Abstract
Celery (Apium graveolens L.) is one of the most economically important vegetables worldwide, but genetic and genomic resources supporting celery molecular breeding are quite limited, thus few studies on celery have been conducted so far. In this study we made use of simple sequence repeat (SSR) markers generated from previous celery transcriptome sequencing and attempted to detect the genetic diversity and relationships of commonly used celery accessions and explore the efficiency of the primers used for cultivars identification. Analysis of molecular variance (AMOVA) of Apium graveolens L. var. dulce showed that approximately 43% of genetic diversity was within accessions, 45% among accessions, and 22% among horticultural types. The neighbor-joining tree generated by unweighted pair group method with arithmetic mean (UPGMA), and population structure analysis, as well as principal components analysis (PCA), separated the cultivars into clusters corresponding to the geographical areas where they originated. Genetic distance analysis suggested that genetic variation within Apium graveolens was quite limited. Genotypic diversity showed any combinations of 55 genic SSRs were able to distinguish the genotypes of all 30 accessions.
1. Introduction
Celery (Apium graveolens L.) is a biennial species from the family of Apiaceae with 2n = 2x = 22 chromosomes. It originated from the Mediterranean basin and several cultivated types are grown worldwide for consumption. Besides the wild (Apium chilanse) species and celeriac (Apium graveolens L. var. rapaceum) species, both coming from Western countries, common celery (Apium graveolens L. var. dulce) cultivars are generally classification based on their origin as celery (cultivars introduced from Western countries), local celery (Chinese celery) and the middle type (hybrids of celery and local celery).
Several types of biochemical and molecular markers have been applied for celery genotyping, such as isozymes [1], restriction fragment length polymorphism (RFLP) [2], random amplified polymorphic DNA (RAPD) [3,4], amplified fragment length polymorphism (AFLP) [5,6], sequence-related amplified polymorphism (SRAP) [7], expressed sequence tag based SSR (EST-SSR) [8,9]. Genotyping with molecular markers is used for identification of cultivars [10,11], cultivar fingerprinting [12,13], detection of genetic variation and genetic diversity [14,15,16], construction of linkage maps [17,18,19], mapping genes of interest, and for marker assisted selection (MAS) [20,21,22,23]. These researches are frequently carried out with SSR markers for their co-dominant and multi-allelic nature, which makes them more informative than dominant-types of markers. However, development of SSR markers is costly and time consuming and therefore research on celery was quite limited. Next-generation transcriptome sequencing provides an efficient means to develop SSR markers and it has been applied to many organisms [24,25,26]. In our previous work, we developed a set of EST-SSR markers [27] through transcriptome sequencing.
The objectives of the present work were to: (1) test marker polymorphism on a set of celery cultivars; (2) assess the genetic variation existing in the materials used; (3) detect the genetic diversity and population structure of these materials and (4) explore the efficiency of the primers used for cultivar identification.
2. Results and Discussion
A list of the samples investigated in this study is given in Table 1. This set of accessions comprised 28 common cultivars, one celeriac and one wild species. The 28 common cultivars can be further divided into 16 celery accessions, nine local celery accessions and three middle type accessions.

Table 1.
List of the 30 accessions genotyped with genic SSR markers.
| Code | Variety | Type | Species |
|---|---|---|---|
| C1 | Xuebaiqincai | Local celery | Apium graveolens L. var. dulce |
| C3 | Jinhuangqincai | Local celery | Apium graveolens L. var. dulce |
| C5 | Jincuifuqin | Local celery | Apium graveolens L. var. dulce |
| C6 | Huangxinqin | Local celery | Apium graveolens L. var. dulce |
| C8 | Shanghaichunqin | Local celery | Apium graveolens L. var. dulce |
| C13 | Tieganqing | Local celery | Apium graveolens L. var. dulce |
| C29 | Wuqiangshiganqincai | Local celery | Apium graveolens L. var. dulce |
| C37 | Shixinqin | Local celery | Apium graveolens L. var. dulce |
| C53 | Duolunshiganqincai | Local celery | Apium graveolens L. var. dulce |
| C111 | Lino | Celery | Apium graveolens L. var. dulce |
| C67 | Jiazhouwang | Celery | Apium graveolens L. var. dulce |
| C87 | Ventura | Celery | Apium graveolens L. var. dulce |
| C114 | Huanghou | Celery | Apium graveolens L. var. dulce |
| C62 | Introduced from USA(fertile) | Celery | Apium graveolens L. var. dulce |
| C63 | Introduced from USA(sterile ) | Celery | Apium graveolens L. var. dulce |
| C65 | - | Celery | Apium graveolens L. var. dulce |
| C68 | Jiahuang | Celery | Apium graveolens L. var. dulce |
| C74 | Dore Golden Spartan | Celery | Apium graveolens L. var. dulce |
| C79 | Kangnaier | Celery | Apium graveolens L. var. dulce |
| C83 | Bailixiqin | Celery | Apium graveolens L. var. dulce |
| C89 | TU52-75 | Celery | Apium graveolens L. var. dulce |
| C97 | Kaifengbolicui | Celery | Apium graveolens L. var. dulce |
| C118 | Guihe | Celery | Apium graveolens L. var. dulce |
| C121 | Qianfang | Celery | Apium graveolens L. var. dulce |
| C123 | Ventura (yellow mutant) | Celery | Apium graveolens L. var. dulce |
| C58 | Majiagouqincai | Middle type | Apium graveolens L. var. dulce |
| C99 | Jinnanhuangxinqin | Middle type | Apium graveolens L. var. dulce |
| C159 | Jinnanshiqin | Middle type | Apium graveolens L. var. dulce |
| C163 | - | Celeriac | Apium graveolens L. var. rapaceum |
| C130 | - | Wild | Apium chilanse |
2.1. Marker Development
In the previous work [27] we mined approximately 3,000 SSRs using the MISA software. Among the SSRs, mononucleotide motifs were discarded since it was difficult to distinguish genuine mononucleotide repeats from those generated by polyadenylation products, base mismatches or sequencing errors. In present research, we randomly selected 140 SSRs for polymorphism analysis (Table 2).
Table 2.
