Easy Access to a Cyclic Key Intermediate for the Synthesis of Trisporic Acids and Related Compounds


Abstract
:1. Introduction
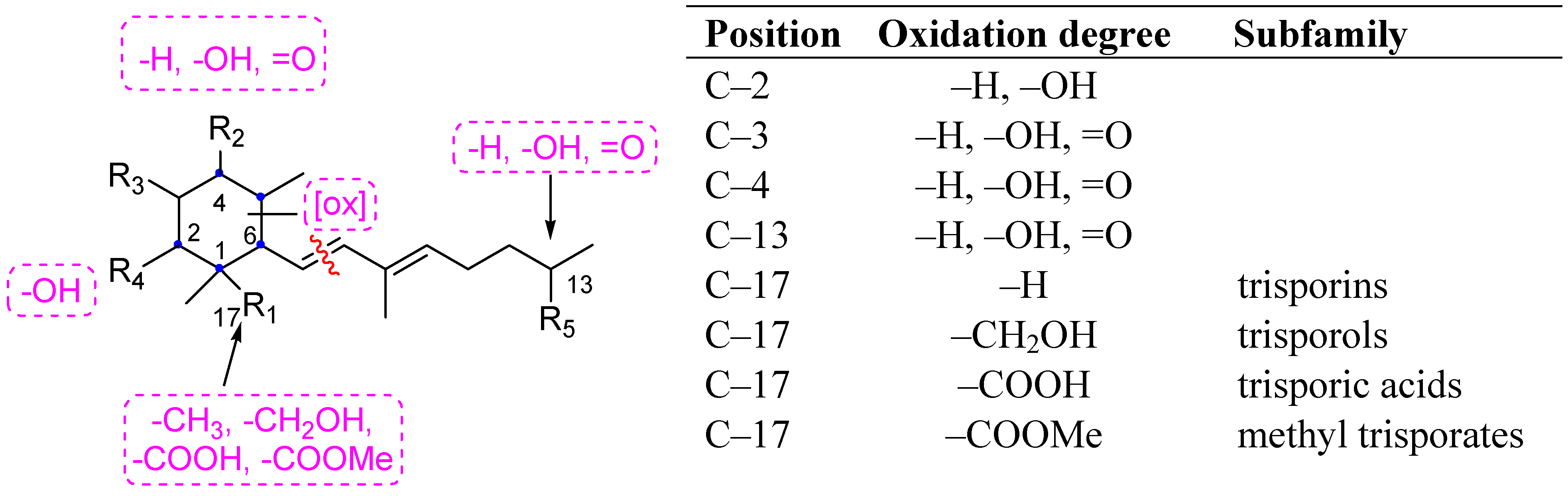
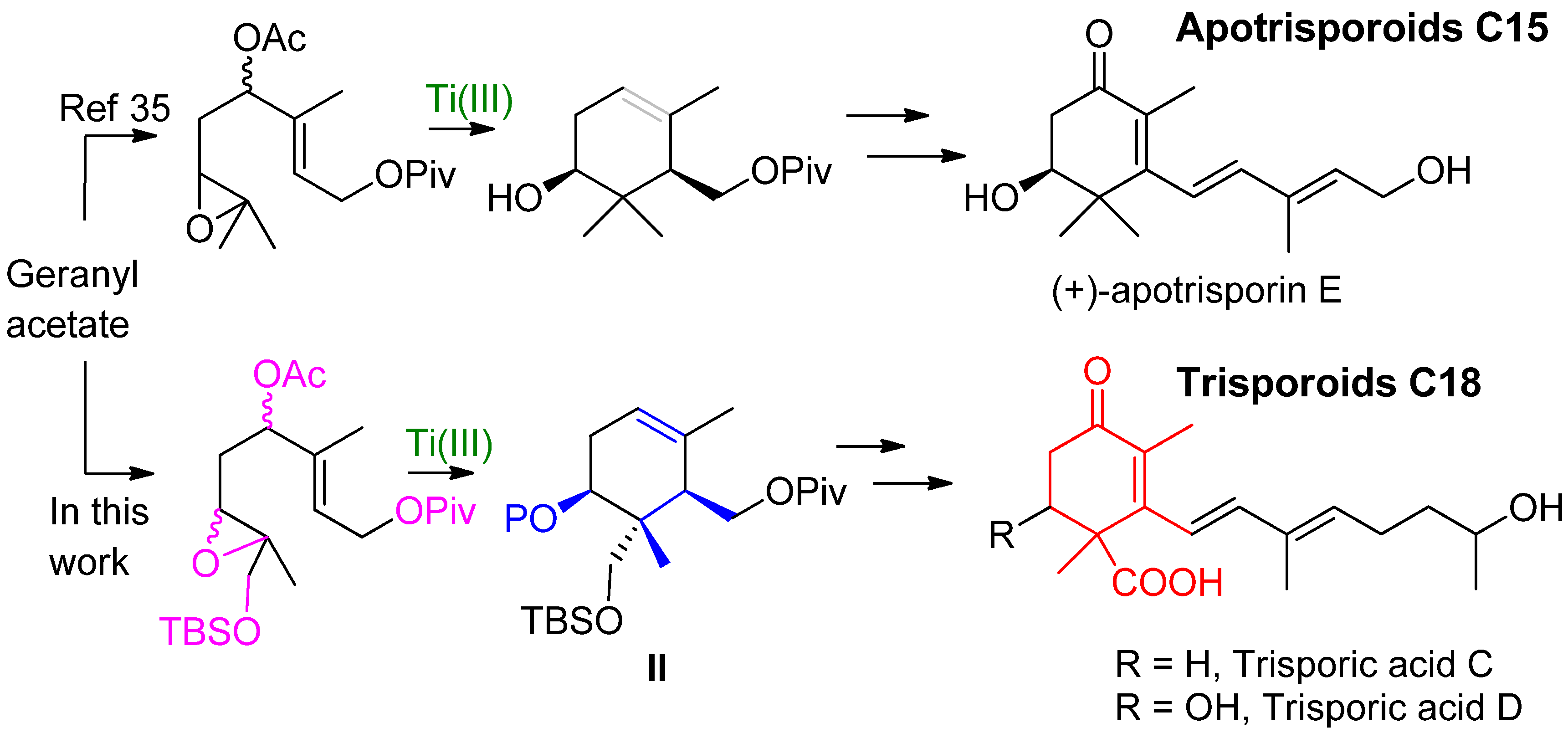
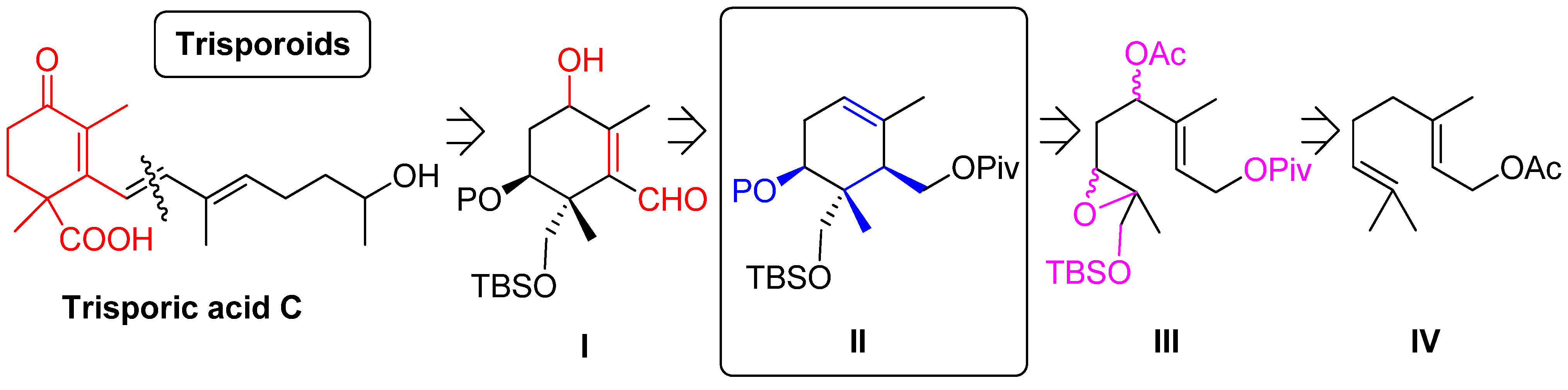
2. Results and Discussion
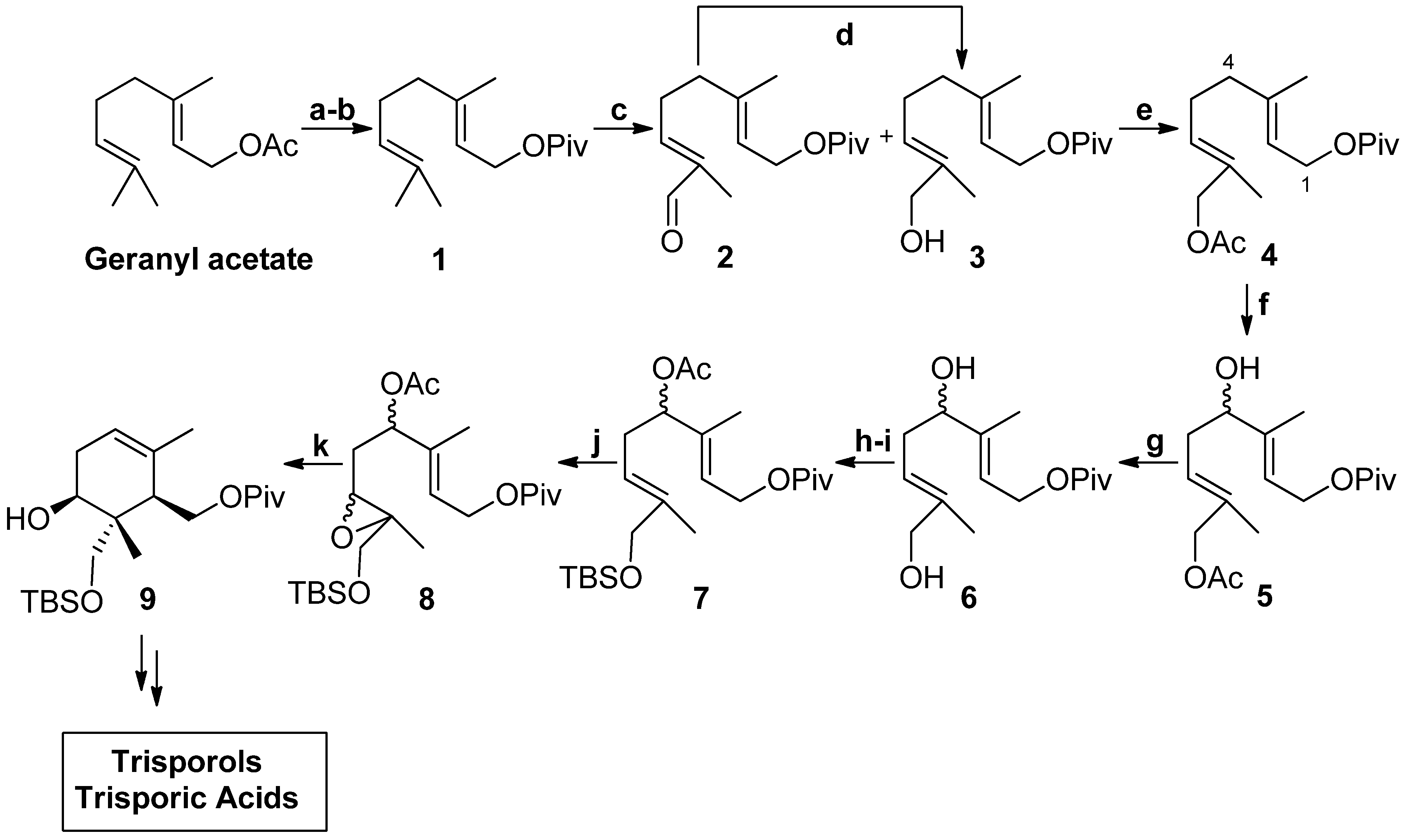

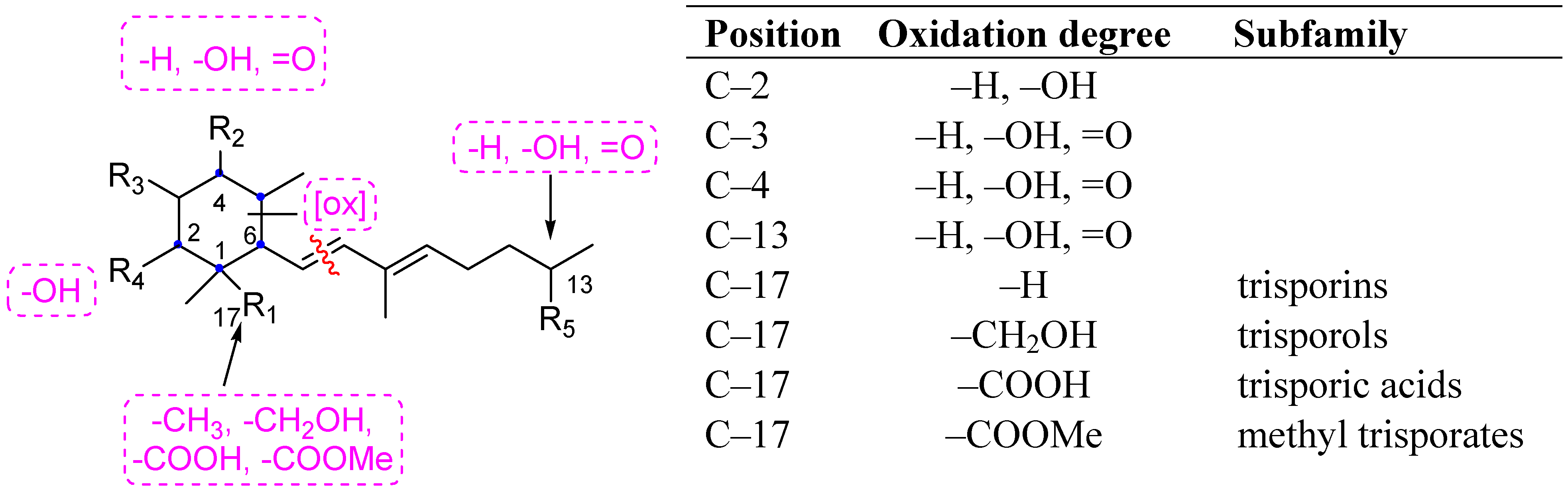{kind=link}
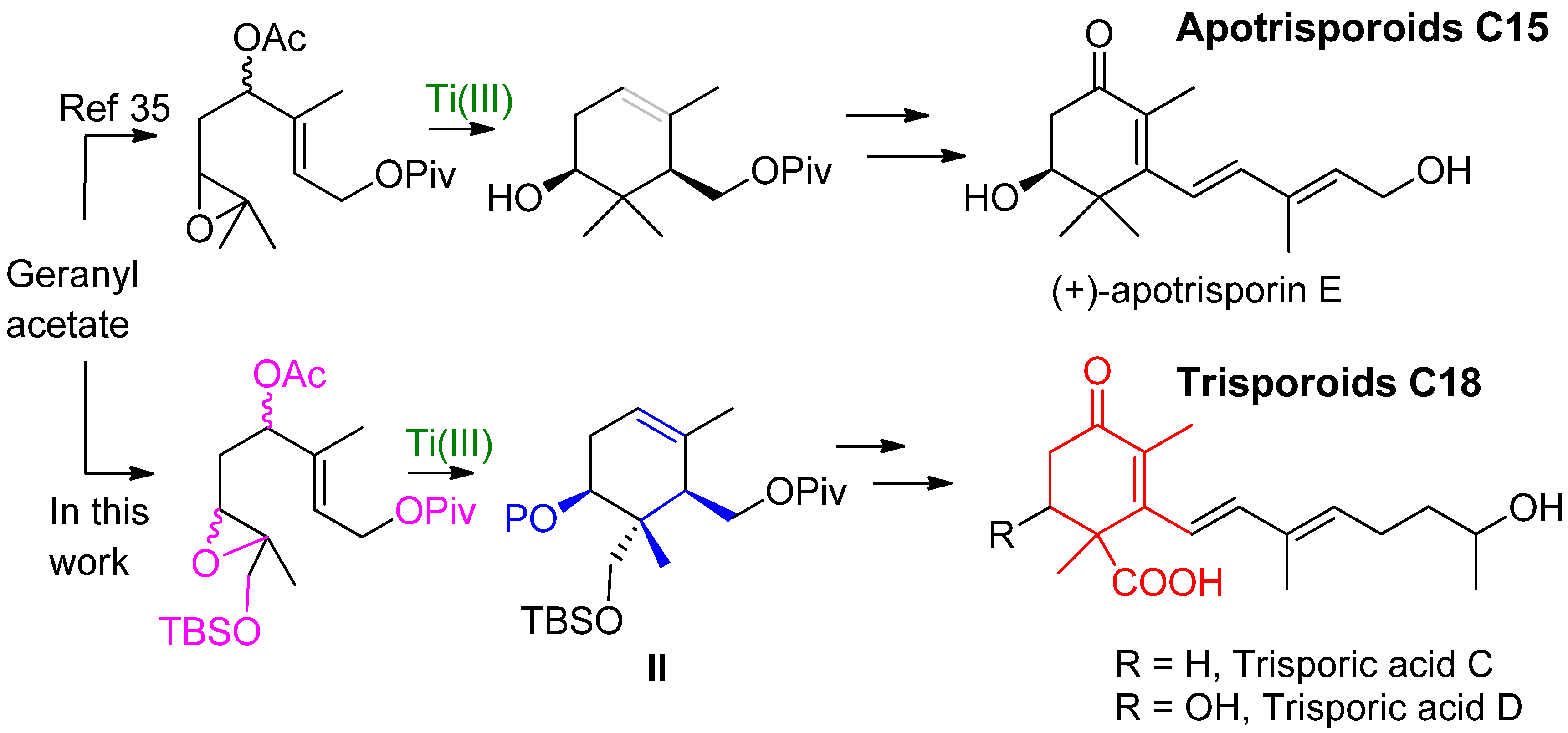{kind=link}
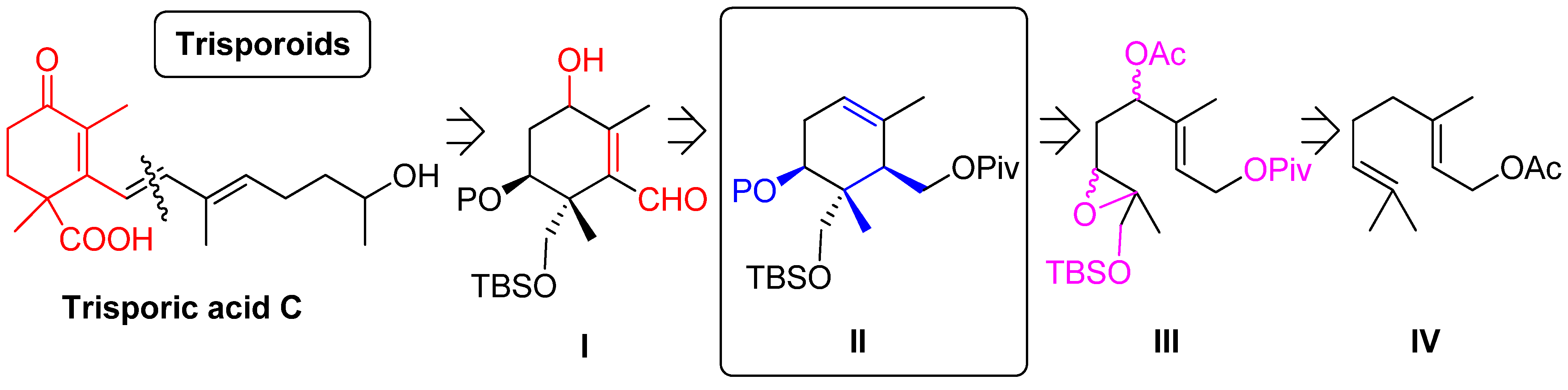{kind=link}
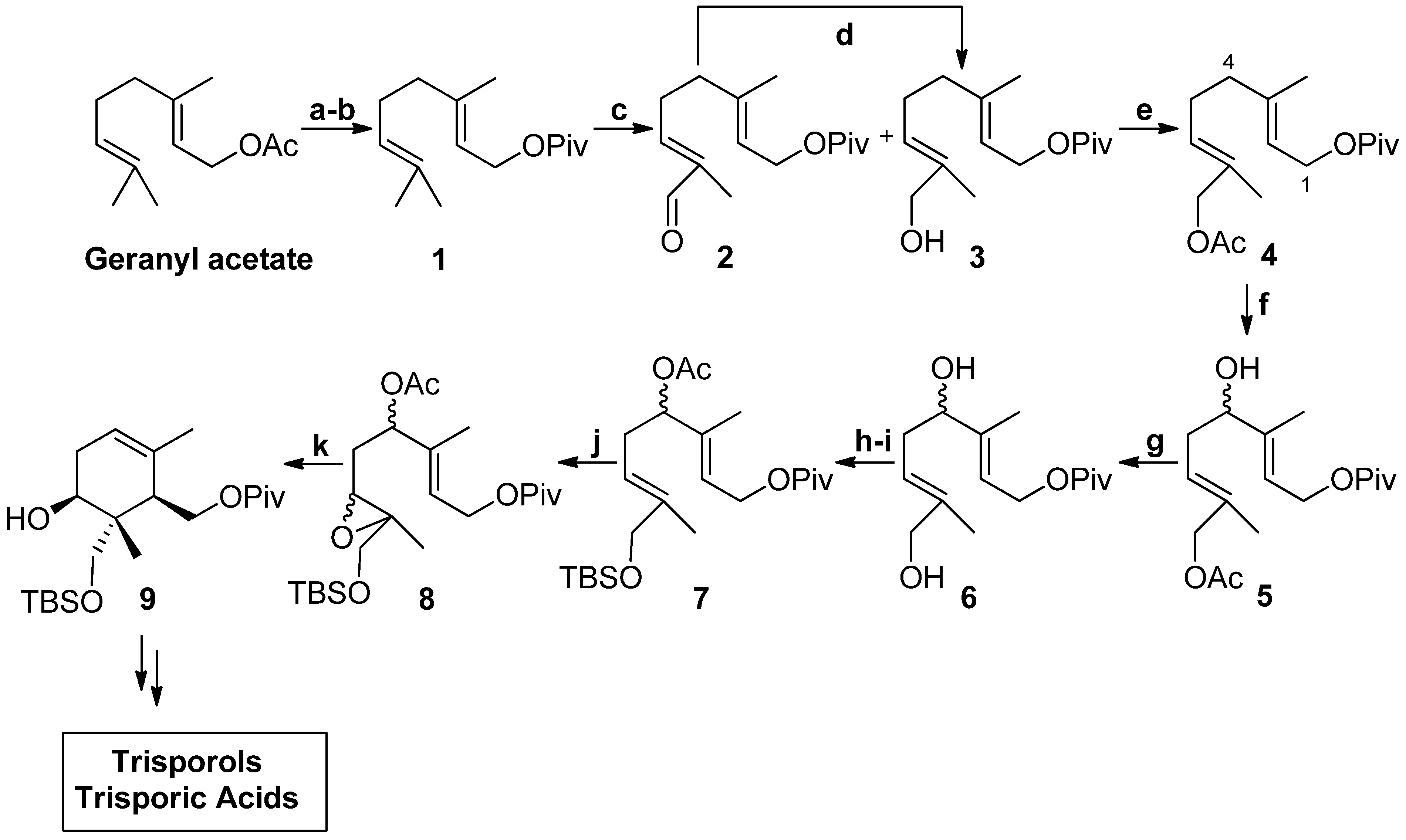{kind=link}
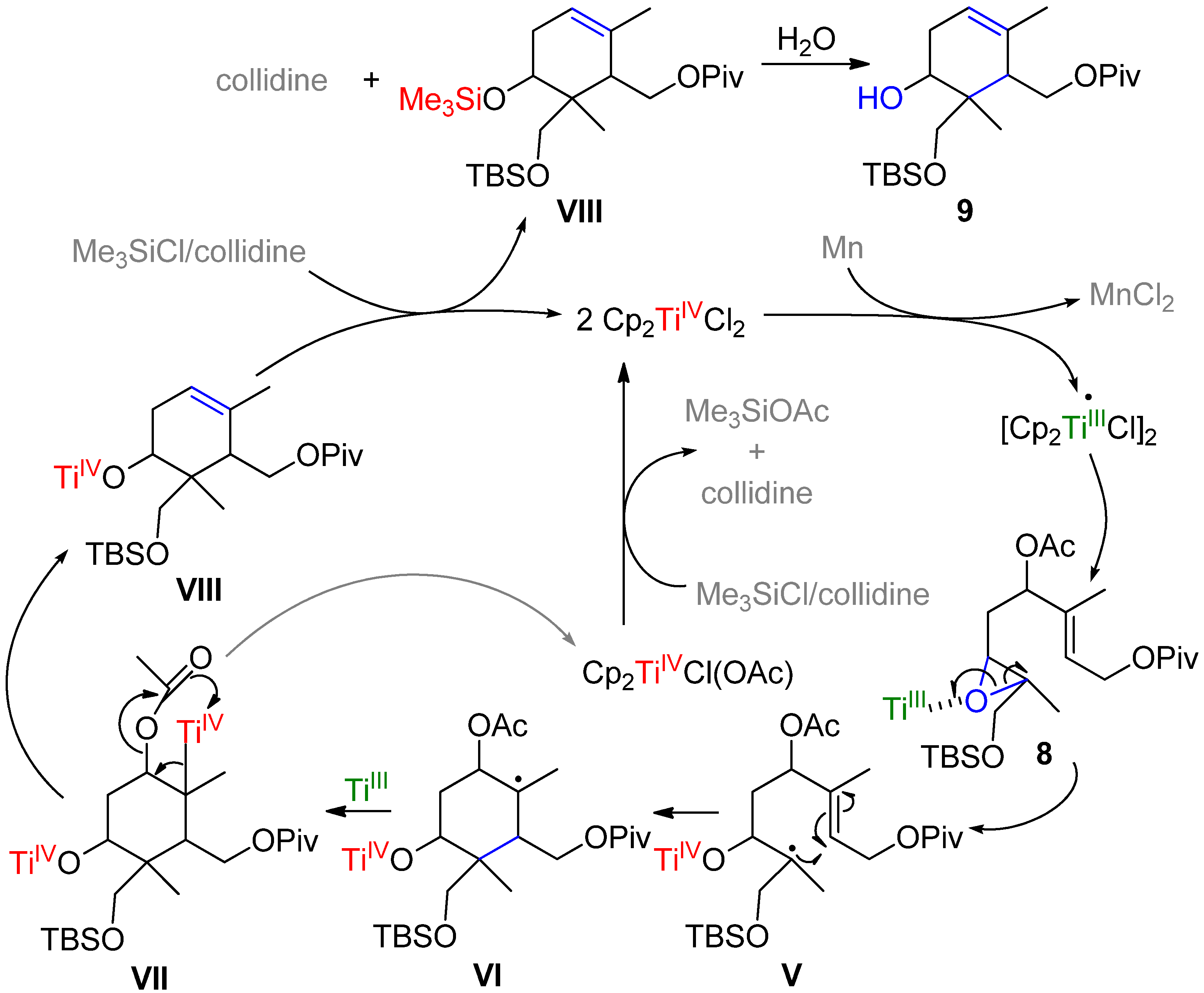
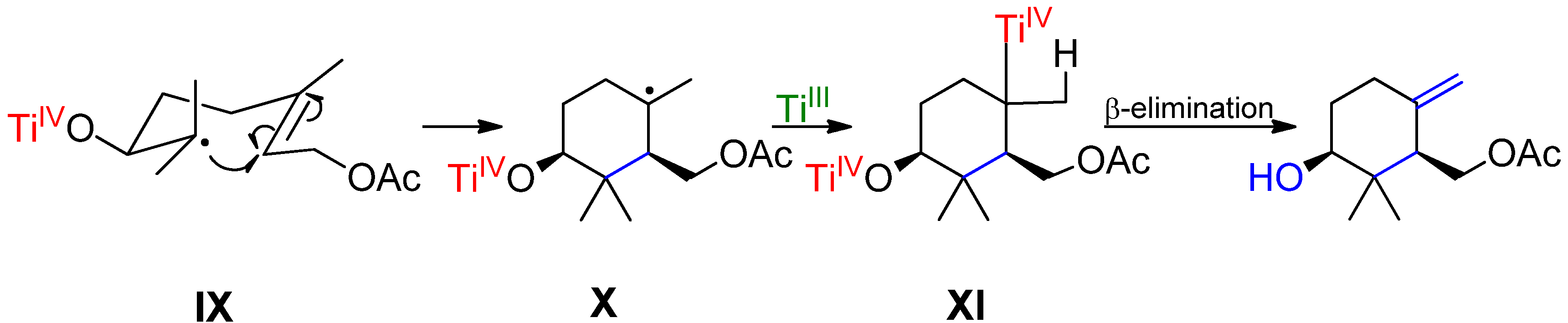
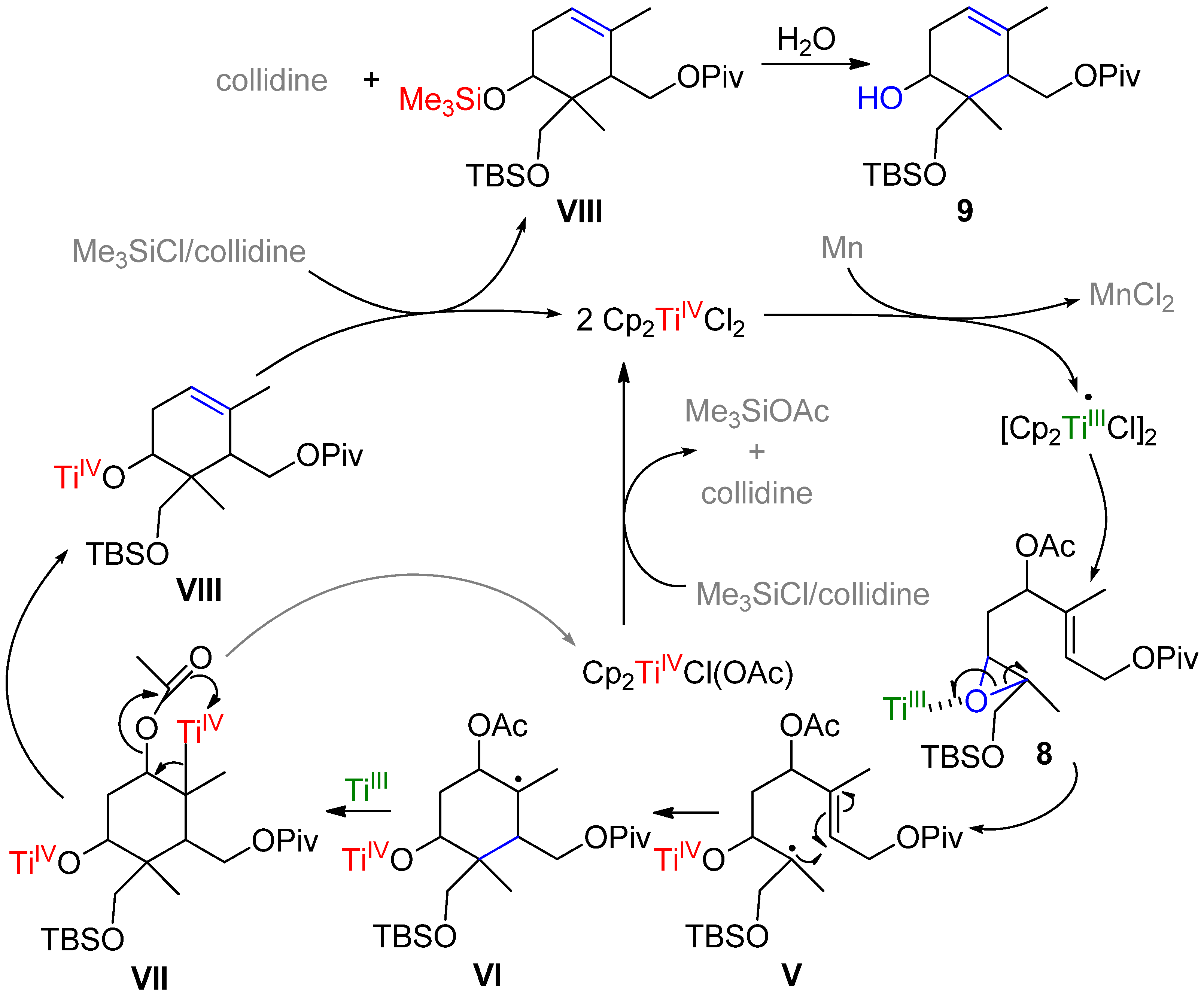{kind=link}
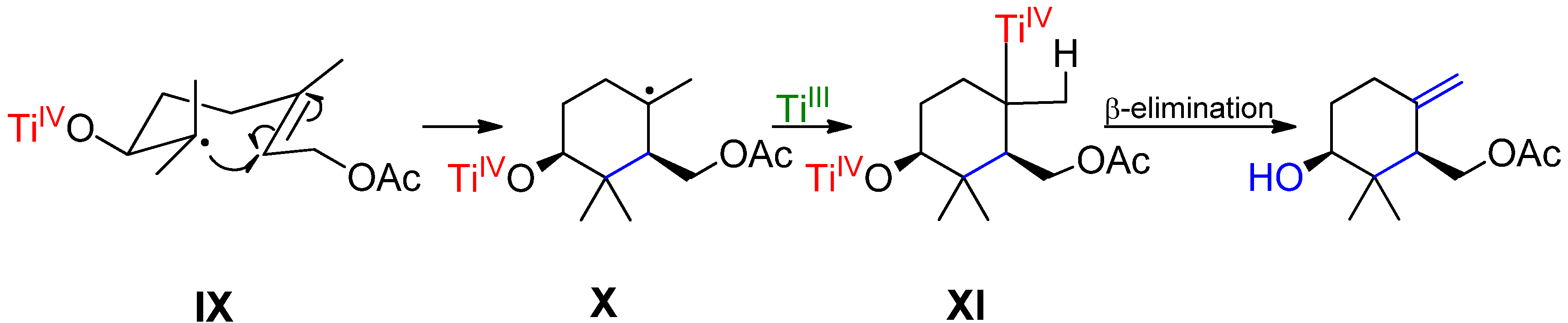{kind=link}
| Entry | EtOH purity [%] | Temperature [°C] | Time [min] | Ratio [5:4] | Yield [%] |
|---|---|---|---|---|---|
| 1 | 40 | Reflux | 360 | 1.0:10 | 5 |
| 2 | 65 | Reflux | 360 | 1.0:7.0 | 7 |
| 3 | 80 | Reflux | 360 | 1.0:4.5 | 15 |
| 4 | 90 | Reflux | 120 | 1.0:4.0 | 36 |
| 5 | 90 | 60 | 240 | 1.0:3.2 | 24 |
| 6 | 99.8 | Reflux | 100 | 1.0:1.7 | 51 a |
| 7 | 95.0 | 60 | 35 | 1.0:1.0 | 49 a |
| 8 | 97.5 | 60 | 120 | 1.0:1.0 | 67 a |
| 9 | 97.5 | Reflux | 120 | 1.1:1.0 | 72 a |
| Position[NOE] | 1H | 13C | Representative NOE effects found in 9. | |||
|---|---|---|---|---|---|---|
| δ [ppm] | signal | J [Hz] | δ [ppm] | |||
| 1 | 5.39 | 1H | bs | 121.1 |  | |
| 2 | 2.22 | 2H | m | 30.8 | ||
