Anticancer and Multidrug Resistance-Reversal Effects of Solanidine Analogs Synthetized from Pregnadienolone Acetate

 ,
, 
Abstract
:1. Introduction
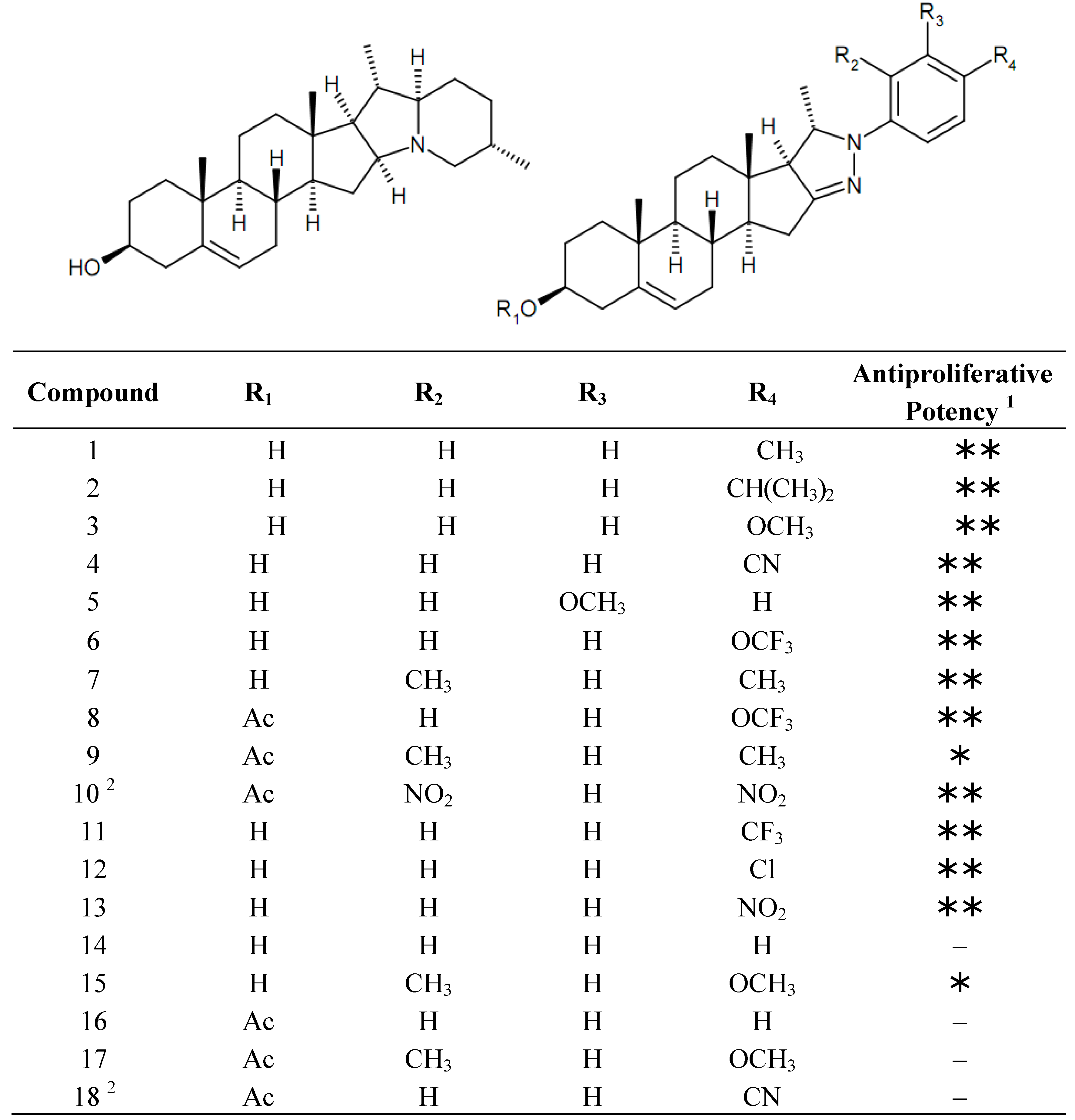
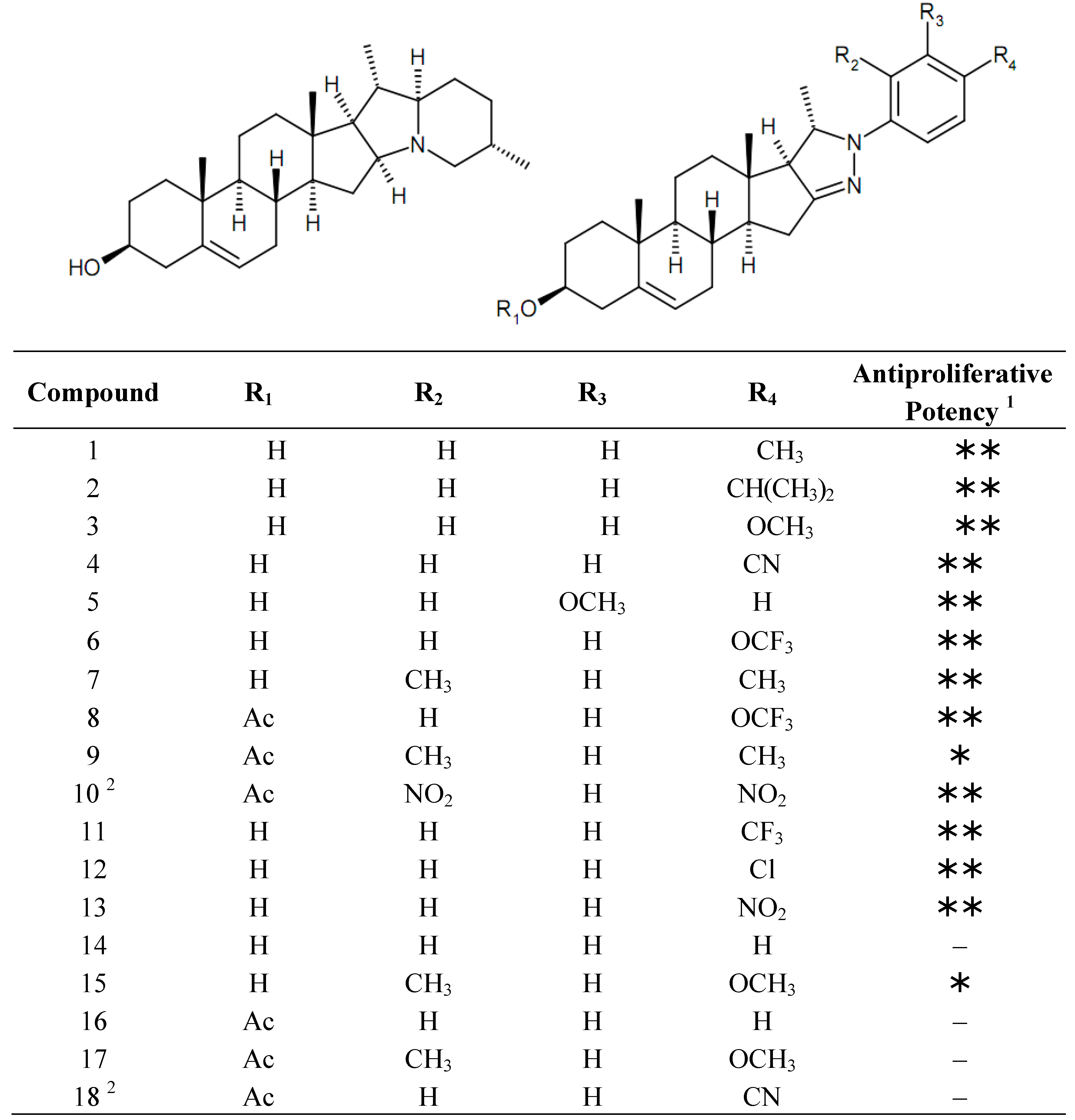
2. Results and Discussion
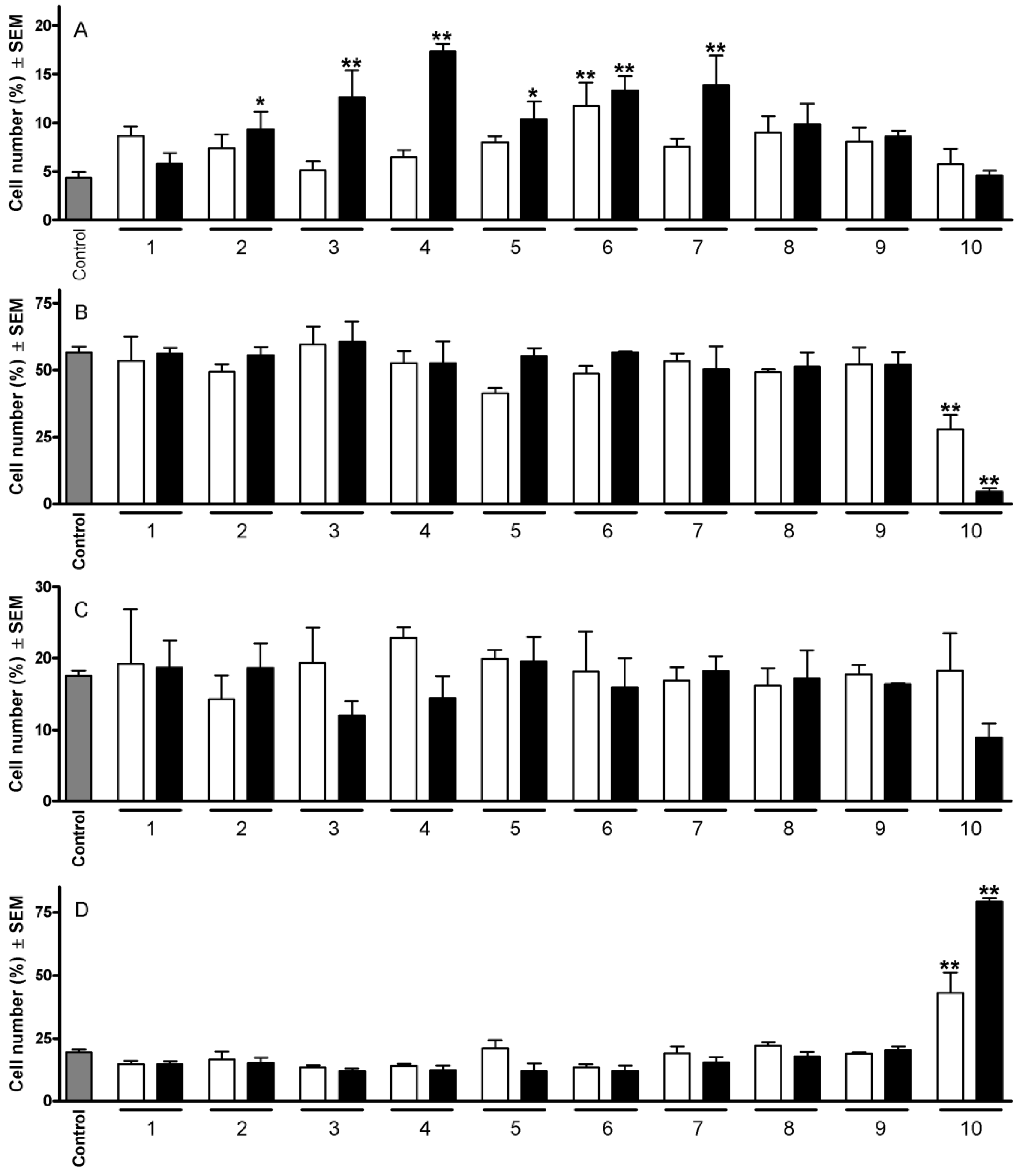
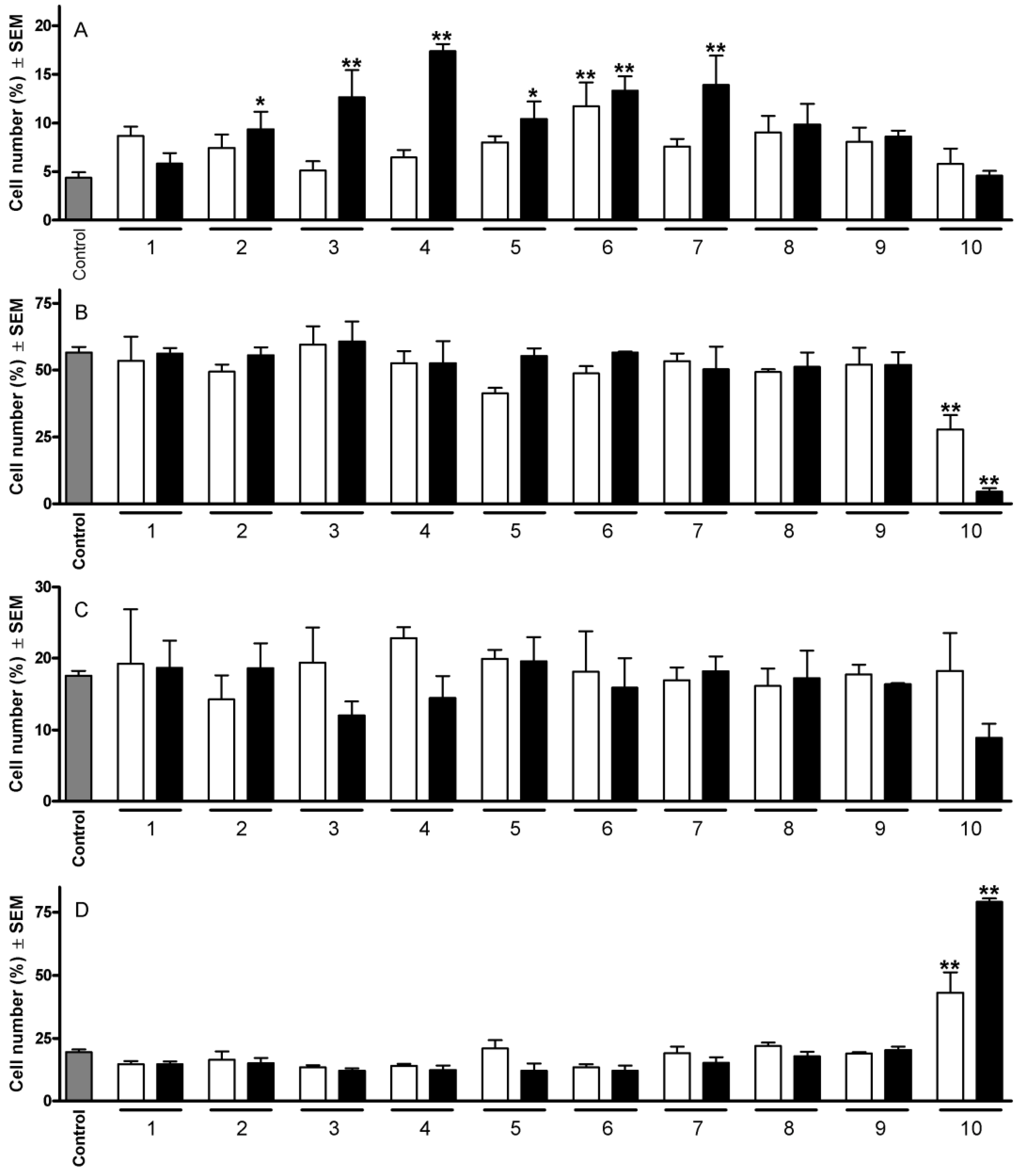
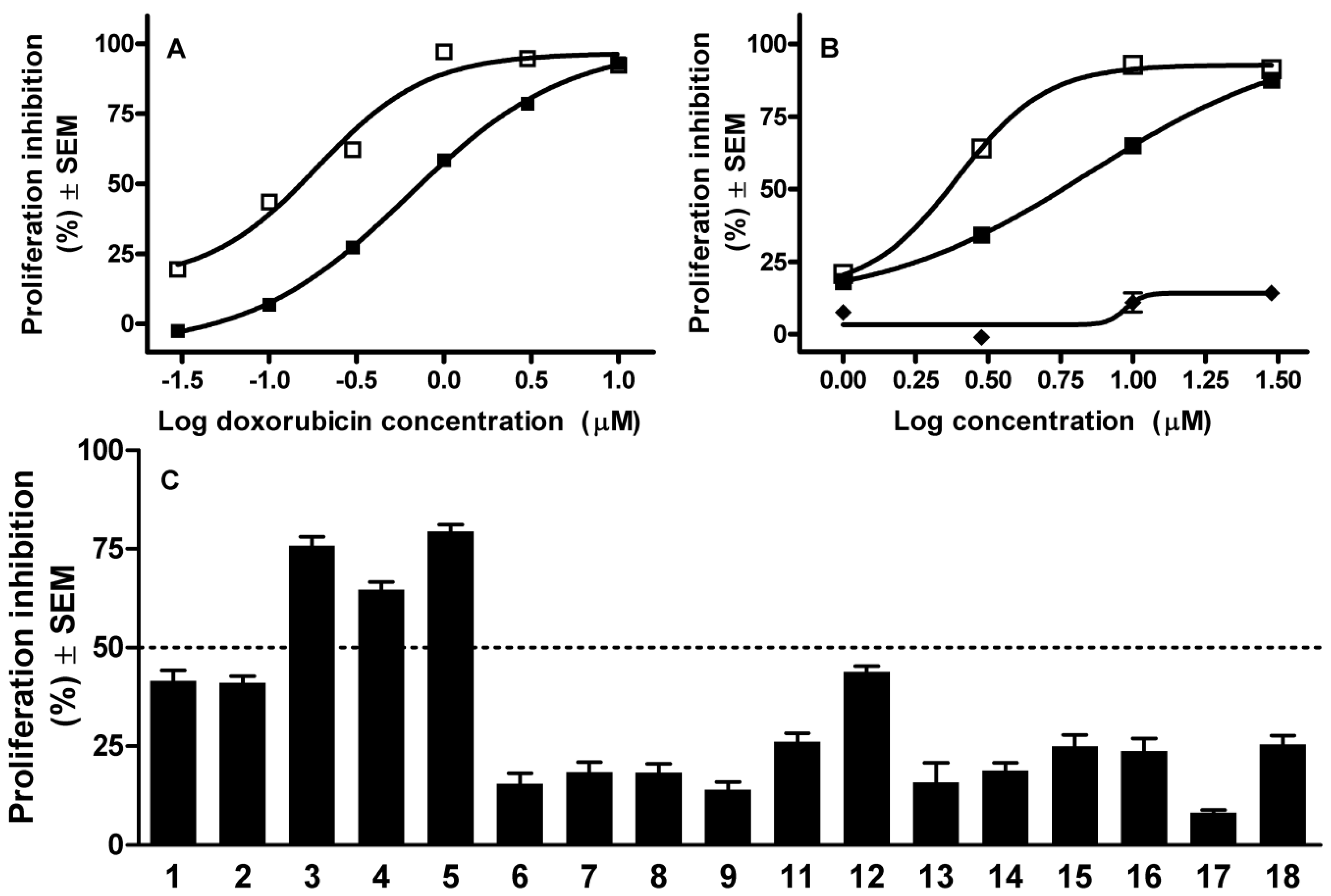
2.1. Cell Cycle Analysis
2.2. Plasmid Supercoil Relaxation Assay

2.3. RNA Profiling with High-Throughput Nanocapillary QRT-PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 10 | ||||||||||
| 6-h incubation | 12-h incubation | 24-h incubation | ||||||||
| GeneName | RefSeqID | 10 M | 20 M | 30 M | 10 M | 20 M | 30 M | 10 M | 20 M | 30 M |
| TNFα; tumor necrosis factor alpha | NM_000594 | −1.02 | −1.08 | −1.23 | 1.51 | −3.90 | −8.41 | 1.72 | −1.87 | −4.00 |
| GADD45alpha | NM_001924 | 1.59 | 1.04 | 1.24 | 1.16 | 1.28 | 1.72 | 1.14 | 5.19 | 3.65 |
| TXNRD1; thioredoxin reductase 1 | NM_003330 | 1.24 | −1.73 | −1.11 | −1.40 | −1.39 | −1.42 | 1.78 | 1.90 | 1.28 |
| SKP2; S-phase kinase-assoc. prot. 2 | NM_005983 | 1.87 | −1.55 | −1.43 | −1.06 | −1.53 | 2.11 | −4.58 | −7.60 | −8.04 |
| APC4; anaphase-promoting complex 4 | NM_013367 | 1.31 | 1.25 | 1.22 | 1.27 | 1.45 | 1.03 | −2.45 | −2.16 | −2.23 |
| CCND1; cyclin D1 | NM_053056 | −1.42 | 1.05 | −1.04 | 1.25 | 1.57 | −1.30 | −1.43 | −2.14 | −4.36 |
| CCNE2; cyclin E2 | NM_057749 | 1.04 | −1.12 | 1.09 | 1.24 | 1.23 | −1.08 | 1.23 | 1.13 | −1.23 |
| CDKN2B; cyclin-dependent kinase inhib. 2B | NM_078487 | −1.22 | −1.26 | 1.27 | 1.05 | −1.50 | 1.02 | −2.74 | −7.11 | −9.36 |
| 16 | ||||||||||
| 6-h incubation | 12-h incubation | 24-h incubation | ||||||||
| GeneName | RefSeqID | 10 M | 20 M | 30 M | 10 M | 20 M | 30 M | 10 M | 20 M | 30 M |
