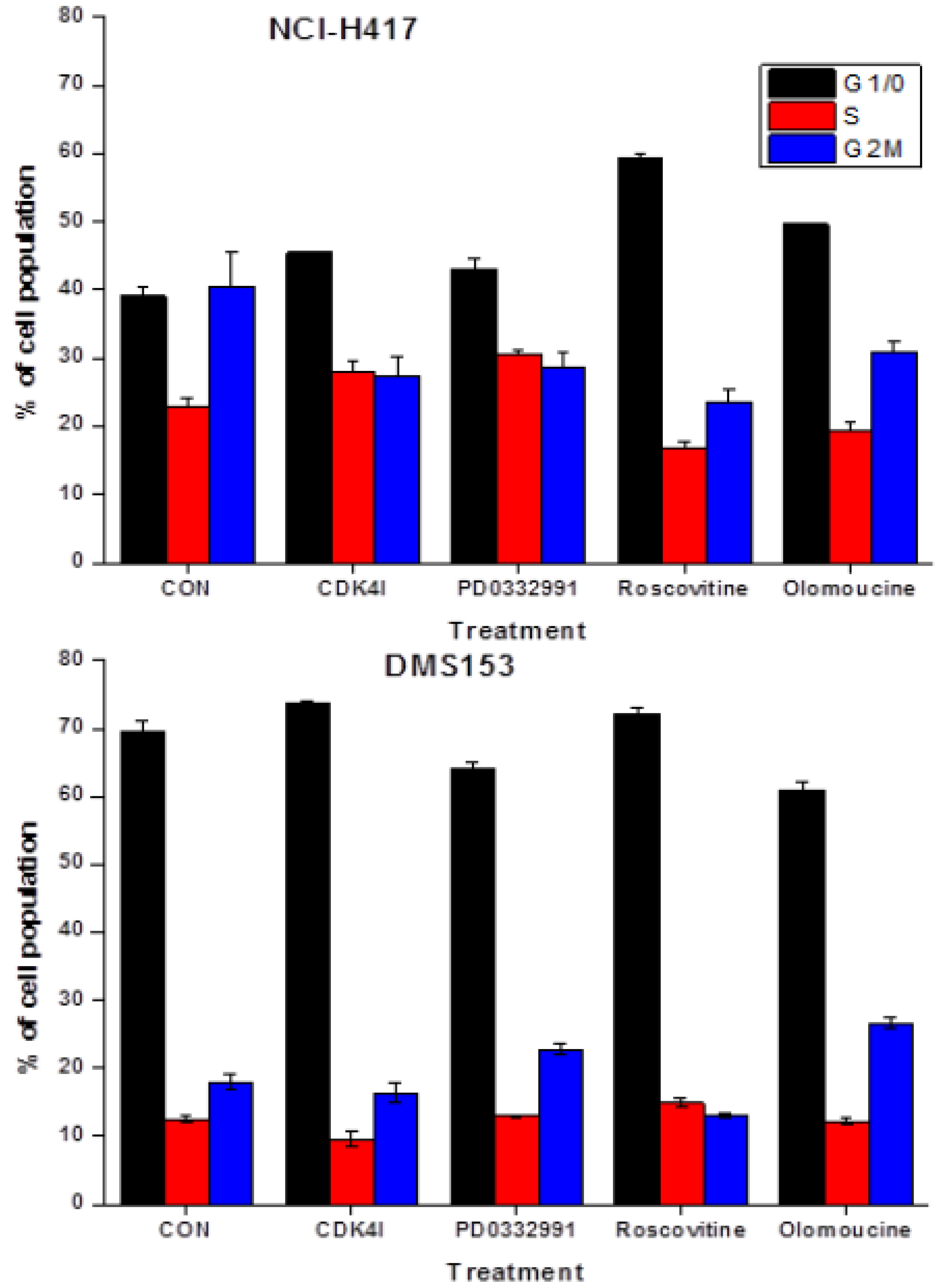
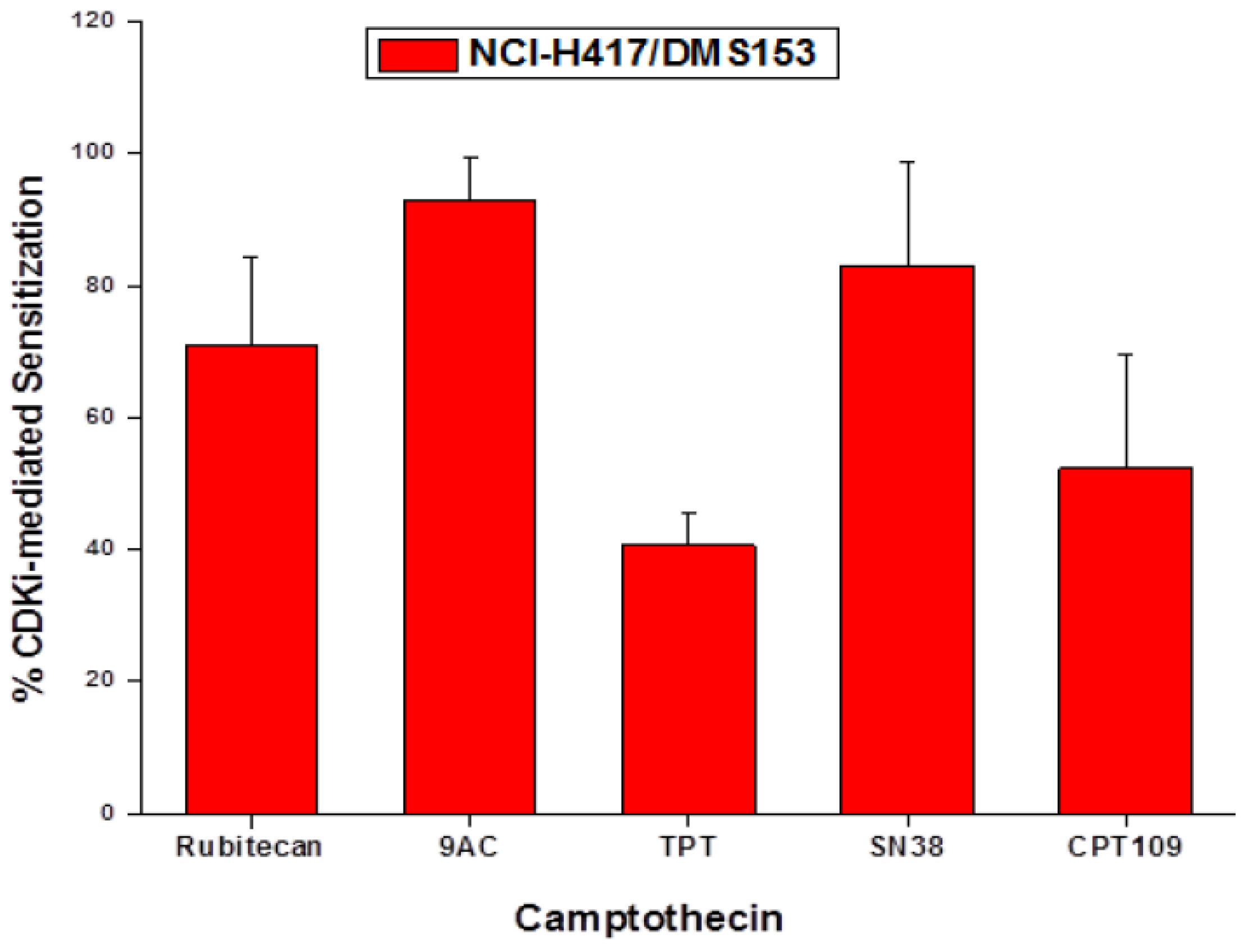
2.5. Chemosensitizing Effects of CDK Inhibitors on CPT Analogs in NCI-H417 and DMS153 Cells
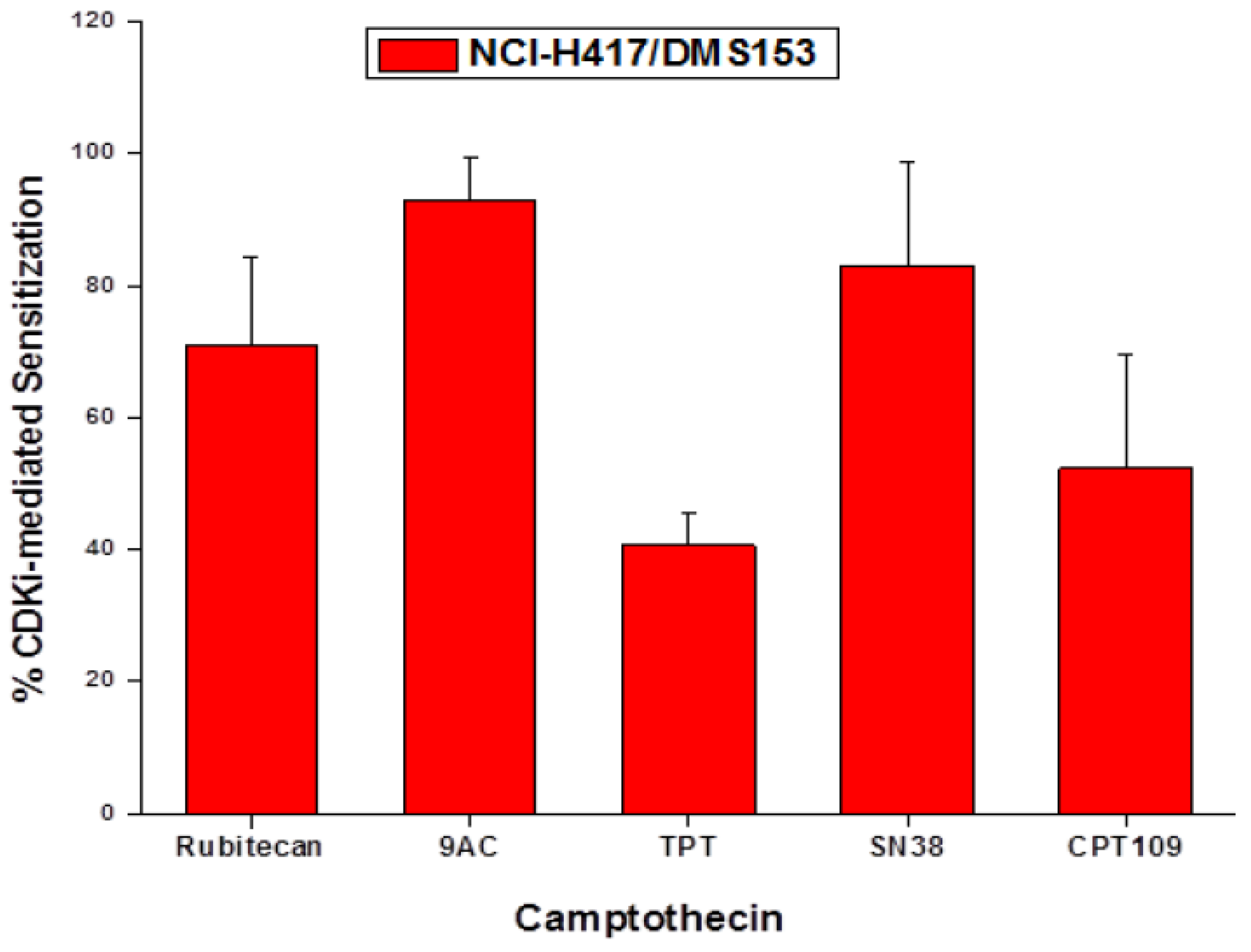
2.6. Effects of the CDK Inhibitors on Topoisomerase I Expression of DMS153 Cells
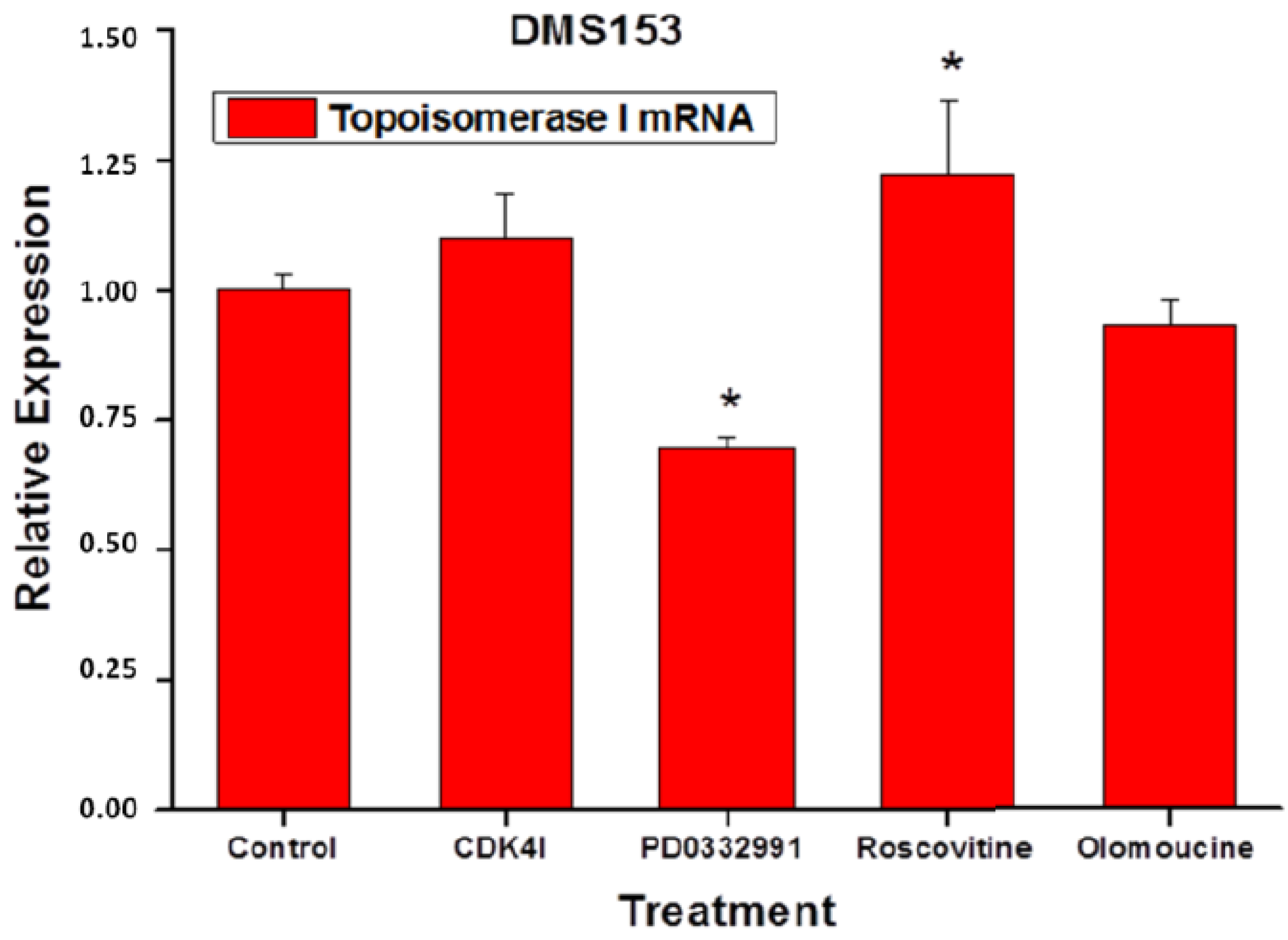
2.7. Discussion
3. Experimental
3.1. Reagents and Cell Lines
3.2. Chemosensitivity Assay
3.3. Measurement of Cell Cycle Distribution
3.4. RT-PCR of Topoisomerase I in DMS153 Cells
3.5. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Youlden, D.R.; Cramb, S.M.; Baade, P.D. The International Epidemiology of Lung Cancer: Geographical distribution and secular trends. J. Thoracic Oncol. 2008, 3, 819–831. [Google Scholar] [CrossRef]
- Califano, A.; Abidin, Z.; Peck, R.; Faivre-Finn, C.; Lorigan, P.R. Management of small cell lung cancer: Recent developments for optimal care. Drugs 2012, 72, 471–490. [Google Scholar] [CrossRef]
- William, W.N.; Glisson, B.S. Novel strategies for the treatment of small-cell lung carcinoma. Nat. Rev. Clin. Oncol. 2011, 8, 611–619. [Google Scholar] [CrossRef]
- Schmittel, A. Second-line therapy for small-cell lung cancer. Expert Rev. Anticancer Ther. 2011, 11, 631–637. [Google Scholar] [CrossRef]
