A New and Efficient Method for the Synthesis of Novel 3-Acetyl Coumarins Oxadiazoles Derivatives with Expected Biological Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion

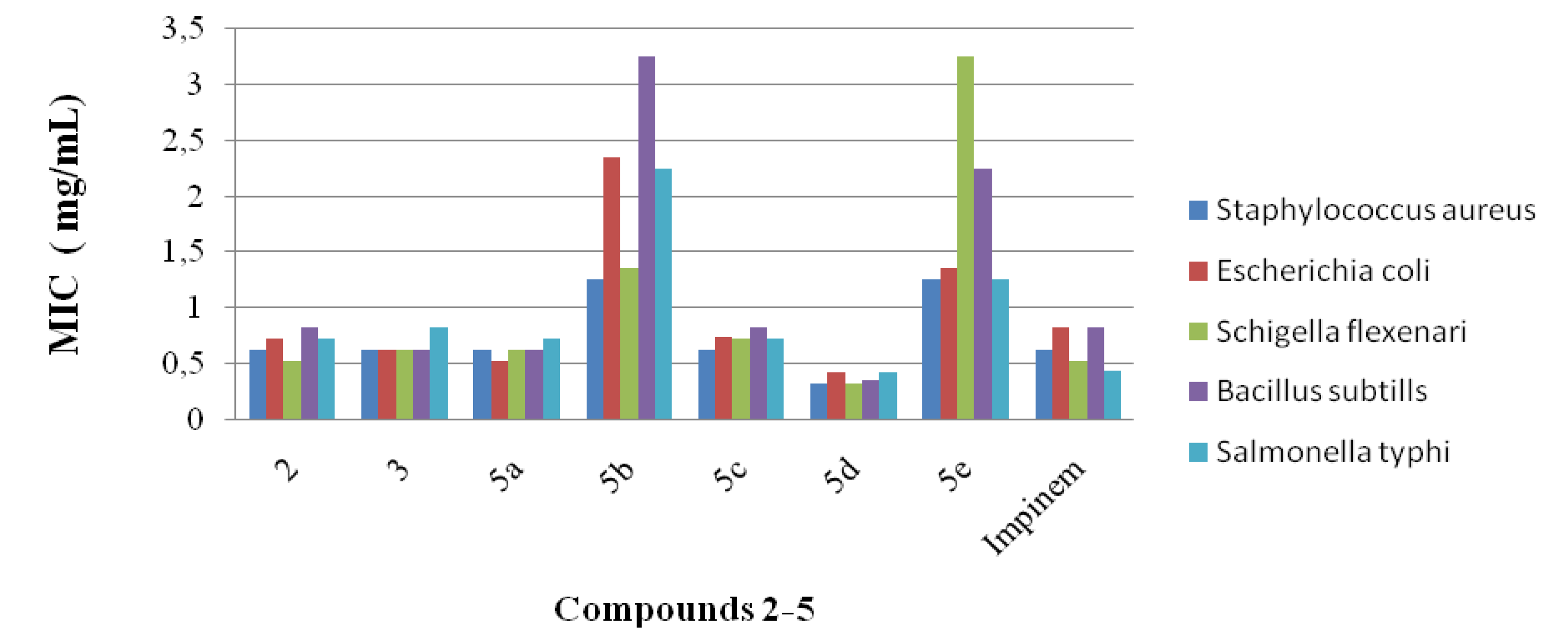
2.1. Antibacterial Studies
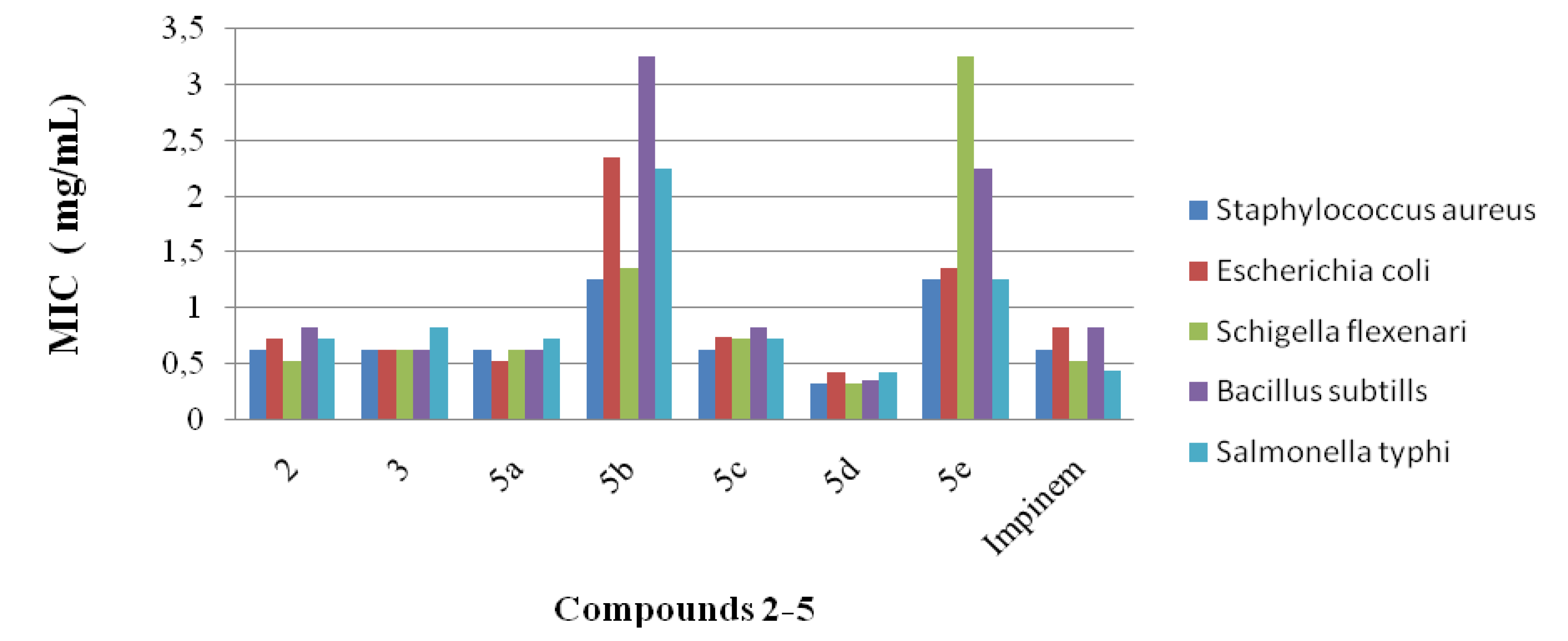
2.2. Statistical Analysis
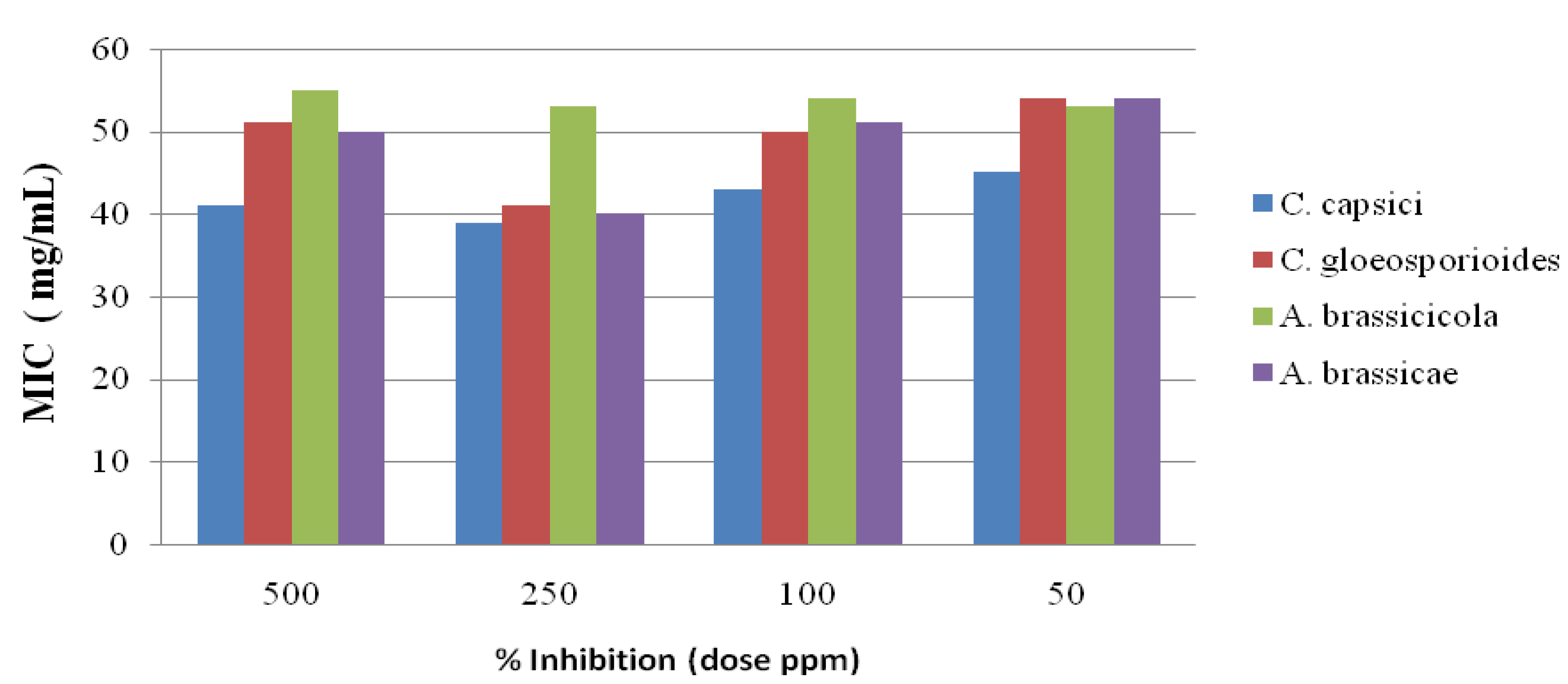
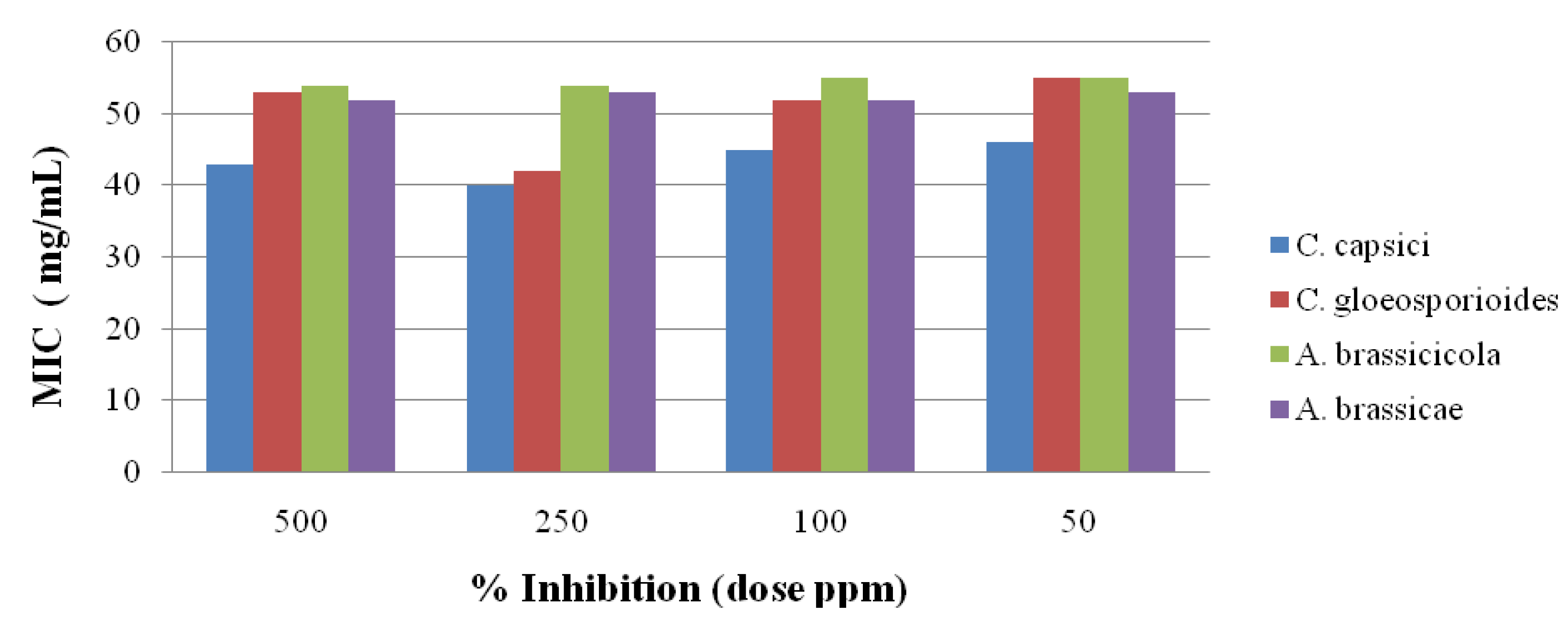
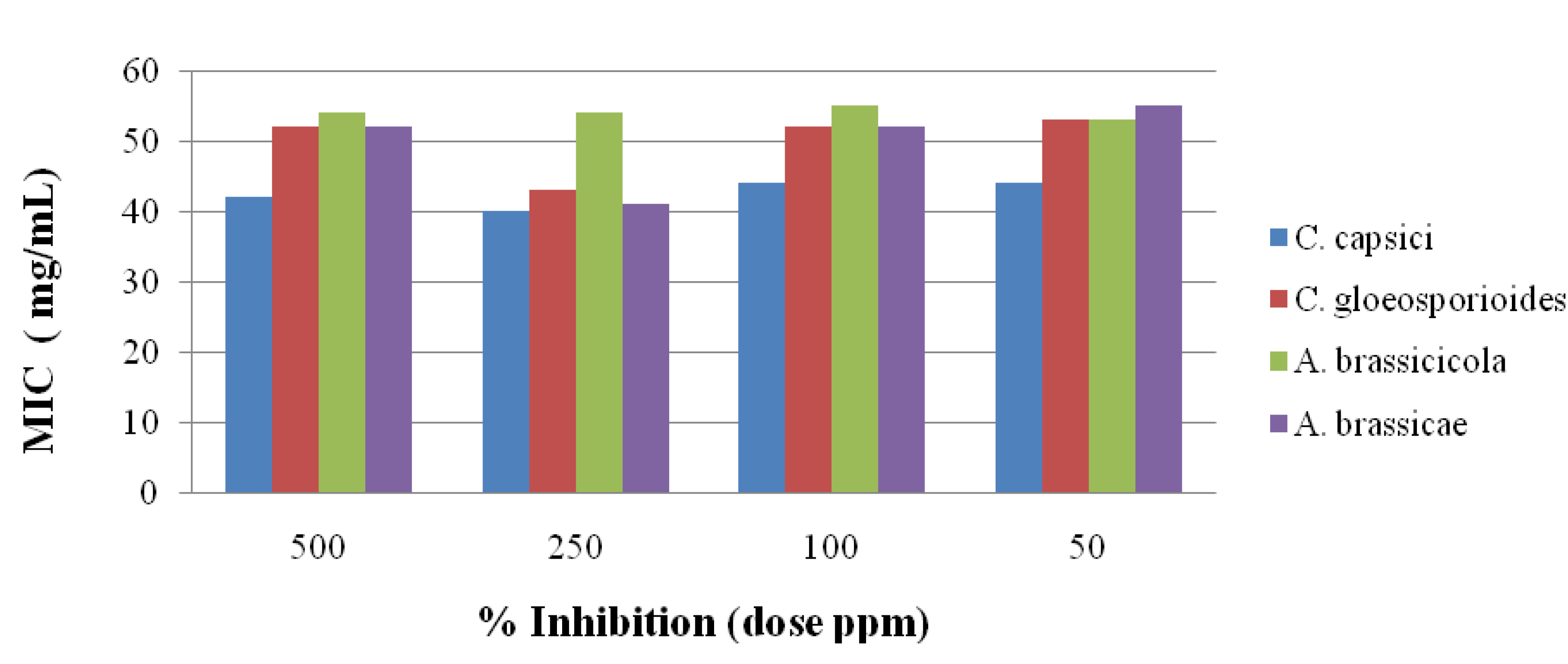
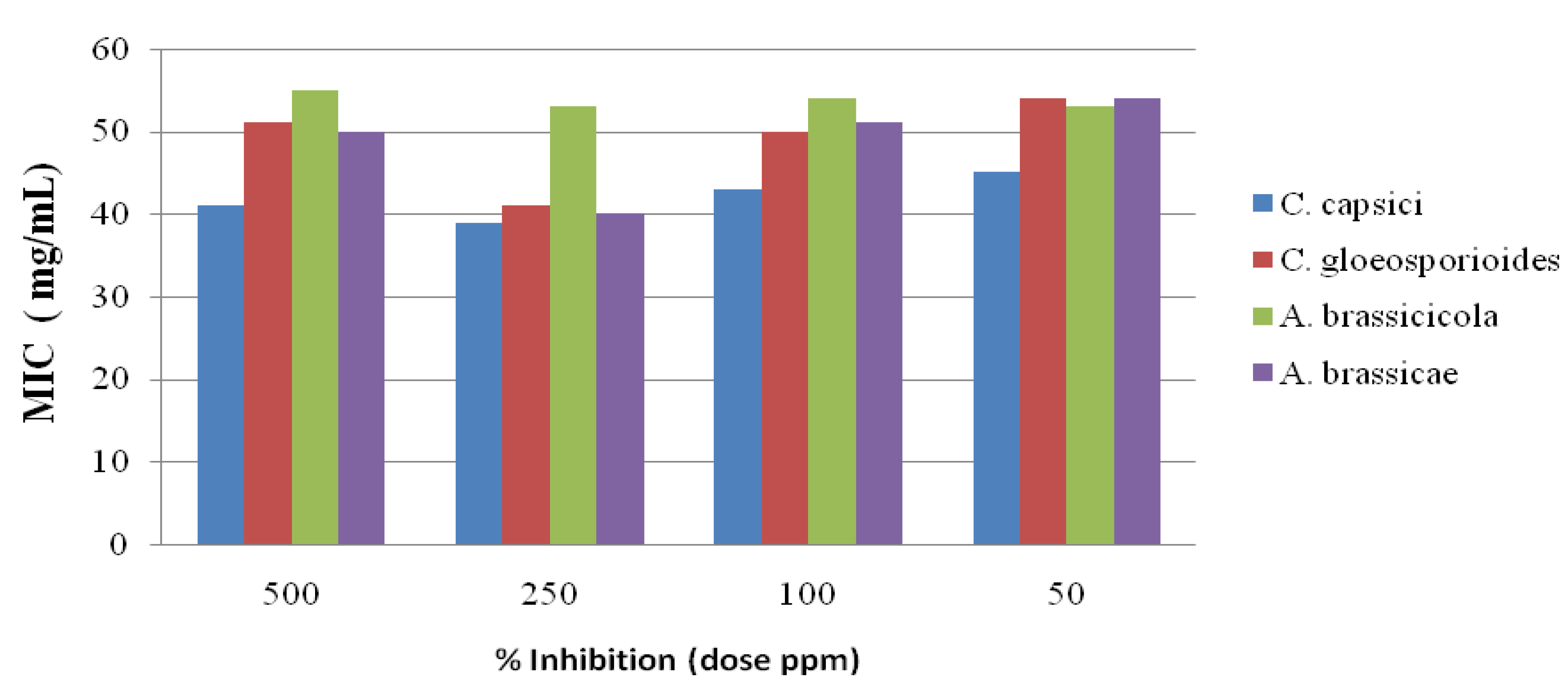
2.3. Antifungal Activity


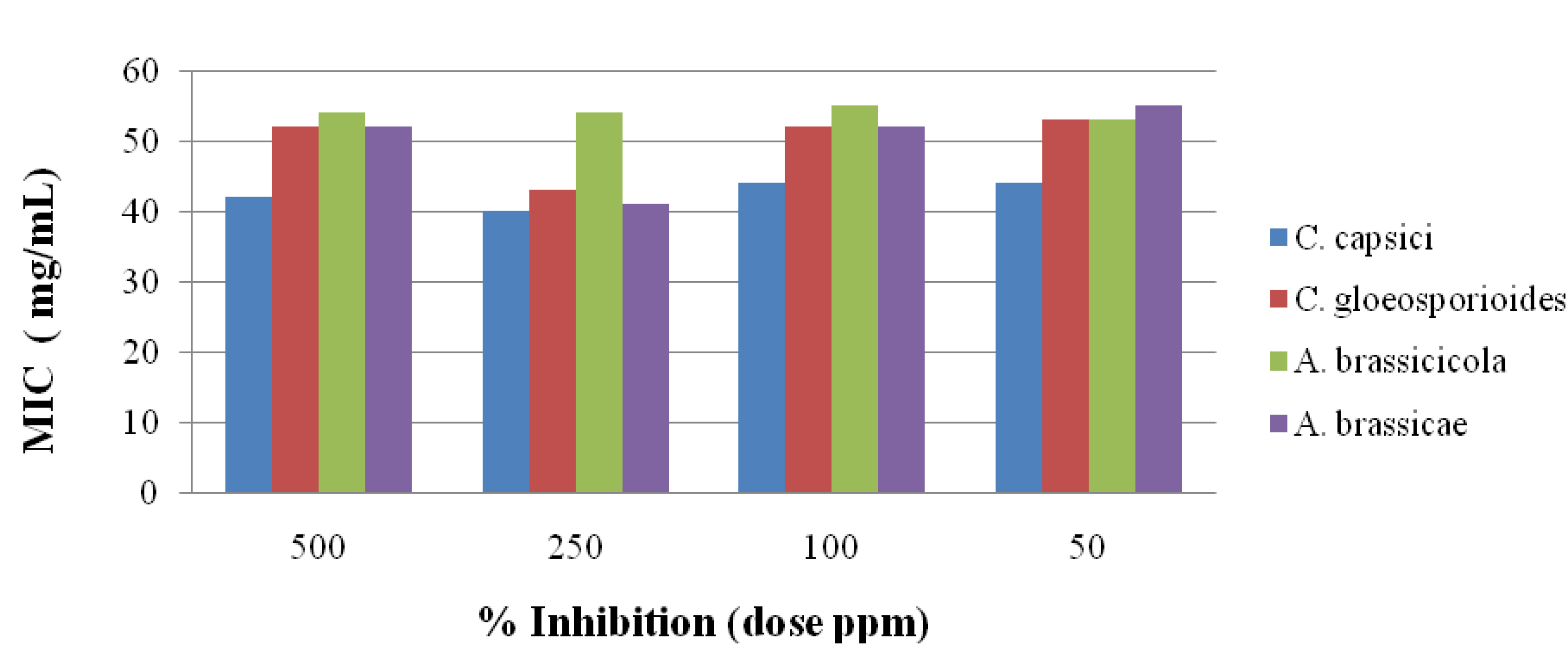

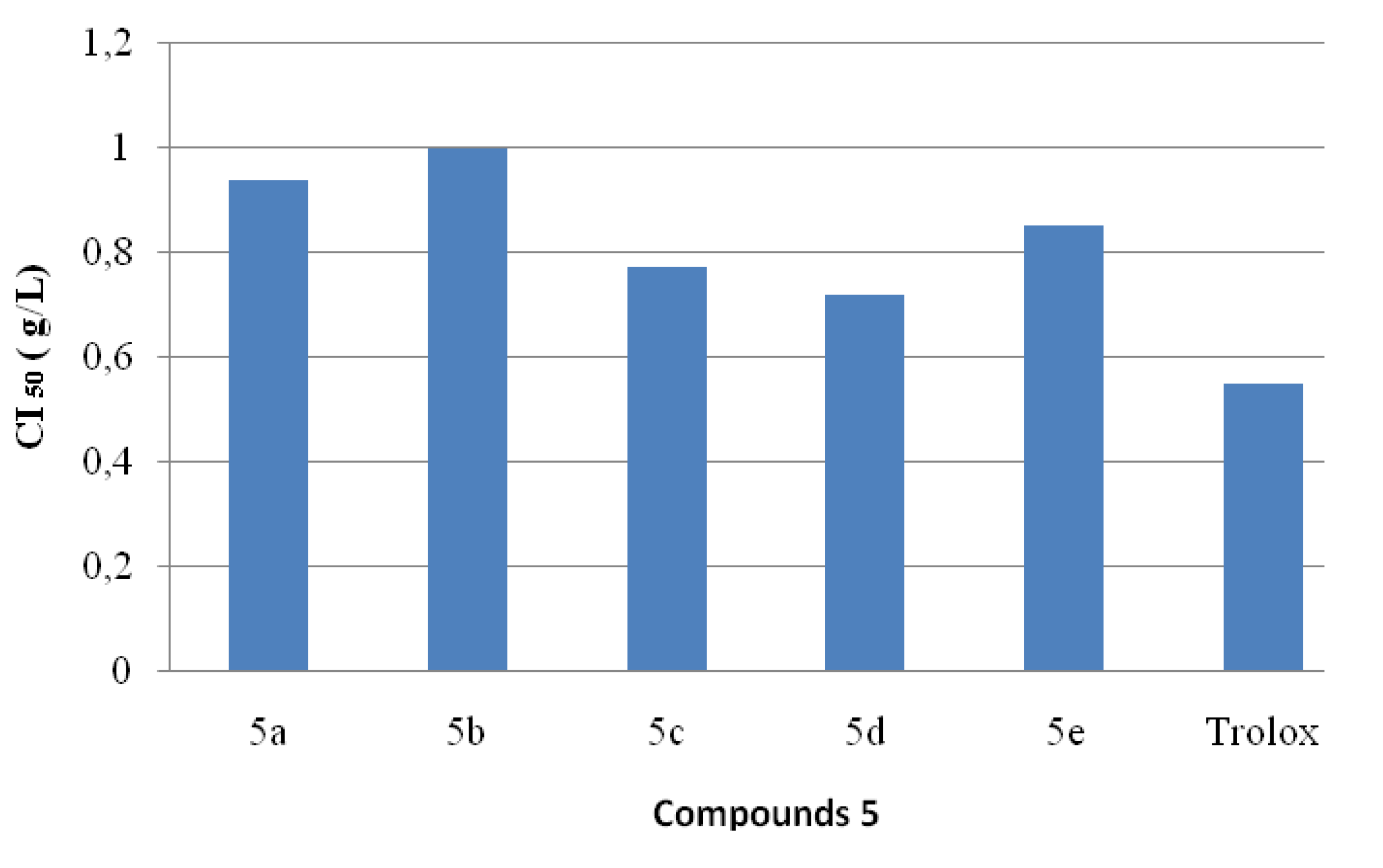
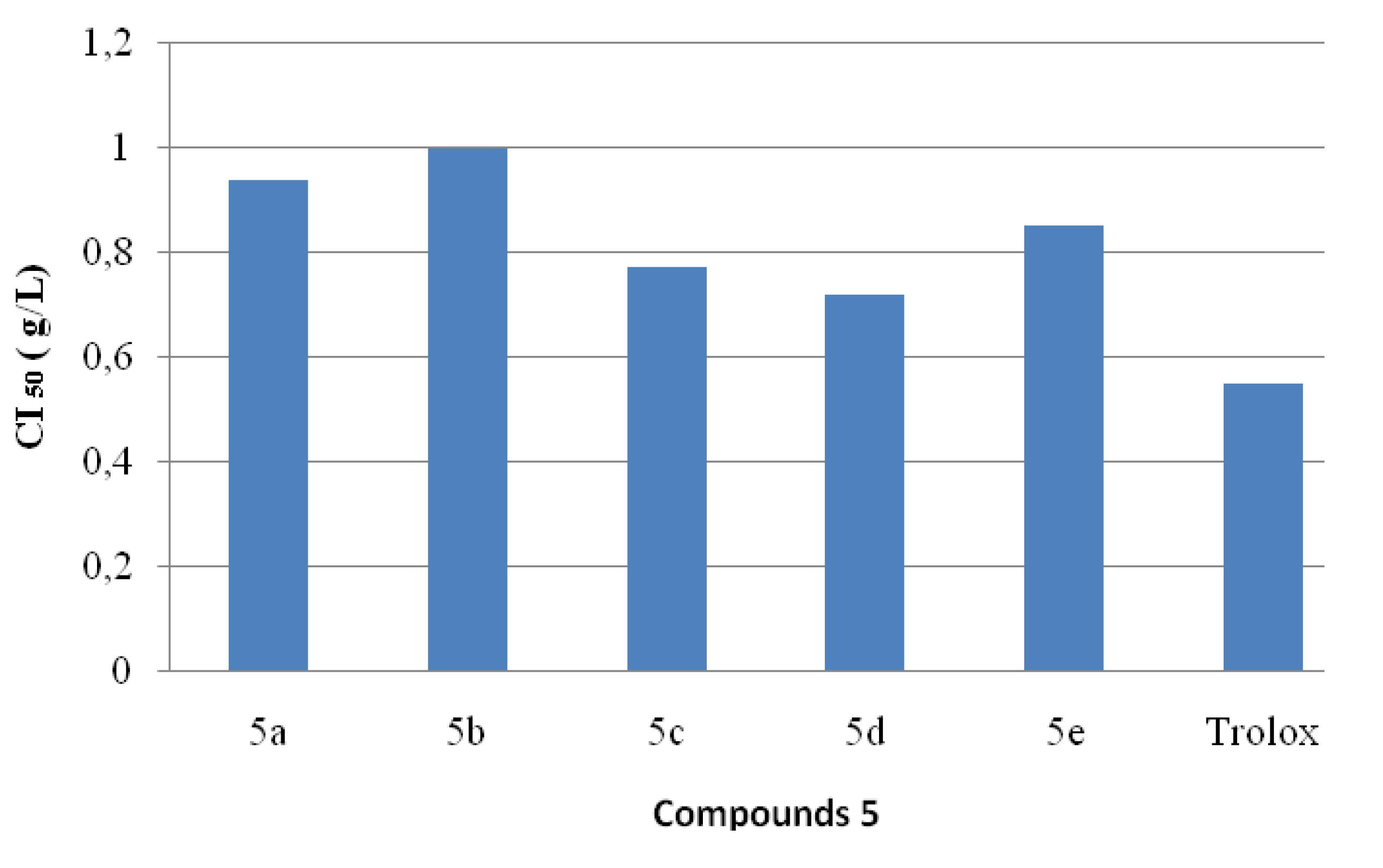