| TNFα; tumor necrosis factor alpha | NM_000594 | 1.56 | -1.76 | −1.69 | −1.13 | −1.72 | −1.75 | −2.23 | 1.16 | 1.45 |
| GADD45alpha | NM_001924 | −1.02 | 1.12 | 1.27 | 1.67 | 1.38 | −1.07 | 1.78 | 1.86 | 2.63 |
| TXNRD1; thioredoxin reductase 1 | NM_003330 | 1.01 | 1.14 | 1.01 | 1.68 | −1.18 | 1.37 | 2.42 | 2.01 | 2.60 |
| SKP2; S-phase kinase-assoc. prot. 2 | NM_005983 | 1.03 | −1.08 | 1.67 | 1.74 | 1.34 | 2.10 | −1.40 | −1.87 | −1.30 |
| APC4; anaphase-promoting complex 4 | NM_013367 | −1.10 | 1.26 | 1.43 | 1.75 | 1.08 | 1.11 | −1.28 | −1.90 | −1.43 |
| CCND1; cyclin D1 | NM_053056 | 1.15 | 1.11 | 1.63 | 1.07 | 1.22 | 1.09 | −1.60 | −1.01 | −1.19 |
| CCNE2; cyclin E2 | NM_057749 | 1.09 | 1.30 | 1.48 | 1.94 | 1.44 | 1.83 | −1.33 | −2.62 | −2.33 |
| CDKN2B; cyclin-dependent kinase inhib. 2B | NM_078487 | 1.69 | 1.18 | 2.27 | -1.61 | 1.96 | 1.88 | −6.22 | −7.86 | −14.83 |
2.4. Rhodamine-123 Accumulation Assay
| Compound | Concentration (µM) | Fluorescence activity ratio | Compound | Concentration (µM) | Fluorescence activity ratio | ||
|---|---|---|---|---|---|---|---|
| 1 | 40 | 3.71 ± 0.96 | 11 | 40 | 4.72 ± 0.53 | ||
| 400 | 50.15 ± 7.62 | 400 | 84.03 ± 14.22 | ||||
| 2 | 40 | 1.38 ± 0.11 | 12 | 40 | 28.74 ± 8.31 | ||
| 400 | 6.86 ± 0.96 | 400 | 49.07 ± 19.09 | ||||
| 3 | 40 | 40.68 ± 13.82 | 13 | 40 | 2.52 ± 0.06 | ||
| 400 | 187.6 ± 51.67 | ||||||
| 400 | 2.27 ± 0.37 | ||||||
| 4 | 40 | 169.6 ± 43.77 | 14 | 40 | 36.31 ± 9.56 | ||
| 400 | 106.1 ± 17.11 | 400 | 14.33 ± 1.20 | ||||
| 5 | 40 | 51.60 ± 12.57 | 15 | 40 | 135.1 ± 48.35 | ||
| 400 | 114.4 ± 26.51 | 400 | 4.58 ± 0.58 | ||||
| 6 | 40 | 2.91 ± 1.76 | 16 | 40 | 12.25 ± 3.66 | ||
| 400 | 10.16 ± 2.77 | 400 | 50.24 ± 3.75 | ||||
| 7 | 40 | 30.59 ± 10.05 | 17 | 40 | 1.72 ± 0.46 | ||