| 3α | 3.85 | 1H | dd | 8.4 5.6 | 71.5 | |
| 4 | 41.4 | |||||
| 5 | 2.02 | 1H | m | 43.1 | ||
| 6 | 132.6 | |||||
| 7 | 1.68 | 3H | 22.0 | |||
| 8β | 4.07 | 1H | dd | 11.8 4.4 | 62.7 | |
| 4.30 | 1H | dd | 11.8 4.3 | |||
| 9α | 3.53 3.68 | 1H 1H | d d | 10.0 10.0 | 68.9 |  |
| 10β | 0.88 | 1H | s | 12.0 | ||
| 11 | 178.5 | |||||
| 12 | 38.8 | |||||
| 13 | 1.19 | 9H | s | 27.2 | ||
| 14 | 0.07 0.06 | 3H 3H | s s | –5.5 | ||
| 15 | 5.39 | 1H | bs | 18.2 | ||
| 16 | 2.22 | 2H | m | 25.9 | ||
3. Experimental
3.1. General
3.2. Experimental Procedures
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Austin, D.J.; Bulock, J.D.; Gooday, G.W. Trisporic Acids: Sexual Hormones from Mucor mucedo and Blakeslea trispora. Nature 1969, 223, 1178–1179. [Google Scholar] [CrossRef]
- Sahadevan, Y.; Richter-Fecken, M.; Kaerger, K.; Voigt, K.; Boland, W. Early and late trisporoids differentially regulate β-carotene production and gene transcript levels in the mucoralean fungi Blakeslea trispora and Mucor mucedo. Appl. Environ. Microbiol. 2013, 79, 7466–7475. [Google Scholar] [CrossRef]
- Schimek, C.; Wöstemeyer, J. Biosynthesis, extraction, purification, and analysis of trisporoid sexual communication compounds from mated cultures of Blakeslea trispora. Meth. Mol. Biol. 2012, 898, 61–74. [Google Scholar] [CrossRef]
- Miller, M.L.; Sutter, R.P. Methyl trisporate E, a sex pheromone in Phycomyces blakesleeanus. J. Biol. Chem. 1984, 259, 6420–6422. [Google Scholar]