- Tomicic, M.T.; Kaina, B. Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochim. Biophys. Acta 2013, 1835, 11–27. [Google Scholar]
- Wal, M.E.; Wani, M.C.; Cooke, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar]
- Pommier, Y. Camptothecins and topoisomerase I: A foot in the door. Targeting the genome beyond topoisomerase I with camptothecins and novel anticancer drugs: Importance of DNA replication, repair and cell cycle checkpoints. Curr. Med. Chem. Anticancer Agents 2004, 4, 429–434. [Google Scholar] [CrossRef]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef]
- Wani, M.C.; Wall, M.E. Plant antitumor agents. II. Structure of two new alkaloids from Camptotheca acuminata. J. Org. Chem. 1969, 34, 1364–1367. [Google Scholar] [CrossRef]
- Hertzberg, R.; Caranfa, M.J.; Holden, K.G.; Jakas, D.R.; Gallagher, G.; Mattern, M.R.; Mong, S.M.; Bartus, J.O.; Johnson, R.K.; Kingsbury, W.D. Modification of the hydroxylactone ring of camptothecin: Inhibition of mammalian topoisomerase I and biological activity. J. Med. Chem. 1989, 32, 715–720. [Google Scholar] [CrossRef]
- Supko, J.G.; Malspeis, L. Pharmacokinetics of the 9-amino and 10,11-methylenedioxy derivatives of camptothecin in mice. Cancer Res. 1993, 53, 3062–3069. [Google Scholar]
- Hinz, H.R.; Harris, N.J.; Natelson, E.A.; Giovanella, B.C. Pharmacokinetics of the in vivo and in vitro conversion of 9-nitro-20(S)-camptothecin to 9-amino-20(S)-camptothecin in humans, dogs and mice. Cancer Res. 1994, 54, 3096–3100. [Google Scholar]
- Hamilton, G.; Olszewski, U.; Klameth, L.; Ulsperger, E.; Geissler, K. Synergistic anticancer activity of topotecan— Cyclin-dependent kinase inhibitor combinations against drug-resistant Small Cell Lung Cancer (SCLC) cell lines. J. Cancer Ther. 2013, 4, 47–53. [Google Scholar]
- Gallorini, M.; Cataldi, A.; di Giacomo, V. Cyclin-dependent kinase modulators and cancer therapy. BioDrugs 2012, 26, 377–391. [Google Scholar]
- Schutte, B.; Nieland, L.; van Engeland, M.; Henfling, M.E.; Meijer, L.; Ramaekers, F.C. The effect of the cyclin-dependent kinase inhibitor olomoucine on cell cycle kinetics. Exp. Cell Res. 1997, 236, 4–15. [Google Scholar] [CrossRef]
- Fukuoka, K.; Adachi, J.; Nishio, K.; Arioka, H.; Kurokawa, H.; Fukumoto, H.; Ishida, T.; Nomoto, T.; Yokote, H.; Tomonari, A.; et al. p16INK4 expression is associated with the increased sensitivity of human non-small cell lung cancer cells to DNA Topoisomerase I Inhibitors. Jpn. J. Cancer Res. 1997, 88, 1009–1016. [Google Scholar] [CrossRef]