SSR markers used in this study.
| Primer Name | Forward Primer | Reverse Primer |
|---|---|---|
| Fn1 | GCGCTTGGTGTATCTCCACT | AGTGCGTCGAAATATCGCTT |
| Fn2 | GCTTCCGCTGTGTATTTTGA | GGGAAGAAACTGCAACTTGG |
| Fn3 | TGAGCTCCACCAACTGACAC | GCATGAGCAGTTCCAAGACA |
| Fn4 | AATTTACCGCTCTTAGCCTCG | ATAGGCAGAATTTGCGACGA |
| Fn5 | TGAAACCCAAGATCACCCAT | TCATATTGACAGGCAACCGA |
| Fn6 | CCAATCTGGGACTGTGTAAGC | TTCCTGGAGGTGAAGGACTG |
| Fn7 | TGGTGTTGCAGTGTGAATCC | ACCGAAGCATCCTTGAACAG |
| Fn8 | TGGTGTTGCAGTGTGAATCC | ACCGAAGCATCCTTGAACAG |
| Fn9 | CATAGGCTAACGCAGCTTCC | AGTACTCCTTCAGCCGACGA |
| Fn10 | CAGGAGGCTGCAATAACACA | GAGTCGCCGGAATATCAAGA |
| Fn11 | CACACAGACGACTGCTGCTT | ACCATGCATGCTCAACTGAT |
| Fn12 | CACACAGACGACTGCTGCTT | ACCATGCATGCTCAACTGAT |
| Fn13 | CGATCAGGGTACTTGGCAAT | TTTCTATATCCGTCTCATTTCTGTT |
| Fn14 | AACCCTAGCTGCCTCTCTCC | CCATGCCACGAATAGCATTA |
| Fn15 | TGTGTTCTCGCATCTCCAAC | CCAATCTCAACATCGCACAG |
| Fn16 | GTTGGTCAATGCTGCTTCCT | TGTGCCAGGGATACCTTCTC |
| Fn17 | TCACTCACTCCCTTGAGCCT | TGAATCAACACCGTCCATTG |
| Fn18 | CAACCTGAACATCGTTGGTG | TCAACTTGATCTCACGGCAG |
| Fn19 | CTCATACGGTCCAGATCGGT | ATGTCCTGGTGAAGGAGGTG |
| Fn20 | GAGATTGCGATAATGGTGGC | CGCATCACATCACTTAACGG |
| Fn21 | CTGCTCTGAAAGGCTCTGCT | ACAGCTGACATCCTTACCGC |
| Fn22 | TTCACTTGTTCAGCGAGACG | CCTAACCCTAGCTCGTCGTG |
| Fn23 | TCCCATCTCCAATTCCAATC | TTCCTTGCAAGACCATAGGC |
| Fn24 | CATGTCACTGTCGAAGCACC | TGACAATTGCCATTCTCCTG |
| Fn25 | GCCTGAGCATCATAAGAGCC | TATTCACCTTCGTATCCGGC |
| Fn26 | TGTTCCATTATGTGTTGCAGTG | GCGAGATGATGTCAGAACGA |
| Fn27 | ACCATGTCCACCACCTTCTC | GCTGGTTATGGTGGTGCTG |
| Fn28 | ATCGCCAACACCTTCTCAAG | AAGGGTGATTCTGATGGTGC |
| Fn29 | CATATCCAGCACCTCCACCT | TCCAATGGCAACTCACAGAG |
| Fn30 | CTCATTCTTCTTCTTGGCCG | TGCTGAAACGCTACCTCCTT |
| Fn31 | ACATGGAATCTTTCACCTTCA | CATGGCCTAGGAGGAGACAA |
| Fn32 | TTCCTGTCCAGCAGTATCCC | GAATTGAGGGGTGAAAAAGC |
| Fn33 | AAATGAGGTGGTGGTGGAAG | CAATGGGTATGGAATCAGGC |
| Fn34 | AGTACGGTGTCTACGACGGG | CCTCCACCATGATTACCACC |
| Fn35 | CATACTTCTTTGGGGGCTCA | ACACAAACTTTCGGCCAGAT |
| Fn36 | AAGGTCAAGGTCCTGTGGTG | GGTTTAGGCCTCCAATAGCC |
| Fn37 | ACAGTACGTGTCTCCCCCTG | AACAACCCTATGATGGCTGC |
| Fn38 | GTTTGAGCCTCCGCTTACAG | TGCCAGTGACACTCTTCACC |
| Fn39 | GTGACGAAGGAATTGACCGT | ATTTGTTGTCGGGTTCCAAA |
| Fn40 | CTGGCACTTGTACGAAACCA | TATGGGCTGTTGATGACAGG |
| Fn41 | TTCAACCCAGACTTCAACCC | GCAGCCTTCAAATCCAGTTC |
| Fn42 | CCCAGCCCTATCAATCTTCA | CCCCTGCCAAGTCTGTTAAT |
| Fn43 | GAGACAGAGACCATGGGGAA | CGGTTTCGGTTTCGATTTTA |
| Fn44 | TCTTGTCCATTAAAAATGTACCCA | TGCGCATAATGAAAGGATCA |
| Fn45 | AGCAGCACAACAACACTTGG | TTAGGGTCTCTTGTCGAGGC |
| Fn46 | GCAAGTTACCACCCCAGAAA | CCTTTTTCAAAAGCTTCCCC |
| Fn47 | AATTGGCCAGAGCAGAGAAA | TCCTTTATCCCTGACCATGC |
| Fn48 | AATGAGGTTGTTTTTCCCCC | GTAAAGGCCCAAACTCCTCC |
| Fn49 | ACAACAATTTCAGGGCCAAG | TCTTGATATCGGCTTCCTGG |
| Fn50 | ACATTTGTTGCTAGGGTGGC | GCACGAATAGCCGTCCTAAA |
| Fn51 | TCCAATCTCCGGAGCAATAC | GCGGTGGACGAGTAAAAGAG |
| Fn52 | AAACCACCAAACAAGGTGCT | TGAAGTGGAGGAGCAGGAGT |
| Fn53 | ATTCCCAGATGGCTGCATAG | AATTCCAGCAAGCTCAAGGA |
| Fn54 | CCCTCTCCCTATCTTCCTCG | TGAGATTGACTCGGTTGCTG |
| Fn55 | AAAGAAGAAACGGGGATGCT | AACAGCAAGCAGTTCAGTAGTCA |
| Fn56 | CTACACCGCCAATTCAACCT | TAAGCATACACCCCCTCACC |
| Fn57 | TAATGGTGGAAAGAAAGGCG | GGCATACCACTCATTTGGCT |
| Fn58 | CCTAGGCGAACTCTCCTCCT | GGGAATGATCCTCCTTCCAT |
| Fn59 | AGAGGGATAAAATCCCGGTG | TGGAAATGCAAAAGAAGCAA |
| Fn60 | CCACCACCACAACTACAACG | AAGCCGAGTAATGCTGGAAA |
| Fn61 | AGGGTTCGTCGGTTGTAGTG | TCCCCATGTCTATTCTTCGC |
| Fn62 | TCTCGACGAGTTTTCACCTG | GGTCTCTTTGGTGCCATTGT |
| Fn63 | TCAATTAGATCCAGACGCCC | TCTTCTGCCTCCTCTTCGAG |
| Fn64 | CTGTTTTCTCCCTCGTCTGC | CCCCATCTCTGCTATAGCTCC |
| Fn65 | CTTTCTGGGTAAGAAGGCCC | CCAACCCACAACGTCTTACC |
| Fn66 | TCATTGGACAAACAGGACGA | CTGTTTGGCGCTCAATTTTT |
| Fn67 | TACATTTGTGGGATTGGGGT | CCGCCAAAACATTGACAGTA |
| Fn68 | TCAGCTAAGCCACCCTGATT | GTTGCTGCTGGAGAAAGGAG |
| Fn69 | TCCATGAATCTTTCAAGCCC | TCCAAAGTCCAATCCCATTT |
| Fn70 | TAACTGAGTCGGTTGGGTCC | CCTCTCTTTTACCAGCCAAGC |
| Fn71 | TCAACACTCAATTTAAACACCCA | TGATAACGATCGTGACGGAA |
| Fn72 | CATCAACACTCGAAATCGAAA | CAAGATGCTTGTTATCCTTGCT |
| Fn73 | GGATCGGAGGAAGGAAGAAA | GGAGGTGGAGGAGGAGAGTT |