- Gooday, G.W. The extraction of a sexual hormone from the mycelium of Mucor mucedo. Phytochemistry 1968, 7, 2103–2105. [Google Scholar] [CrossRef]
- Ellenberger, S.; Schuster, S.; Wöstemeyer, J. Correlation between sequence, structure and function for trisporoid processing proteins in the model zygomycete Mucor mucedo. J. Theor. Biol. 2013, 320, 66–75. [Google Scholar] [CrossRef]
- Schachtschabel, D.; Schimek, C.; Wöstemeyer, J.; Boland, W. Biological activity of trisporoids and trisporoid analogues in Mucor mucedo (–). Phytochemistry 2005, 66, 1358–1365. [Google Scholar] [CrossRef]
- Schipper, M.A. Introduction of zygospore production in Mucor saximontensis, an agamic strain of Zygorhynchus moelleri. Trans. Br. Mycol. Soc. 1971, 56, 157–159. [Google Scholar] [CrossRef]
- Schimek, C.; Kleppe, K.; Saleem, A.-R.; Voigt, K.; Burmester, A.; Wöstemeyer, J. Sexual reactions in Mortierellales are mediated by the trisporic acid system. Mycol. Res. 2003, 107, 736–747. [Google Scholar] [CrossRef]
- Burmester, A.; Karimi, S.; Wetzel, J.; Wöstemeyer, J. Complementation of a stable Met2-1 mutant of the zygomycete Absidia glauca by the corresponding wild-type allele of the mycoparasite Parasitella parasitica, transferred during infection. Microbiology 2013, 159, 1639–1648. [Google Scholar]
- Schultze, K.; Schimek, C.; Wöstemeyer, J.; Burmester, A. Sexuality and parasitism share common regulatory pathways in the fungus Parasitella parasitica. Gene 2005, 348, 33–44. [Google Scholar]