| 400 | 44.90 ± 16.08 | 400 | 7.27 ± 2.30 | ||||
| 8 | 40 | 1.62 ± 0.15 | 18 | 40 | 6.74 ± 1.70 | ||
| 400 | 2.82 ± 0.94 | 400 | 21.98 ± 4.33 | ||||
| 9 | 40 | 3.16 ± 1.34 | Verapamil | 40.6 | 5.93 ± 1.92 | ||
| 400 | 17.15 ± 3.67 |
2.5. Combination Experiments

2.6. Discussion
3. Experimental
3.1. Cancer Cell Lines
3.2. Cell Cycle Analysis
3.3. Plasmid Supercoil Relaxation Assays
3.4. RNA Isolation, cDNA Conversion
3.5. Profiling of RNAs with High-Throughput, nanOcapillary QRT-PCR
3.6. Rhodamine-123 Exclusion Assay
3.7. Antiproliferative Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burg, V.K.; Grimm, H.S.; Rothhaar, T.L.; Grosgen, S.; Hundsdorfer, B.; Haupenthal, V.J.; Zimmer, V.C.; Mett, J.; Weingartner, O.; Laufs, U.; et al. Plant sterols the better cholesterol in Alzheimer’s disease? A mechanistical study. J. Neurosci. 2013, 33, 16072–16087. [Google Scholar] [CrossRef]
- Aperia, A. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J. Intern. Med. 2007, 261, 44–52. [Google Scholar] [CrossRef]
- Minorics, R.; Szekeres, T.; Krupitza, G.; Saiko, P.; Giessrigl, B.; Wölfling, J.; Frank, E.; Zupkó, I. Antiproliferative effects of some novel synthetic solanidine analogs on HL-60 human leukemia cells in vitro. Steroids 2011, 76, 156–162. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Perjesi, P.; Rivera-Portalatin, N.M.; Simpkins, J.W.; Prokai, L. Mechanistic investigations on the antioxidant action of a neuroprotective estrogen derivative. Steroids 2008, 73, 280–288. [Google Scholar] [CrossRef]