| Fn74 | CCCCCAAACAATAAGTATCCC | TTGGAACTTTTTGTGTCCATTG |
| Fn75 | GCCAGCAGTGTCCCATATTT | GCCCGGAAAAATAACAATCA |
| Fn76 | TTCTACCACTTTCCTTGAATCC | CAGGAGCAGTCTCGATTTCC |
| Fn77 | GGTGTATTTGAATATTAACACCTTTCG | AGAGATGGTGGTCTTGTGGG |
| Fn78 | ACAAGCCCCCTCTACTTTGG | ATGTTGCCAGTTCAGGTTCC |
| Fn79 | TGGGACCCATTTCTTGATTT | AAAATTGCTCCGATTTGTGC |
| Fn80 | CCAGGTAAGCCCAGTCTCAA | TTTTTCTCAATTAAAACTTGCTCATTT |
| Fn81 | AATCCTTGAACTAACCGGGG | CTCTTCGCCACCAGATTCAT |
| Fn82 | GGACGCCCAAGAGAGGTAGT | AGTGGTCTCGACATTTTCCC |
| Fn83 | CCACACCTTGATCGTTGAGA | TTGCTTCTTCCGGCTCTTTA |
| Fn84 | GTTACTTGACGGCACCGTTT | ATCAGTTCTTCATCCGTGGG |
| Fn85 | TCACCCTCTCATCCACATCA | GCAGTGGGTGGATCTAGGAA |
| Fn86 | TCAAATGGACGACGAATCAA | TGCAATGATTTATCCCCCTC |
| Fn87 | CACACACAGGACACACATATTTC | GTAAAGCCGTCTTGGACGAG |
| Fn88 | CGGCATCTTCTCTTCCTCAC | TGTTTGGATCTTTTCTGTTTTCA |
| Fn89 | CAGAAGCGGCTCCTTCTCTA | CCCATTTGAGCTTCACCACT |
| Fn90 | CTAAACGACGCCGTTACCAT | GCTTCTCTCCGCCTTGTATG |
| Fn91 | GGCATACATCGGACGCTAAT | TTGACCCTTTATCTCAATACACACA |
| Fn92 | CCCTCTCTCTCCCTCCTGTC | ACGATTAGCCATTGGTGAGC |
| Fn93 | TGTGTGCTGATTTGAAACCC | ACCGACACTCCACCTTCATC |
| Fn94 | CACCTCTGCTTTCACGGAAC | GTCCAAGAGTGGTCCTCACC |
| Fn95 | ATGGTAACACCACCCTGGAA | GCTTCAACCAGGCAAAGACT |
| Fn96 | TACTTACACCCCTCCCTCCC | TGCAGCACAAGGGATTCATA |
| Fn97 | AAGAGCGATCAAGAACAGGG | TCCCATCTCTCTCCCTCGTA |
| Fn98 | TGCGAACAATACAGTCCCAA | CAGATCCAAACACAGAATTAGCA |
| Fn99 | GAAGAAAGAGGAGAAGGCCG | TCTCGAAACCACCCATCTTC |
| Fn100 | GCGATCCCTAATCAATCCAA | CTTTGAGAGTTACGACGGGC |
| Fn101 | TCAATGGTGTAGAACCAGAACAA | CCCAGATGCTTAAAAGAACCA |
| Fn102 | ACAGGAGGCACTGGTCTCAC | CATTAAAATCCCACAAAAACTTCA |
| Fn103 | TCCCATTCCATTTCAACCTC | AGAGGTGTGGGGAGATTGTG |
| Fn104 | GCGGGGACACTCCACTACT | TTGATCATCAGCAGACTGGC |
| Fn105 | CAAAAATTTAACCCCATACCC | ACATGTACGGACGTTGTGGA |
| Fn106 | GCTTTGCACACACACACACA | CTTTCCCTCGACCTCATCCT |
| Fn107 | GATTTTTCCGATGCAGCACT | GGCATGCACCAAACGTTATC |
| Fn108 | GAGGAGGCTGTTACGTGGAG | TCCCTTTTCTCACTCCATTCC |
| Fn109 | GTAGAAGGCCTGCAGATGCT | GTCTTTGCTTCTCCTCACCG |
| Fn110 | GCACCAGCAAGAGGAGACTT | TTGTTGCTTGCCAGTGAAAC |
| Fn111 | AAGCGAGTAGCTGAAAGGCA | CACTACCACCTCCGATTGCT |
| Fn112 | CCAAGCTTCGACCATTGTTT | TTGTACATCGGTGAAACGGA |
| Fn113 | AGCAGAAAGGCGTTCCACTA | GTTGAGCCCTTCCTGCATAA |
| Fn114 | CGCCCTCTTCCTTTATCTCC | CACTTAGGTTTACCGCTGCC |
| Fn115 | CACGTTTGGTGACATTCCAG | ACAATTATCTCCTTCCGCCC |
| Fn116 | TCCTCTCCTCACCAAACCAC | CACAACCCTTCAACATCACG |
| Fn117 | GTGGTTGGTGGGGATCATAG | GCCCAAAGTTCTTCCAAACA |
| Fn118 | CAATCATATTAATCATCCCCAAA | GAGTTGGTCTGCAGGAGGAG |
| Fn119 | TAAGATGCATGAGGCACGAC | GACTTTGATGCGCACTTTCA |
| Fn120 | TGATTTGTGCACCAAAAGGA | GGAGAGTCGACCGATTCAGA |
| Fn121 | AACTCAGCAACCGGAATCAC | ATACGTAGACGCATCGGAGG |
| Fn122 | GCACACAATAAGCCTCCCAT | CACATGCTACAAAACAGGCG |
| Fn123 | CCACTGGACATTTCTTGGCT | TTTACAAGCCCCAACAGAGC |
| Fn124 | CTGGAACCGGAGTAGGTGAA | AACAGCCTTTACCCTTCATCA |
| Fn125 | ATCTGCCTGTAGCCGAACAG | CTCTTAGTTGGCGCTGCTCT |
| Fn126 | ATAATTTGCCCAACGCTCTG | CTCTCTTGAAAAACACGGGC |
| Fn127 | CAACACAAACACCAAAACCCT | CGTGCCTCATTGGGTTCTAT |
| Fn128 | GTTGTACTTGGTGCGGAGGT | CAAAATTCCAAAAGCCCAAA |
| Fn129 | TCTTTCGATTTGGATTTGGG | TAGAGCTCTCGGCCTCTTCA |
| Fn130 | TTGGTGCCATTGTTGTTGTT | AACGCCTTTCTTCCCAATTT |
| Fn131 | CACCGCGATTCTTCTCTCTC | CGACATCGTCTCTCTCCCTC |
| Fn132 | TTCTTTTTCTGTTCCGCCC | CCGCCGTTAGAGACAAACTC |
| Fn133 | ATTGAAACCCCACCACTGAA | AACGGCCAGAAAAAGCTGTA |
| Fn134 | TGGTTGGGGGAGAATTGTAA | TGAGTTTGCCACAACTGACA |
| Fn135 | TCCCGATAACAAGAGAGAGACT | TGGAGATGAACAAGGGAAGG |
| Fn136 | AGTCCTCAGTTCTCCTGGCA | CAGAATGGTGATGCTGATGG |
| Fn137 | CCAGGACATACATACGTTCTCAA | GACGGACTTAGCCCCCTTAT |
| Fn138 | TAGCTGCGGTTGATTCAGTG | ATTATCAGCGGAAGGCACAC |
| Fn139 | TGCACCACCAAAAACACCTA | GAGGAGGGGTTGAGTGATGA |
| Fn140 | TCACCACCCCTAATTACCGA | AGATAAACCGGGGAGCTTGT |
2.2. Marker Informative Analysis of Accessions
When the 140 developed SSR markers were used for genotyping the set of 30 accessions, 23 markers had no clear bands or were amplified only in very few accessions. These markers were excluded from further analyses, reducing the number of good quality markers to 117 (83.57%). Among those good quality markers, 54 were monomorphic and 63 (53.85%) were polymorphic on the 30 accessions. The successful amplification rate and polymorphism rate were similar to those of our previous work (81.25% and 59.57%, respectively) [27], though the set of accessions previously genotyped with EST-SSRs was not identical to the current set of accessions.