- Halary, S.; Daubois, L.; Terrat, Y.; Ellenberger, S.; Wöstemeyer, J.; Hijri, M. Mating Type Gene Homologues and Putative Sex Pheromone-Sensing Pathway in Arbuscular Mycorrhizal Fungi, a Presumably Asexual Plant Root Symbiont. PLos One 2013, 8, 1–12. [Google Scholar]
- Polaino, S.; Herrador, M.M.; Cerdá-Olmedo, E.; Barrero, A.F. Splitting of β-carotene in the sexual interaction of Phycomyces. Org. Biomol. Chem. 2010, 8, 4229–4231. [Google Scholar] [CrossRef]
- Barrero, A.F.; Herrador, M.M.; Arteaga, P.; Gil, J.; González-Delgado, J.A.; Alcalde, E.; Cerdá-Olmedo, E. New apocarotenoids and β-carotene cleavage in Blakeslea trispora. Org. Biomol. Chem. 2011, 9, 7190–7195. [Google Scholar] [CrossRef]
- Medina, H.; Cerdá-Olmedo, E.; Al-Babili, S. Cleavage oxygenases for the biosynthesis of trisporoids and other apocarotenoids in Phycomyces. Mol. Microbiol. 2011, 8, 199–208. [Google Scholar]
- Polaino, S.; González-Delgado, J.A.; Arteaga, P.; Herrador, M.M.; Barrero, A.F.; Cerdá-Olmedo, E. Apocarotenoids in the sexual interaction of Phycomyces blakesleeanus. Org. Biomol. Chem. 2012, 10, 3002–3009. [Google Scholar]
- Edwards, J.A.; Schwarz, V.; Fajkos, J.; Maddox, M.L.; Fried, J.H. Fungal sex hormones. The synthesis of (±)-7(E),9(E)-trisporic acid B methyl ester. The stereochemistry at C-9 of the trisporic acids. J. Chem. Soc. D: Chem. Commun. 1971, 292–293. [Google Scholar]