2.4. Antioxidant Activities
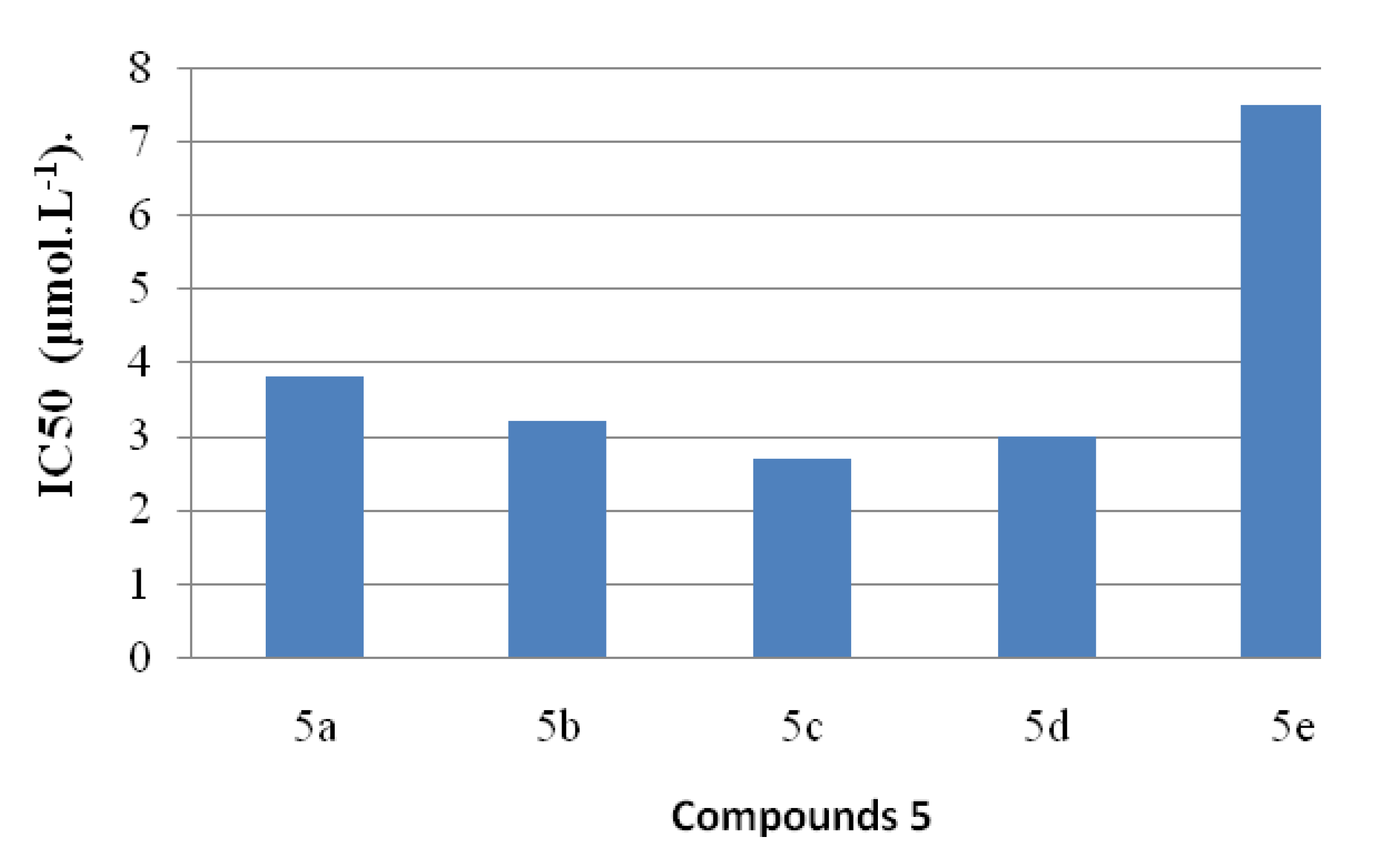
2.5. ABTS Radical Cation Decolourization Assay
3. Experimental
3.1. General Information
3.2. Preparation of 4-O-Acetylcoumarin (a1) and 3-Acetyl-4-hydroxycoumarin (a2)
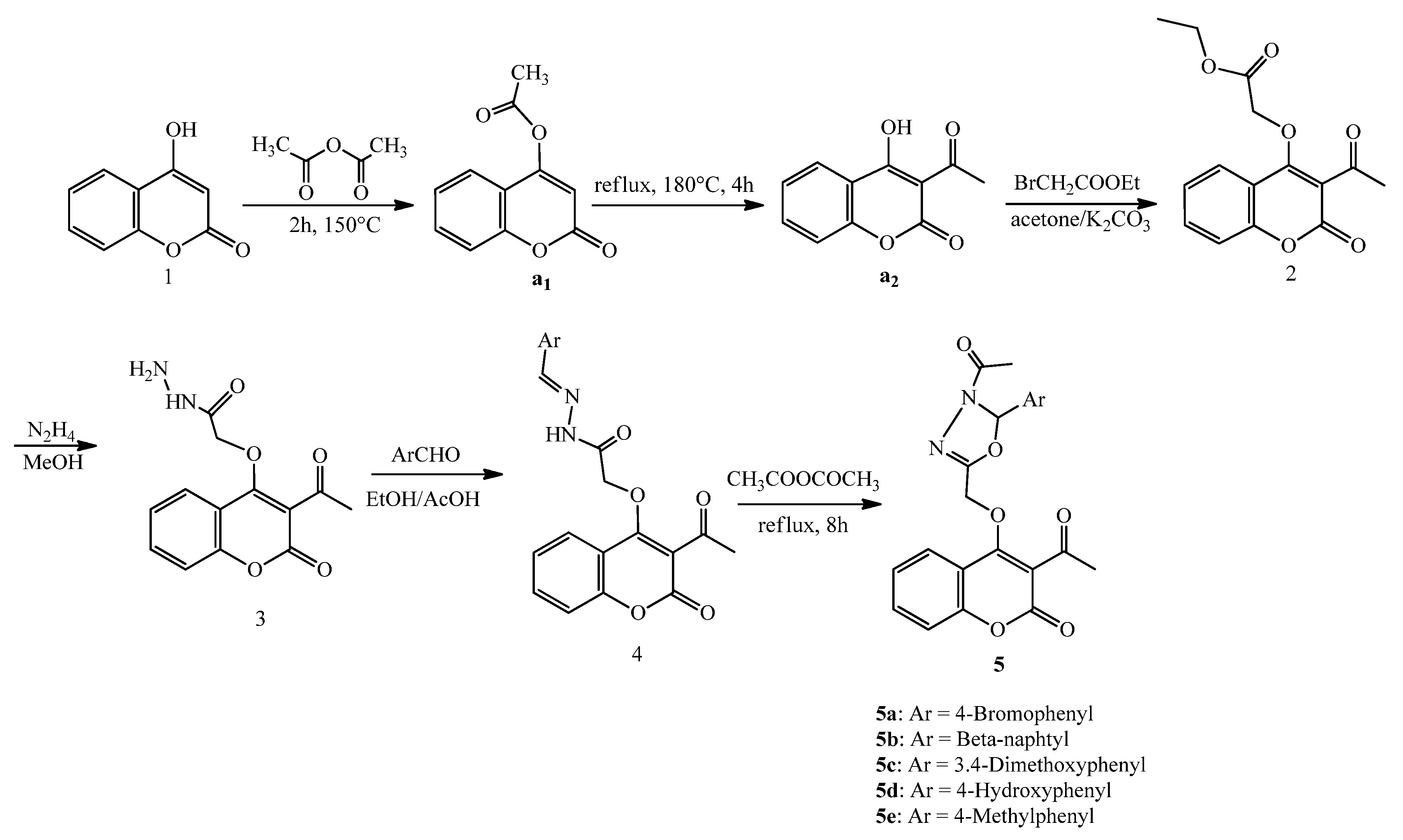
3.3. Preparation of Compounds 2–5
3.3.1. Preparation of 3-Acetyl Ethyl (coumarin-4-oxy) Acetate (2)
3.3.2. Preparation of 3-Acetylcoumarin-4-oxyacetic Hydrazide (3)
3.3.3. Preparation of 3-Acetyl-(E)-N-arylidenecoumarin-4-oxyacetic Hydrazones 4
3.3.4. Preparation of 3-Acetyl-2-[(coumarin-4-oxy)methyl]-4-acetyl-5-substitued-1,3,4-oxodiazolines 5