- Shu, K.X.; Li, B.; Wu, L.X. The p53 network: p53 and its downstream genes. Colloids Surf. B Biointerfaces 2007, 55, 10–18. [Google Scholar] [CrossRef]
- Otterson, G.A.; Kratzke, R.A.; Coxon, A.; Kim, Y.W.; Kaye, F.J. Absence of p16INK4 protein is restricted to the subset of lung cancer lines that retains wildtype RB. Oncogene 1994, 9, 3375–3378. [Google Scholar]
- Wikman, H.; Kettunen, E. Regulation of the G1/S phase of the cell cycle and alterations in the RB pathway in human lung cancer. Exp. Rev. Anticancer Ther. 2006, 6, 515–530. [Google Scholar] [CrossRef]
- Okamoto, A.; Hussain, S.P.; Hagiwara, K.; Spillare, E.A.; Rusin, M.R.; Demetrick, D.J.; Serrano, M.; Hannon, G.J.; Shiseki, M.; Zariwala, M.; et al. Mutations in the p16INK4/MTS1/CDKN2, p15INK4B/MTS2, and p18 genes in primary and metastatic lung cancer. Cancer Res. 1995, 55, 1448–1451. [Google Scholar]
- Zandi, R.; Selivanova, G.; Christensen, C.L. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef]