- Isoe, S.; Hayase, Y.; Sakan, T. Sexual hormones of Mucorales. The synthesis of methyl trisporate B and C. Tetrahedron Lett. 1971, 12, 3691–3694. [Google Scholar] [CrossRef]
- Bulock, J.D.; Jones, B.E.; Winskill, N. The Apocarotenoid System of Sex Hormones and Prohormones in Mucorales. Pure Appl. Chem. 1976, 47, 191–202. [Google Scholar] [CrossRef]
- Secrist, J.A.; Hickey, C.J.; Norris, R.E. A convenient total synthesis of (±)-(7E,9E)-trisporic acid B methyl ester. J. Org. Chem. 1977, 42, 525–527. [Google Scholar] [CrossRef]
- Prisbylla, M.P.; Takabe, K.; White, J.D. Stereospecific synthesis of (±)-trisporol B, a prohormone of Blakeslea trispora, and a facile synthesis of (±)-trisporic acids. J. Am. Chem. Soc. 1979, 101, 762–763. [Google Scholar]
- Trost, B.M.; Ornstein, P.L. Bifunctional cyclopropyl reagents: a total synthesis of (7E,9Z)-methyl trisporate B. Tetrahedron Lett. 1983, 24, 2833–2836. [Google Scholar] [CrossRef]
- Takabe, K.; White, J.D. Synthesis of methyl 4-dihydrotrisporate B, a prohormone of Blakeslea trispora. Tetrahedron Lett. 1983, 24, 3709–3712. [Google Scholar] [CrossRef]
- White, J.D.; Takabe, K.; Prisbylla, M.P. Stereoselective Synthesis of Trisporic Acids A and B, Their Methyl Esters, and Trisporols A and B, Hormones and Prohormones of Mucoraceous Fungi. J. Org. Chem. 1985, 50, 5233–5244. [Google Scholar] [CrossRef]