- Rocha, M.; Banuls, C.; Bellod, L.; Jover, A.; Victor, V.M.; Hernandez-Mijares, A. A review on the role of phytosterols: New insights into cardiovascular risk. Curr. Pharm. Des. 2011, 17, 4061–4075. [Google Scholar] [CrossRef]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Mijatovic, T.; van Quaquebeke, E.; Delest, B.; Debeir, O.; Darro, F.; Kiss, R. Cardiotonic steroids on the road to anti-cancer therapy. Biochim. Biophys. Acta 2007, 1776, 32–57. [Google Scholar]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef]
- Koduru, S.; Grierson, D.S.; van de Venter, M.; Afolayan, A.J. Anticancer activity of steroid alkaloids isolated from Solanum aculeastrum. Pharm. Biol. 2007, 45, 613–618. [Google Scholar] [CrossRef]
- Punjabi, S.; Cook, L.J.; Kersey, P.; Marks, R.; Cerio, R. Solasodine glycoalkaloids: A novel topical therapy for basal cell carcinoma. A double-blind, randomized, placebo-controlled, parallel group, multicenter study. Int. J. Dermatol. 2008, 47, 78–82. [Google Scholar]
- Saha, P.; Debnath, C.; Berube, G. Steroid-linked nitrogen mustards as potential anticancer therapeutics: A review. J. Steroid Biochem. Mol. Biol. 2013, 137, 271–300. [Google Scholar] [CrossRef]
- Kádár, Z.; Molnár, J.; Schneider, G.; Zupkó, I.; Frank, É. A facile ‘click’ approach to novel 15β-triazolyl-5α-androstane derivatives, and an evaluation of their antiproliferative activities in vitro. Bioorg. Med. Chem. 2012, 20, 1396–1402. [Google Scholar] [CrossRef]
- Kádár, Z.; Kovács, D.; Frank, É.; Schneider, G.; Huber, J.; Zupkó, I.; Bartók, T.; Wölfling, J. Synthesis and in vitro antiproliferative activity of novel androst-5-ene triazolyl and tetrazolyl derivatives. Molecules 2011, 16, 4786–4806. [Google Scholar] [CrossRef]
- Kádár, Z.; Baji, A.; Zupkó, I.; Bartók, T.; Wölfling, J.; Frank, É. Efficient approach to novel 1α-triazolyl-5α-androstane derivatives as potent antiproliferative agents. Org. Biomol. Chem. 2011, 9, 8051–8057. [Google Scholar] [CrossRef]