The number of loci per SSR ranged from two to five, with a mean value of 2.71, which was similar to that of previous studies on celery using EST-SSRs (2.68) [27], but lower than the number of ISSR markers reported by Qing-Kuo (5.05) [9]. This indicated that primer sequences designed from SSR flanking regions were highly conserved and SSR markers were more specific than ISSR markers. Polymorphism information content (PIC) values ranged from 0.06 to 0.67, with an average of 0.33. The largest group of markers (27.42%) was in a range from 0.4 to 0.5, followed by the group with PIC values ranging from 0 to 0.1 (Figure 1). The second largest group was made up of markers polymorphic between the wild and cultivated species, but monomorphic within cultivated species.
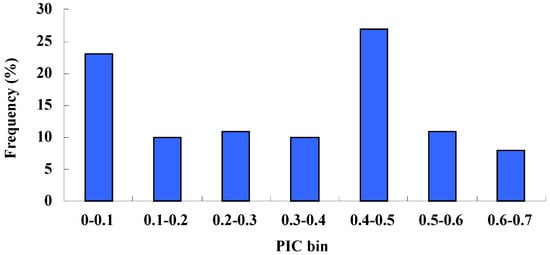
Figure 1.
Distribution of polymorphism information content (PIC) values of the SSR markers used for genotyping the 30 studied varieties.
The number of loci per SSR and the PIC values in our study were low. Generally, it was believed that these indexes for genomic-derived SSRs were significantly higher than EST-SSRs, as indicated by the reports on flax [28], wheat [29], levant cotton [30], sunflower [31] and sugar beet [32]. The lower polymorphism of EST-SSR markers than genomic SSRs was likely due to the conserved nature of genome coding regions [33]. However, it has been reported in some other studies on sorghum [34] and apple [35] that EST-SSR markers have greater discriminating power than the genomic SSRs. The higher average number of alleles per EST-SSR marker reported may be primarily attributed to the difference of species used or the selection of multiple-locus SSRs or compound SSRs, since we usually believe that single-locus SSRs provided less polymorphism. In addition, genotypes may also influence the number of alleles detected at each SSR locus.
Based on the polymorphic marker data, we made an analysis of the observed heterozygosity (Ho) and expected heterozygosity (He). The former varied from 0 to 0.73 (mean 0.13), while the latter varied from 0.07 to 0.68 (mean 0.33). The mean Ho and He values were similar to the previous results of 0.14 and 0.36, respectively [27]. The distribution of He values showed that 85.48% of the markers were in the range from 0 to 0.3, which was a very low heterozygosity (Figure 2). The fact that observed heterozygosity was lower than expected may be due to the small sample size or the results of inbreeding.
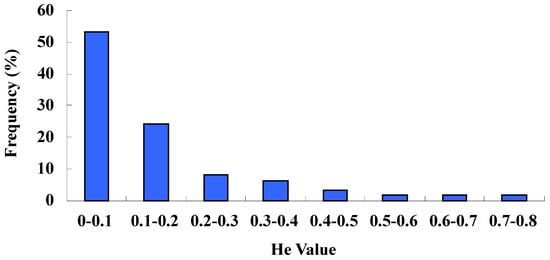
Figure 2.
Distribution of the estimate of genetic heterozygosity (He).
2.3. Analysis of Molecular Variance (AMOVA)
AMOVA analysis indicated that approximately 35% of the genetic diversity was within individuals, 43% among individuals, and the remaining 22% among horticultural types (Table 3). This was consistent with the findings from other organisms like faba bean [36], grape [37], Haematococcus pluvialis [38], olive [39], apple [35] and lettuce [22] showing that considerable genetic diversity was partitioned within, rather than among populations. On the contrary, low levels of genetic diversity within populations and significant genetic differentiation among populations were detected in Omphalogramma souliei, barely and Chinese-grown pecan [40,41,42].
Table 3.
Analysis of molecular variance (AMOVA).
| Source of variation | Percentage of variation | p-value |
|---|---|---|
| Among horticultural types | 22% | 0.001 |
| Among Individuals | 43% | 0.001 |
| Within Individuals | 35% |
We also calculated pairwise differentiation (Fst) for all pairs of horticultural types with at least two accessions per type (Table 4). The variation of the Fst values ranged from 0.086 to 0.261. Obviously, differentiation between celery and the other two types were significantly higher than that between local celery and the middle type.
Table 4.