- Miyaura, N.; Satoh, Y.; Hara, S.; Suzuki, A. Stereospecific Synthesis of the Fungal Prohormone (–)-Trisporol B via the Palladium-Catalyzed Cross-Coupling Reaction of 1-Alkenylborane with 1-Haloalkene. Bull. Chem. Soc. Jpn. 1986, 59, 2029–2031. [Google Scholar] [CrossRef]
- Takahashi, S.; Oritani, T.; Yamashita, K. Enantioselective Synthesis of (+)-Methyl Trisporate B. Agric. Biol. Chem. 1987, 51, 2291–2293. [Google Scholar] [CrossRef]
- Bacigaluppo, J.A.; Colombo, M.I.; Preite, M.D.; Zinczuk, J.; Rúveda, E.A.; Sieler, J. Enantioselective synthesis and resolution of the key white intermediate for the synthesis of trisporic acid. Tetrahedron Asym. 1996, 7, 1041–1057. [Google Scholar] [CrossRef]
- Reeder, M.R.; Meyers, A.I. Asymmetric routes to the trisporic acids via chiral bicyclic lactams. Tetrahedron Lett. 1999, 40, 3115–3118. [Google Scholar] [CrossRef]
- Jansen, F.J.H.M.; Lugtenburg, J. Synthesis and characterization of all-E-(4,4'-13C2)-astaxanthin strategies for labelling the C15-end groups of carotenoids. Eur. J. Org. Chem. 2000, 829–836. [Google Scholar] [CrossRef]
- Colombo, M.I.; Zinczuk, J.; Mischne, M.; Rúveda, E.A. Studies directed toward the preparation of key intermediates for the synthesis of trisporic acids and cassiol. Pure Appl. Chem. 2001, 73, 623–626. [Google Scholar] [CrossRef]
- Schachtschabel, D.; Boland, W. Efficient Generation of a Trisporoid Library by Combination of Synthesis and Biotransformation. J. Org. Chem. 2007, 72, 1366–1372. [Google Scholar] [CrossRef]