- Abal, M.; Bras-Goncalves, R.; Judde, J.G.; Fsihi, H.; de Cremoux, P.; Louvard, D.; Magdelenat, H.; Robine, S.; Poupon, M.F. Enhanced sensitivity to irinotecan by Cdk1 inhibition in the p53-Deficient HT29 human colon cancer cell line. Oncogene 2004, 23, 1737–1744. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hamilton, G.; Klameth, L.; Rath, B.; Thalhammer, T. Synergism of Cyclin-Dependent Kinase Inhibitors with Camptothecin Derivatives in Small Cell Lung Cancer Cell Lines. Molecules 2014, 19, 2077-2088. https://doi.org/10.3390/molecules19022077
Hamilton G, Klameth L, Rath B, Thalhammer T. Synergism of Cyclin-Dependent Kinase Inhibitors with Camptothecin Derivatives in Small Cell Lung Cancer Cell Lines. Molecules. 2014; 19(2):2077-2088. https://doi.org/10.3390/molecules19022077
Chicago/Turabian StyleHamilton, Gerhard, Lukas Klameth, Barbara Rath, and Theresia Thalhammer. 2014. "Synergism of Cyclin-Dependent Kinase Inhibitors with Camptothecin Derivatives in Small Cell Lung Cancer Cell Lines" Molecules 19, no. 2: 2077-2088. https://doi.org/10.3390/molecules19022077
APA StyleHamilton, G., Klameth, L., Rath, B., & Thalhammer, T. (2014). Synergism of Cyclin-Dependent Kinase Inhibitors with Camptothecin Derivatives in Small Cell Lung Cancer Cell Lines. Molecules, 19(2), 2077-2088. https://doi.org/10.3390/molecules19022077