Pairwise differentiation (Fst) among horticultural types.
| Horticultural type | Local celery | Celery | Middle type |
|---|---|---|---|
| Local celery | - | ||
| Celery | 0.231 | - | |
| Middle type | 0.086 | 0.261 | - |
2.4. PCA Analysis
The PCA results revealed that accessions of the same horticultural types clustered together. Accessions of local celery were well separated from those of celery. The middle type celery accessions were scattered among celery accessions and were closer to celery cluster than local celery cluster (Figure 3).
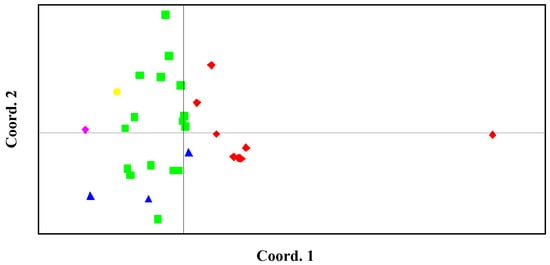
Figure 3.
Principal component analysis (PCA) of the 30 accessions genotyped with 62 polymorphic SSRs. Red, local celery; Green, celery; Blue, the middle type; Yellow, celeriac; Pink, the wild type.
PCA analysis unambiguously separated the wild and the var. rapaceum (celeriac species) from var. dulce (cultivated celery). Compared to the wild species, var. rapaceum was closer to var. dulce, which may be due to the fact that the studied var. rapaceum belonged to cultivars. The observed distances between wild species, celeriac species and var. dulce accessions corresponded to the sexual compatibility of the two species with var. dulce accessions, which can be further proved by the fact that marker transfer rate was 100% in var. rapaceum, but only 54.84% in wild type. What’s more, both celeriac and wild species were the closest to the cluster of celery accessions, but the most distant from the cluster of local celery accessions. This result was supported by the differences in their origins.
2.5. Genetic Distance Analysis
The average Nei genetic distance for the 29 accessions of var. dulce and var. rapaceum was 0.34, with a range from 0.26 (C123) to 0.72 (C1). The largest genetic distance (0.72) was between C1 and C83, while the least genetic distance of 0.02 was found between C29 and C97. The average genetic distance of wild species was 2.42 with a range from 2.06 to 3.22, which was much larger than cultivated accessions (less than 0.7). Overall, seventy percent of the genetic distance between any two cultivars was no more than 0.4 and only thirty percent were larger than 0.4 (Figure 4). These results suggested that genetic variation within Apium graveolens was limited, while the wild species had wider genetic diversity and could serve as a valuable resource.
2.6. Cluster and Population Structure Analysis
In order to see the relationship of the materials used in this study, a dendrogram was constructed from the pairwise distance matrices (Figure 5). UPGMA cluster analysis indicated that at the genetic distance of 0.38, cultivated and wild species were separated. The statistical analysis based on the allele frequencies separated most of the cultivars, both in the trees (Figure 5) and in the PCA (Figure 3), into two main clusters corresponding to the geographical areas where they originated. At the distance of 0.72, most local celery accessions formed a cluster and all celery formed a large cluster with three local varieties scattered in. What’s more, the three middle type accessions (C58, C99, and C159) were well clustered together.
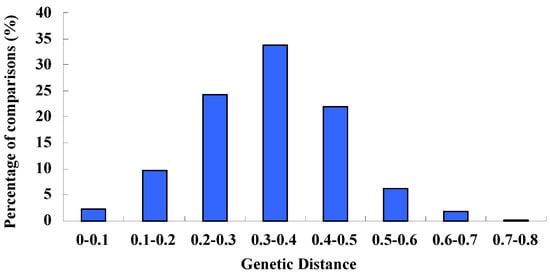
Figure 4.
Distribution of genetic distance values obtained from pairwise comparisons of 29 cultivars using SSR marker data.
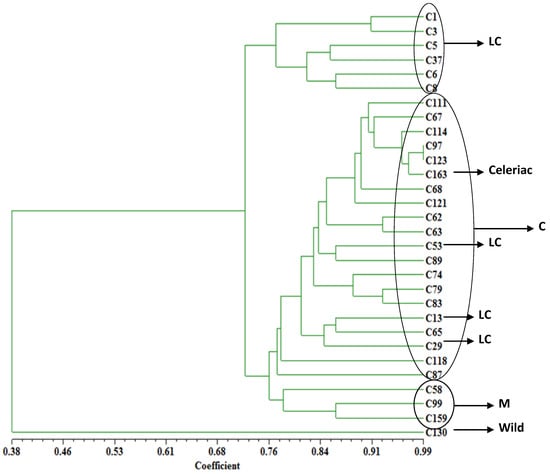
Figure 5.
Dendrogram of 30 accessions based on SSR marker data generated from Nei’s genetic distance matrix by UPGMA in NTSYSpc 2.11a. LC: local celery; C: celery; M: middle type.
We also estimated the number of genetic clusters of 30 accessions using Structure software without specifying prior information concerning sample class and allowing for admixed individuals. In order to choose an appropriate value of K for modeling the data, we ran a series of independent runs of the data at a range of values of K from 1 to 7.
When K ranged from 2 to 7, the wild species were separated from the cultivated species and when K was larger than 3, the wild species stood alone. When K = 3, three populations were obtained (Figure 6).
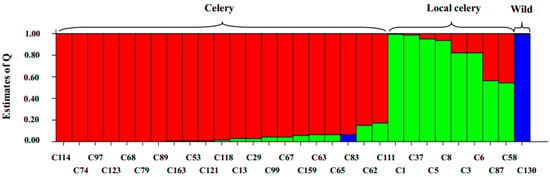
Figure 6.
Bar plot of population structure estimates for 30 Apium varieties by SSR markers. Each accession is represented by a single vertical bar broken into three colored segments, with lengths proportional to Q of the three inferred populations (K = 3). The sum of Q values for each bar is 1. Classes of the materials are shown at the top.
The first population mainly comprised local celery accessions. The second population contained all celery accessions, the celeriac accessions and three local celery varieties. The third population only included the wild species. This structure was identical with the obtained dendrogram and supported the accuracy of the clustering.
2.7. Genotypic Diversity
Genotypic diversity is defined as the probability that two individuals taken at random have different genotypes. This value is 0 if every individual is the same, and 1 if every individual is different. We used the Multilocus program to calculate the number of different genotypes and the genotypic diversity on the set of 30 accessions. On average five markers were needed to identify 50% of genotypes, 14 markers to identify 90% of genotypes, and 29 markers to identify 99% of genotypes (Figure 7). Our analysis showed that any combinations of 55 SSR markers were able to distinguish genotypes of all 30 accessions unambiguously. This was a relatively high number of markers that were needed for genotyping.