- López-Sánchez, C.; Hernández-Cervantes, C.; Rosales, A.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. A convenient cross-Metathesis approach to trisporins. Tetrahedron 2009, 65, 9542–9549. [Google Scholar] [CrossRef]
- Wojtkielewicz, A.; Maj, J.; Dzieszkowska, A.; Morzycki, W. Cross metathesis approach to retonoids and other β-apocarotenoids. Tetrahedron 2011, 67, 6868–6875. [Google Scholar] [CrossRef]
- Dubberke, S.; Abbas, M.; Westerman, B. Oxidative allylic rearrangement of cycloalkenols: Formal total synthesis of enantiomerically pure trisporic acid B. Beilstein J. Org. Chem. 2011, 7, 421–425. [Google Scholar] [CrossRef]
- González-Delgado, J.A.; Arteaga, J.F.; Herrador, M.M.; Barrero, A.F. First total synthesis of (+)-apotrisporin E and (+)-apotrientriols A–B: A cyclization approach to apocarotenoids. Org. Biomol. Chem. 2013, 11, 5404–5408. [Google Scholar] [CrossRef]
- The single electron transfer bis(cyclopentadienyl)-Ti(III) chloride, can be generated in situ by stirring commercial Cp2TiCl2 with dust Mn. For pionnering reports on the use of this reagent, see: RajanBabu, T.V.; Nugent, W.A. Selective generation of free radicals from epoxides using a transition-metal radical. A powerful new tool for organic synthesis. J. Am. Chem. Soc. 1994, 116, 986–997. [Google Scholar]and references cited therein.