3.4. Antioxidant Activity Tests
3.4.1. Free Radical Scavenging Activity Evaluation by DPPH Test
3.4.2. Free Radical Scavenging Activity Evaluation by ABTS Radical Cation Test
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hall, A.; Brown, S.H.; Chowdhury, A.; Giblin, G.M.P.; Gibson, M.; Healy, M.P.; Livermore, D.G.; Wilson, R.J.M.; Naylor, A.; Rawlings, D.A.; et al. Identification and optimization of novel 1,3,4-oxadiazole EP1 receptor antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 4450–4455. [Google Scholar] [CrossRef]
- Abdel-Hamid, M.K.; Abdel-Hafez, A.A.; El-Koussi, N.A.; Mahfouz, N.M.; Innocenti, A.; Supuran, C.T. Design, synthesis, and docking studies of new 1,3,4-thiadiazole-2-thione derivatives with carbonic anhydrase inhibitory activity. Bioorg. Med. Chem. 2007, 15, 6975–6984. [Google Scholar] [CrossRef]
- Amir, M.; Kumar, H.; Javed, S.A. Synthesis and pharmacological evaluation of condensed heterocyclic 6-substituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives of naproxen. Bioorg. Med. Chem. Lett. 2007, 17, 4504–4508. [Google Scholar] [CrossRef]