- Frank, É.; Mucsi, Z.; Szécsi, M.; Zupkó, I.; Wölfling, J.; Schneider, G. Intramolecular approach to some new D-ring-fused steroidal isoxazolidines by 1,3-dipolar cycloaddition: Synthesis, theoretical and in vitro pharmacological studies. New J. Chem. 2010, 34, 2671–2681. [Google Scholar] [CrossRef]
- Frank, É.; Mucsi, Z.; Zupkó, I.; Réthy, B.; Falkay, G.; Schneider, G.; Wölfling, J. Efficient approach to androstene-fused arylpyrazolines as potent antiproliferative agents. Experimental and theoretical studies of substituent effects on BF3-catalyzed intramolecular [3 + 2] cycloadditions of olefinic phenylhydrazones. J. Am. Chem. Soc. 2009, 131, 3894–3904. [Google Scholar] [CrossRef]
- Zhang, A.L.; He, L.Y.; Gao, J.M.; Xu, X.; Li, S.Q.; Bai, M.S.; Qin, J.C. Metabolites from an endophytic fungus sphaceloma sp. LN-15 isolated from the leaves of Melia azedarach. Lipids 2009, 44, 745–751. [Google Scholar] [CrossRef]
- Choi, C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Lavie, Y.; Harel-Orbital, T.; Gaffield, W.; Liscovitch, M. Inhibitory effect of steroidal alkaloids on drug transport and multidrug resistance in human cancer cells. Anticancer Res. 2001, 21, 1189–1194. [Google Scholar]
- Chan, C.H.; Lee, S.W.; Wang, J.; Lin, H.K. Regulation of Skp2 expression and activity and its role in cancer progression. ScientificWorldJournal 2010, 10, 1001–1015. [Google Scholar] [CrossRef]
- Wang, Z.; Fukushima, H.; Inuzuka, H.; Wan, L.; Liu, P.; Gao, D.; Sarkar, F.H.; Wei, W. Skp2 is a promising therapeutic target in breast cancer. Front. Oncol. 2012, 1, 57. [Google Scholar]
- Yokoi, S.; Yasui, K.; Iizasa, T.; Takahashi, T.; Fujisawa, T.; Inazawa, J. Down-regulation of SKP2 induces apoptosis in lung-cancer cells. Cancer Sci. 2003, 94, 344–349. [Google Scholar] [CrossRef]