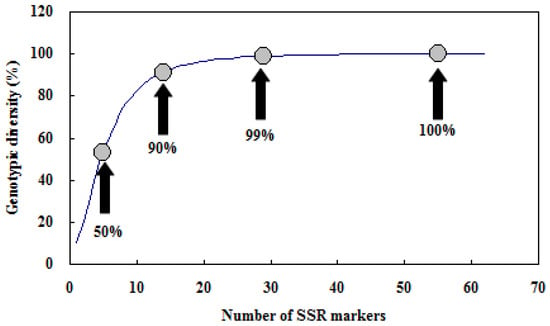
Figure 7.
Effect of the increasing number of SSR markers on the estimate of genotyping diversity. Circles indicate genotypic diversity of 50%, 90%, 99%, and 100%, respectively. The value of 100% was reached with 55 and more markers.
For example, 32 SSR markers were sufficient to distinguish genotypes of all 36 lettuce accessions [22], only 17 SSR markers on average were required to identify 54 sugar beet hybrid varieties [43] and eight SSR markers were enough to distinguish 35 asparagus varieties [44]. In general, genetic similarity among accessions of the same type is high. So it is more difficult to distinguish closely related or less diverse materials. In this study, a total of 50 unique genotypes specific to some accessions were identified by different markers. The number of unique genotype indentified by one primer ranged from 1 to 4. Of these 50 unique genotypes, 23 (46%) exclusively presented in wild species, suggesting the low diversity of the materials used. Therefore more molecular markers were needed to distinguish these closely related materials with high genetic similarity. In addition, the polymorphism of the markers was another important factor affecting whether we can distinguish more genotypes or not. So it is a must to develop higher polymorphic markers to distinguish accessions more efficiently.
3. Experimental
3.1. Plant Materials and DNA Isolation
A set of 30 accessions (Table 1) was used to test polymorphism of the developed SSR markers. This set comprised 28 common cultivars, one celeriac and one wild species. All materials were grown at the experimental station of China Agricultural University (Beijing, China). Genomic DNA was extracted from celery tender leaves using a modified version of the cetyltrimethylammonium bromide (CTAB) method [45]. Quality of DNA was checked by electrophoresis in 1% agarose gel. The genomic DNA was diluted 10-fold for PCR analysis.
3.2. Development of Genic SSR Markers and Genotyping with Markers
The SSR markers were developed through celery transcripotme sequencing [27]. Primers were designed using Primer 3 [46] with default parameters and synthesized at Sangon Biotech Co., Ltd. (Shanghai, China). PCR amplifications were conducted in a final volume of 10 μL containing 3.5 μL 2× Taq PCR MasterMix (Beijing Biomed Co., Ltd., Beijing, China), 4.5 μL double distilled (dd) H2O, 0.5 μL of each primer (5 μM) and 1 μL of template (aprox. 20 ng/μL). PCR was performed as follows: denaturation at 94 °C for 5 min, followed by 38 cycles of 30 s at 94 °C, 30 s at Tm (annealing temperature), 1 min at 72 °C and a final step at 72 °C for 10 min. PCR products were firstly detected by agarose gel electrophoresis and the products possessing single band or only a few bands were subjected to 7% polyacrylamide gel to separate alleles. With regard to those had no bands or multiple bands, we optimized the PCR condition to get better products for separation of alleles. PCR products were mixed with a volume of loading buffer and then denatured at 95 °C for 10min before being loaded on the polyacrylamide gel.
3.3. Analysis of Marker Polymorphism and Genetic Heterozygosity
SSR alleles were scored manually starting from the smallest to the largest-sized bands. The presence or absence of each single fragment was coded as 1 or 0, respectively, and scored for a binary data matrix. Scored data from polymorphic loci were used to calculate the polymorphism information content (PIC) according to Equation (1):
where pi is the frequency of ith allele for each locus [47]. Observed heterozygosity (Ho) and expected heterozygosity (He) were calculated using the Popgene software version 1.31 [48]. Ho represents the estimated proportion of observed heterozygotes at a given locus for co-dominant markers. He, estimated using the Levene algorithm [49], represents the estimated proportion of expected heterozygotes under random mating for co-dominant markers.
[PIC = 1 − ∑pi2]
3.4. AMOVA and PCA Analysis
Analysis of molecular variance (AMOVA) [50] between all the pairs of horticultural types with at least two accessions, and principal components analysis (PCA) of all accessions were performed using GenAlEx 6.5 [51].
3.5. Genetic Diversity and Population Structure Analysis
A genetic similarity matrix was constructed and Nei’s genetic distance [52] was calculated for each pair of all accessions using the NTSYSpc 2.1 software [53]. Unweighted pair group method with arithmetic mean (UPGMA) cluster analysis was performed to develop a dendrogram. Population structure was analyzed using the free software package STRUCTURE 2.3.4 [54,55,56]. A model without prior population information was used to assign individuals to populations.
3.6. Identification of Genotypes
In order to see whether scoring more loci is likely to increase the genotypic diversity, or whether one has reached a plateau, we used the software MultiLocus ver. 1.3b [57] to estimate the number of different genotypes that can be identified in a set of 30 accessions with a gradually increasing number of markers. The program randomly sampled from 1 to m−1 loci from the dataset and calculated the number of different genotypes identified.
4. Conclusions
This was the first attempts at celery genetic and genotypic diversity analysis using SSR markers developed from transcriptome sequencing. The AMOVA analysis indicated that the largest part of genetic diversity was within populations, while genetic diversity found among populations was low. The geneetic distance of wild species was much larger than that of cultivated accessions, suggesting the wider genetic diversity of the wild species, while the diversity within cultivars was quite limited. PCA analysis revealed that accessions of the same horticultural types were well clustered together. The UPGMA dendrogram and population structure clearly separated wild species from cultivars, and further divided the cultivars into two clusters, corresponding to the geographical areas from where they originated. Genotypic diversity analysis suggested that 29 markers were needed to identify 99% of genotypes and any combinations of 55 SSR markers were able to distinguish genotypes of all 30 accessions. Given that the genetic similarity of commonly used accessions was high, we need to develop more and higher polymorphic markers to efficiently distinguish closely related varieties. This study would provide a common ground for celery accessions identification, breeding and protection of breeders’ rights.