- Pastorin, G.; da Ros, T.; Bolcato, C.; Montopoli, C.; Moro, S.; Cacciari, B.; Baraldi, P.G.; Varani, K.; Borea, P.A.; Spalluto, G. Synthesis and biological studies of a new series of 5-heteroaryl carbamoylaminopyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidines as human A3 adenosine receptor antagonists. Influence of the heteroaryl substituent on binding affinity and molecular modeling investigations. J. Med. Chem. 2006, 49, 1720–1729. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, X.C. Synthesis and biological screening of some new 2,5-disubstitued 1,3,4-oxadiazoles. Int. J. Pharm. Pharm. Sci. 2011, 3, 110–114. [Google Scholar]
- Amir, M.; Shahani, S. Oxadiazole: A biologically important heterocycle. Indian J. Heterocycl. Chem. 2009, 1, 130–140. [Google Scholar]
- Shah, B.R.; Bhatt, J.J.; Patel, H.H.; Undavia, N.K.; Trivedi, P.B.; Desai, N.C. Synthesis of 2,3-disubstituted-3,1-quinazolin-4(4H)-ones as potential anticancer and anti-HIV agents. Indian J. Chem. 1995, 34B, 201–210. [Google Scholar]
- Hazarika, J.; Kataky, J.C.S. Synthesis of some novel 1,3,4-oxadiazole and its anti-bacterial and anti-fungal activity. Indian J. Heterocycl. Chem. 1998, 7, 83–92. [Google Scholar]