- Gansauer, A.; Bluhm, H. Reagent-Controlled Transition-Metal-Catalyzed Radical Reactions. Chem. Rev. 2000, 100, 2771–2788. [Google Scholar] [CrossRef]
- Gansauer, A.; Lauterbach, T.; Narayan, S. Strained heterocycles in radical chemistry. Angew. Chem. Int. Ed. 2003, 42, 5556–5573. [Google Scholar] [CrossRef]
- Barrero, A.F.; Quílez del Moral, J.F.; Sánchez, E.M.; Arteaga, J.F. Titanocene-mediated radical cyclization: An emergent method towards the synthesis of natural products. Eur. J. Org. Chem. 2006, 1627–1641. [Google Scholar]
- Justicia, J.; Álvarez de Cienfuegos, L.; Campaña, A.G.; Miguel, D.; Jakoby, V.; Gansäuer, A.; Cuerva, J.M. Bioinspired terpene synthesis: A radical approach. Chem. Soc. Rev. 2011, 40, 3525–3537. [Google Scholar] [CrossRef]
- Barrero, A.F.; Quílez del Moral, J.F.; Herrador, M.M.; Sánchez, E.M.; Arteaga, J.F. Regio- and enantioselective functionalization of acyclic polyprenoids. J. Mex. Chem. Soc. 2006, 50, 149–156. [Google Scholar]
- Rapoport, H.; Bhalerao, U.T. Stereochemistry of allylic oxidation with selenium dioxide. Stereospecific oxidation of gem-dimethyl olefins. J. Am. Chem. Soc. 1971, 93, 4835–4840. [Google Scholar] [CrossRef]
- Umbreit, M.A.; Sharpless, K.B. Allylic oxidation of olefins by catalytic and stoichiometric selenium dioxide with tert-butyl hydroperoxide. J. Am. Chem. Soc. 1977, 99, 5526–5527. [Google Scholar] [CrossRef]
- Kovylyaeva, G.I.; Sharipova, R.R.; Strobykina, I.Y.; Militsina, O.I.; Musin, R.Z.; Beskrovnyi, D.V.; Gubaidullin, A.T.; Alfonsov, V.A.; Kataev, V.E. Reaction of isosteviol diterpenoid with selenium dioxide. Russ. J. Gen. Chem. 2009, 79, 2663–2667. [Google Scholar] [CrossRef]
- Barrero, A.F.; Cuerva, J.M.; Herrador, M.M.; Valdivia, M.V. A new strategy for the synthesis of cyclic terpenoids based on the radical opening of acyclic epoxypolyenes. J. Org. Chem. 2001, 66, 4074–4078. [Google Scholar] [CrossRef]
- Barrero, A.F.; Herrador, M.M.; Quílez del Moral, J.F.; Arteaga, P.; Sánchez, E.M.; Arteaga, J.F.; Piedra, M. Transannular cyclization of epoxycaryophyllenes catalyzed by TiIII: An efficient synthesis of tricyclo[6.3.0.02,5]undecanes. Eur. J. Org. Chem. 2006, 2006, 3434–3441. [Google Scholar]
- Arteaga, J.F.; Domingo, V.; Quílez del Moral, J.F.; Barrero, A.F. Org. Lett. 2008, 9, 1723–1726.
- Domingo, V.; Silva, L.; Diéguez, H.R.; Arteaga, J.F.; Quílez del Moral, J.F.; Barrero, A.F. Enantioselective total synthesis of the potent anti-inflammatory (+)-myrrhanol A. J. Org. Chem. 2009, 74, 6151–6156. [Google Scholar] [CrossRef]
- Arteaga, J.F.; Diéguez, H.R.; González-Delgado, J.A.; Quílez del Moral, J.F.; Barrero, A.F. Control of the regio- and diastereoselectivity for the preparation of highly functionalized terpenic cyclopentanes through radical cyclization. Eur. J. Org. Chem. 2011, 5002–5011. [Google Scholar]
- Domingo, V.; Arteaga, J.F.; López Pérez, J.L.; Peláez, R.; Quílez del Moral, J.F.; Barrero, A.F. Total synthesis of (+)-seco-C-oleanane via stepwise controlled radical cascade cyclization. J. Org. Chem. 2012, 77, 341–350. [Google Scholar] [CrossRef]
- Justicia, J.; Rosales, A.; Buñuel, E.; Oller-Lopez, J.L.; Valdivia, M.V.; Haidour, A.; Oltra, J.E.; Barrero, A.F.; Cardenas, D.J.; Cuerva, J.M. Titanocene-catalyzed cascade cyclization of epoxypolyprenes: Straightforward synthesis of terpenoids by free-radical chemistry. Chem. Eur. J. 2004, 10, 1778–1788. [Google Scholar] [CrossRef]
- Daasbjerg, K.; Svith, H.; Grimme, S.; Gerenkamp, M.; Muck-Lichtenfeld, C.; Gansäuer, A.; Barchuk, A.; Keller, F. Elucidation of the mechanism of titanocene-mediated epoxide opening by a combined experimental and theoretical approach. Angew. Chem. Int. Edit. 2006, 45, 2041–2044. [Google Scholar] [CrossRef]
- Barrero, A.F.; Quilez del Moral, J.F.; Herrador, M.M.; Loayza, I.; Sanchez, E.M.; Arteaga, J.F. Synthesis of five- to seven-membered polyfunctional terpenic carbocycles via Ti(III)-catalyzed radical cyclizations of epoxypolyprenes. Tetrahedron 2006, 62, 5215–5222. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–9 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
González-Delgado, J.A.; Escobar, G.; Arteaga, J.F.; Barrero, A.F. Easy Access to a Cyclic Key Intermediate for the Synthesis of Trisporic Acids and Related Compounds. Molecules 2014, 19, 1748-1762. https://doi.org/10.3390/molecules19021748
González-Delgado JA, Escobar G, Arteaga JF, Barrero AF. Easy Access to a Cyclic Key Intermediate for the Synthesis of Trisporic Acids and Related Compounds. Molecules. 2014; 19(2):1748-1762. https://doi.org/10.3390/molecules19021748
Chicago/Turabian StyleGonzález-Delgado, José A., Gustavo Escobar, Jesús F. Arteaga, and Alejandro F. Barrero. 2014. "Easy Access to a Cyclic Key Intermediate for the Synthesis of Trisporic Acids and Related Compounds" Molecules 19, no. 2: 1748-1762. https://doi.org/10.3390/molecules19021748
APA StyleGonzález-Delgado, J. A., Escobar, G., Arteaga, J. F., & Barrero, A. F. (2014). Easy Access to a Cyclic Key Intermediate for the Synthesis of Trisporic Acids and Related Compounds. Molecules, 19(2), 1748-1762. https://doi.org/10.3390/molecules19021748