Acknowledgments
This study was funded by Chinese Universities Scientific Fund (No. 2013QJ094). The authors thank all our lab mates for the field sampling and DNA extraction.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arus, P.; Ortan, T. Inheritance patterns and linkage relationships of eight genes of celery. J. Hered. 1984, 75, 11–14. [Google Scholar]
- Huestis, G.; McGrath, J.M.; Quiros, C.F. Development of genetic markers in celery based on restriction fragment length polymorphisms. Theor. Appl. Genet. 1993, 85, 889–896. [Google Scholar]
- Yang, X.; Quiros, C. Identification and classification of celery cultivars with RAPD markers. Theor. Appl. Genet. 1993, 86, 205–212. [Google Scholar]
- Domblides, A.; Domblides, H.; Kharchenko, V. Discrimination between celery cultivars with the use of RAPD markers. Proc. Latv. Acad. Sci. B 2008, 62, 219–222. [Google Scholar]
- Li, G.; Quiros, C.F. Use of amplified fragment length polymorphism markers for celery cultivar identification. HortScience 2000, 35, 726–728. [Google Scholar]
- Ju, J. An analysis of celery genetic diversity by AFLP. Chin. Agric. Sci. Bull. 2007, 23, 120–123. [Google Scholar]
- Wang, S.; Yang, W.; Shen, H. Genetic diversity in Apium graveolens and related species revealed by SRAP and SSR markers. Sci. Hortic. 2011, 129, 1–8. [Google Scholar] [CrossRef]
- Ince, A.; Karaca, M.; Turgut, K. Development of new set of EST-SSR primer pairs for celery (Apium graveolens L.). Planta Med. 2010, 76, P036. [Google Scholar]
- Qing-kuo, L.; Ou-jing, L.; Ji-bo, Z.; Xin, Z.; Zhu, Z.; Rui, C.; Shi-yong, X.; Yong, W.; Yong-ze, G. Construction of SSR-based molecular fingerprinting and analysis of genetic diversity for celery varieties from Tianjin. Tianjin Agric. Sci. 2012, 18, 7–11. [Google Scholar]
- Yu, M.; Chu, J.; Ma, R.; Shen, Z.; Fang, J. A novel strategy for the identification of 73 Prunus domestica cultivars using random amplified polymorphic DNA (RAPD) markers. Afr. J. Agric. Res. 2013, 8, 243–250. [Google Scholar]
- Zhang, J.; Shu, Q.; Liu, Z.; Ren, H.; Wang, L.; de Keyser, E. Two EST-derived marker systems for cultivar identification in tree peony. Plant Cell Rep. 2012, 31, 299–310. [Google Scholar] [CrossRef]
- Figueiredo, E.; Canhoto, J.; Ribeiro, M. Fingerprinting and genetic diversity of Olea europaea L. ssp. Europaea accessions from the cultivar Galega using RAPD markers. Sci. Hortic. 2013, 156, 24–28. [Google Scholar] [CrossRef]
- Hameed, U.; Pan, Y.; Muhammad, K.; Afghan, S.; Iqbal, J. Use of simple sequence repeat markers for DNA fingerprinting and diversity analysis of sugarcane (Saccharum spp) cultivars resistant and susceptible to red rot. Genet. Mol. Res. 2012, 11, 1195–1204. [Google Scholar] [CrossRef]
- Baldwin, S.; Pither-Joyce, M.; Wright, K.; Chen, L.; McCallum, J. Development of robust genomic simple sequence repeat markers for estimation of genetic diversity within and among bulb onion (Allium cepa L.) populations. Mol. Breed. 2012, 30, 1401–1411. [Google Scholar] [CrossRef]
- Park, Y.-J.; Lee, J.K.; Kim, N.-S. Simple sequence repeat polymorphisms (SSRPs) for evaluation of molecular diversity and germplasm classification of minor crops. Molecules 2009, 14, 4546–4569. [Google Scholar] [CrossRef]
- Dos Santos, J.M.; Duarte Filho, L.S.C.; Soriano, M.L.; da Silva, P.P.; Nascimento, V.X.; de Souza Barbosa, G.V.; Todaro, A.R.; Ramalho Neto, C.E.; Almeida, C. Genetic diversity of the main progenitors of sugarcane from the RIDESA germplasm bank using SSR markers. Ind. Crops Prod. 2012, 40, 145–150. [Google Scholar] [CrossRef]
- Zhang, W.W.; Pan, J.S.; He, H.L.; Zhang, C.; Li, Z.; Zhao, J.L.; Yuan, X.J.; Zhu, L.H.; Huang, S.W.; Cai, R. Construction of a high density integrated genetic map for cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2012, 124, 249–259. [Google Scholar] [CrossRef]
- Schouten, H.J.; van de Weg, W.E.; Carling, J.; Khan, S.A.; McKay, S.J.; van Kaauwen, M.P.; Wittenberg, A.H.; Koehorst-van Putten, H.J.; Noordijk, Y.; Gao, Z. Diversity arrays technology (DArT) markers in apple for genetic linkage maps. Mol. Breed. 2012, 29, 645–660. [Google Scholar] [CrossRef]
- Qin, H.; Feng, S.; Chen, C.; Guo, Y.; Knapp, S.; Culbreath, A.; He, G.; Wang, M.L.; Zhang, X.; Holbrook, C.C. An integrated genetic linkage map of cultivated peanut (Arachis hypogaea L.) constructed from two RIL populations. Theor. Appl. Genet. 2012, 124, 653–664. [Google Scholar] [CrossRef]
- Moriguchi, Y.; Ujino-Ihara, T.; Uchiyama, K.; Futamura, N.; Saito, M.; Ueno, S.; Matsumoto, A.; Tani, N.; Taira, H.; Shinohara, K. The construction of a high-density linkage map for identifying SNP markers that are tightly linked to a nuclear-recessive major gene for male sterility in Cryptomeria japonica D. Don. BMC Genomics 2012, 13, 95. [Google Scholar] [CrossRef]
- Sinha, P.; Tomar, S.; Singh, V.K.; Balyan, H. Genetic analysis and molecular mapping of a new fertility restorer gene Rf8 for Triticum timopheevi cytoplasm in wheat (Triticum aestivum L.) using SSR markers. Genetica 2013, 141, 431–441. [Google Scholar]
- Rauscher, G.; Simko, I. Development of genomic SSR markers for fingerprinting lettuce (Lactuca sativa L.) cultivars and mapping genes. BMC Plant Biol. 2013, 13, 11. [Google Scholar] [CrossRef]
- Raman, H.; Raman, R.; Eckermann, P.; Coombes, N.; Manoli, S.; Zou, X.; Edwards, D.; Meng, J.; Prangnell, R.; Stiller, J. Genetic and physical mapping of flowering time loci in canola (Brassica napus L.). Theor. Appl. Genet. 2013, 126, 119–132. [Google Scholar] [CrossRef]
- Chung, J.-W.; Kim, T.-S.; Suresh, S.; Lee, S.-Y.; Cho, G.-T. Development of 65 Novel Polymorphic cDNA-SSR markers in Common Vetch (Vicia sativa subsp. sativa) using next generation sequencing. Molecules 2013, 18, 8376–8392. [Google Scholar] [CrossRef]
- Mudalkar, S.; Golla, R.; Ghatty, S.; Reddy, A.R. De novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. using Illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol. Biol. 2014, 84, 159–171. [Google Scholar] [CrossRef]