- Pennington, J.D.; Jacobs, K.M.; Sun, L.; Bar-Sela, G.; Mishra, M.; Gius, D. Thioredoxin and thioredoxin reductase as redox-sensitive molecular targets for cancer therapy. Curr. Pharm. Des. 2007, 13, 3368–3377. [Google Scholar] [CrossRef]
- Zhu, N.; Shao, Y.; Xu, L.; Yu, L.; Sun, L. Gadd45-alpha and Gadd45-gamma utilize p38 and JNK signaling pathways to induce cell cycle G2/M arrest in Hep-G2 hepatoma cells. Mol. Biol. Rep. 2009, 36, 2075–2085. [Google Scholar] [CrossRef]
- Alpan, A.S.; Zencir, S.; Zupkó, I.; Coban, G.; Réthy, B.; Gunes, H.S.; Topcu, Z. Biological activity of bis-benzimidazole derivatives on DNA topoisomerase I and HeLa, MCF7 and A431 cells. J. Enzyme Inhib. Med. Chem. 2009, 24, 844–849. [Google Scholar] [CrossRef]
- Ishar, M.P.; Singh, G.; Singh, S.; Sreenivasan, K.K. Design, synthesis, and evaluation of novel 6-chloro-/fluorochromone derivatives as potential topoisomerase inhibitor anticancer agents. Bioorg. Med. Chem. Lett. 2006, 16, 1366–1370. [Google Scholar] [CrossRef]
- Puskás, L.G.; Fehér, L.Z.; Vizler, C.; Ayaydin, F.; Rásó, E.; Molnár, E.; Magyary, I.; Kanizsai, I.; Gyuris, M.; Madácsi, R.; et al. Polyunsaturated fatty acids synergize with lipid droplet binding thalidomide analogs to induce oxidative stress in cancer cells. Lipids Health Dis. 2010, 9, 56. [Google Scholar] [CrossRef]
- Fabian, G.; Farago, N.; Feher, L.Z.; Nagy, L.I.; Kulin, S.; Kitajka, K.; Bito, T.; Tubak, V.; Katona, R.L.; Tiszlavicz, L.; et al. High-density real-time PCR-based in vivo toxicogenomic screen to predict organ-specific toxicity. Int. J. Mol. Sci. 2011, 12, 6116–6134. [Google Scholar] [CrossRef]
- Chellappan, S.P.; Giordano, A.; Fisher, P.B. Role of cyclin-dependent kinases and their inhibitors in cellular differentiation and development. Curr. Top. Microbiol. Immunol. 1998, 227, 57–103. [Google Scholar]