- Suresh, S.; Park, J.-H.; Cho, G.-T.; Lee, H.-S.; Baek, H.-J.; Lee, S.-Y.; Chung, J.-W. Development and molecular characterization of 55 novel polymorphic cDNA-SSR markers in faba bean (Vicia faba L.) using 454 pyrosequencing. Molecules 2013, 18, 1844–1856. [Google Scholar] [CrossRef]
- Fu, N.; Wang, Q.; Shen, H.-L. De novo assembly, gene annotation and marker development using illumina paired-end transcriptome sequences in celery (Apium graveolens L.). PLoS One 2013, 8, e57686. [Google Scholar]
- Cloutier, S.; Miranda, E.; Ward, K.; Radovanovic, N.; Reimer, E.; Walichnowski, A.; Datla, R.; Rowland, G.; Duguid, S.; Ragupathy, R. Simple sequence repeat marker development from bacterial artificial chromosome end sequences and expressed sequence tags of flax (Linum usitatissimum L.). Theor. Appl. Genet. 2012, 125, 685–694. [Google Scholar] [CrossRef]
- Eujayl, I.; Sorrells, M.; Baum, M.; Wolters, P.; Powell, W. Isolation of EST-derived microsatellite markers for genotyping the A and B genomes of wheat. Theor. Appl. Genet. 2002, 104, 399–407. [Google Scholar]
- Jena, S.N.; Srivastava, A.; Rai, K.M.; Ranjan, A.; Singh, S.K.; Nisar, T.; Srivastava, M.; Bag, S.K.; Mantri, S.; Asif, M.H. Development and characterization of genomic and expressed SSRs for levant cotton (Gossypium herbaceum L.). Theor. Appl. Genet. 2012, 124, 565–576. [Google Scholar]
- Pashley, C.H.; Ellis, J.R.; McCauley, D.E.; Burke, J.M. EST databases as a source for molecular markers: Lessons from Helianthus. J. Hered. 2006, 97, 381–388. [Google Scholar]
- Laurent, V.; Devaux, P.; Thiel, T.; Viard, F.; Mielordt, S.; Touzet, P.; Quillet, M. Comparative effectiveness of sugar beet microsatellite markers isolated from genomic libraries and GenBank ESTs to map the sugar beet genome. Theor. Appl. Genet. 2007, 115, 793–805. [Google Scholar] [CrossRef]
- Liewlaksaneeyanawin, C.; Ritland, C.E.; El-Kassaby, Y.A.; Ritland, K. Single-copy, species-transferable microsatellite markers developed from loblolly pine ESTs. Theor. Appl. Genet. 2004, 109, 361–369. [Google Scholar]
- Ramu, P.; Billot, C.; Rami, J.; Senthilvel, S.; Upadhyaya, H.; Reddy, L.A.; Hash, C. Assessment of genetic diversity in the sorghum reference set using EST-SSR markers. Theor. Appl. Genet. 2013, 126, 2051–2064. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Zhao, Y.; Korban, S.S.; Han, Y. Evaluation of genetic diversity in Chinese wild apple species along with apple cultivars using SSR markers. Plant Mol. Biol. Rep. 2012, 30, 539–546. [Google Scholar] [CrossRef]
- Wang, H.F.; Zong, X.X.; Guan, J.P.; Yang, T.; Sun, X.L.; Ma, Y.; Redden, R. Genetic diversity and relationship of global faba bean (Vicia faba L.) germplasm revealed by ISSR markers. Theor. Appl. Genet. 2012, 124, 789–797. [Google Scholar] [CrossRef]
- De Andrés, M.; Benito, A.; Pérez-Rivera, G.; Ocete, R.; Lopez, M.; Gaforio, L.; Muñoz, G.; Cabello, F.; Martínez Zapater, J.M.; Arroyo-García, R. Genetic diversity of wild grapevine populations in Spain and their genetic relationships with cultivated grapevines. Mol. Ecol. 2012, 21, 800–816. [Google Scholar] [CrossRef]
- Mostafa, N.; Omar, H.; Tan, S.G.; Napis, S. Studies on the genetic variation of the green unicellular alga Haematococcus pluvialis (Chlorophyceae) obtained from different geographical locations using ISSR and RAPD molecular marker. Molecules 2011, 16, 2599–2608. [Google Scholar] [CrossRef]
- Belaj, A.; Muñoz-Diez, C.; Baldoni, L.; Porceddu, A.; Barranco, D.; Satovic, Z. Genetic diversity and population structure of wild olives from the north-western Mediterranean assessed by SSR markers. Ann. Bot. 2007, 100, 449–458. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, C.Q.; Li, D.Z. Low genetic diversity and high genetic differentiation in the critically endangered Omphalogramma souliei (Primulaceae): Implications for its conservation. J. Syst. Evol. 2009, 47, 103–109. [Google Scholar] [CrossRef]
- Ozkan, H.; Kafkas, S.; Ozer, M.S.; Brandolini, A. Genetic relationships among South-East Turkey wild barley populations and sampling strategies of Hordeum spontaneum. Theor. Appl. Genet. 2005, 112, 12–20. [Google Scholar] [CrossRef]
- Jia, X.-D.; Wang, T.; Zhai, M.; Li, Y.-R.; Guo, Z.-R. Genetic diversity and identification of chinese-grown pecan using ISSR and SSR markers. Molecules 2011, 16, 10078–10092. [Google Scholar] [CrossRef]
- Simko, I.; Eujayl, I.; van Hintum, T.J. Empirical evaluation of DArT, SNP, and SSR marker-systems for genotyping, clustering, and assigning sugar beet hybrid varieties into populations. Plant Sci. 2012, 184, 54–62. [Google Scholar] [CrossRef]
- Caruso, M.; Federici, C.T.; Roose, M.L. EST–SSR markers for asparagus genetic diversity evaluation and cultivar identification. Mol. Breed. 2008, 21, 195–204. [Google Scholar]
- Kabelka, E.; Franchino, B.; Francis, D. Two loci from Lycopersicon hirsutum LA407 confer resistance to strains of Clavibacter michiganensis subsp. michiganensis. Phytopathology 2002, 92, 504–510. [Google Scholar] [CrossRef]
- Primer3 (version 0.4.0). Available online: http://bioinfo.ut.ee/primer3-0.4.0/primer3/ (accessed on 10 February 2014).
- Weir, B.S. Genetic Data Analysis. In Methods for Discrete Population Genetic Data; Sinauer Associates: Sunderland, MA, USA, 1990. [Google Scholar]
- Yeh, F.; Yang, R.-C.; Boyle, T. PopGene Version 131: Microsoft Window-Based Freeware for Population Genetic Analysis. Available online: http://www.ualberta.ca/~fyeh/popgene.pdf (accessed on 7 February 2014).
- Levene, H. On a matching problem arising in genetics. Ann. Math. Stat. 1949, 20, 91–94. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar]
- Rohlf, F.J. Ntsys-Pc Numerical Taxonomy and Multivariate Analysis System Version 2.1, Exeter Software; Setauket: New York, NY, USA, 2000. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Agapow, P.M.; Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 2001, 1, 101–102. [Google Scholar] [CrossRef]
- Sample Availability: All samples are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).