- Park, M.T.; Lee, S.J. Cell cycle and cancer. J. Biochem. Mol. Biol. 2003, 36, 60–65. [Google Scholar] [CrossRef]
- Pestell, R.G. New roles of cyclin D1. Am. J. Pathol. 2013, 183, 3–9. [Google Scholar] [CrossRef]
- Pastan, I.; Gottesman, M.M.; Ueda, K.; Lovelace, E.; Rutherford, A.V.; Willingham, M.C. A retrovirus carrying an MDR1 cDNA confers multidrug resistance and polarized expression of P-glycoprotein in MDCK cells. Proc. Natl. Acad. Sci. USA 1988, 85, 4486–4490. [Google Scholar] [CrossRef]
- Molnár, J.; Szabó, D.; Mándi, Y.; Mucsi, I.; Fischer, J.; Varga, A.; Konig, S.; Motohashi, N. Multidrug resistance reversal in mouse lymphoma cells by heterocyclic compounds. Anticancer Res. 1998, 18, 3033–3038. [Google Scholar]
- Réthy, B.; Zupkó, I.; Minorics, R.; Hohmann, J.; Ocsovszki, I.; Falkay, G. Investigation of cytotoxic activity on human cancer cell lines of arborinine and furanoacridones isolated from Ruta graveolens. Planta Med. 2007, 73, 41–48. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Reutelingsperger, C. Flow cytometry of apoptotic cell death. J. Immunol. Methods 2000, 243, 167–190. [Google Scholar] [CrossRef]
- Coban, G.; Zencir, S.; Zupkó, I.; Réthy, B.; Gunes, H.S.; Topcu, Z. Synthesis and biological activity evaluation of 1H-benzimidazoles via mammalian DNA topoisomerase I and cytostaticity assays. Eur. J. Med. Chem. 2008, 44, 2280–2285. [Google Scholar]
- Sarikaya, D.; Bilgen, C.; Kamataki, T.; Topcu, Z. Comparative cytochrome P450 -1A1, -2A6, -2B6, -2C, -2D6, -2E1, -3A5 and -4B1 expressions in human larynx tissue analysed at mRNA level. Biopharm. Drug Dispos. 2006, 27, 353–359. [Google Scholar] [CrossRef]
- Vass, L.; Kelemen, J.Z.; Fehér, L.Z.; Lorincz, Z.; Kulin, S.; Cseh, S.; Dormán, G.; Puskás, L.G. Toxicogenomics screening of small molecules using high-density, nanocapillary real-time PCR. Int. J. Mol. Med. 2009, 23, 65–74. [Google Scholar]
- Zupkó, I.; Réthy, B.; Hohmann, J.; Molnár, J.; Ocsovszki, I.; Falkay, G. Antitumor activity of alkaloids derived from Amaryllidaceae species. In Vivo 2009, 23, 41–48. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zupkó, I.; Molnár, J.; Réthy, B.; Minorics, R.; Frank, É.; Wölfling, J.; Molnár, J.; Ocsovszki, I.; Topcu, Z.; Bitó, T.; et al. Anticancer and Multidrug Resistance-Reversal Effects of Solanidine Analogs Synthetized from Pregnadienolone Acetate. Molecules 2014, 19, 2061-2076. https://doi.org/10.3390/molecules19022061
Zupkó I, Molnár J, Réthy B, Minorics R, Frank É, Wölfling J, Molnár J, Ocsovszki I, Topcu Z, Bitó T, et al. Anticancer and Multidrug Resistance-Reversal Effects of Solanidine Analogs Synthetized from Pregnadienolone Acetate. Molecules. 2014; 19(2):2061-2076. https://doi.org/10.3390/molecules19022061
Chicago/Turabian StyleZupkó, István, Judit Molnár, Borbála Réthy, Renáta Minorics, Éva Frank, János Wölfling, Joseph Molnár, Imre Ocsovszki, Zeki Topcu, Tamás Bitó, and et al. 2014. "Anticancer and Multidrug Resistance-Reversal Effects of Solanidine Analogs Synthetized from Pregnadienolone Acetate" Molecules 19, no. 2: 2061-2076. https://doi.org/10.3390/molecules19022061
APA StyleZupkó, I., Molnár, J., Réthy, B., Minorics, R., Frank, É., Wölfling, J., Molnár, J., Ocsovszki, I., Topcu, Z., Bitó, T., & Puskás, L. G. (2014). Anticancer and Multidrug Resistance-Reversal Effects of Solanidine Analogs Synthetized from Pregnadienolone Acetate. Molecules, 19(2), 2061-2076. https://doi.org/10.3390/molecules19022061