- Liszkiewicz, H.; Kowalska, M.W.; Wietrzyk, J.; Opolski, A. Synthesis and anti-proliferative activity in vitro of new 5-(2-amino-3-pyridyl)-2-thioxo-3H-1,3,4-oxadiazole derivatives. Indian J. Chem. 2003, 42B, 2846–2852. [Google Scholar]
- Mogilaiah, K.; Reddy, N.V. Microwave assisted heterocyclization: A rapidand efficient synthesis of 1,8-naphthyridinyl-1,3,4-oxadiazoles. Indian J. Chem. 2003, 42B, 2124–2125. [Google Scholar]
- Kode, N.R.; Eynde, J.J.V.; Mayence, A.; Wang, G.D.; Huang, T.L. Design and Synthesis of N1,N5-bis[4-(5-Alkyl-1,2,4-oxadiazol-3-yl)phenyl]glutaramides as potential antifungal prodrugs. Molecules 2013, 18, 11250–11263. [Google Scholar] [CrossRef]
- Hamdi, N.; Passarelli, V.; Romerosa, A. Synthesis, spectroscopy and electrochemistry of new 4-(4-acetyl-5-substituted-4,5-dihydro-1,3,4-oxodiazol-2-yl)methoxy)-2H-chromen-2-ones as a novel class of potential antibacterial and antioxidant derivatives. C. R. Chim. 2011, 14, 548–555. [Google Scholar] [CrossRef]
- Khan, M.S.Y.; Akhtar, M. Synthesis and analgesic activity of 2,5-disubstitued 1,3,4-oxadiazole. Indian J. Chem. 2003, 42B, 900–904. [Google Scholar]
- Gao, W.Q.; Li, Q.Y.; Chen, J.; Wang, Z.C.; Hua, C.L. Total synthesis of six 3,4-unsubstituted coumarins. Molecules 2013, 18, 15613–15623. [Google Scholar] [CrossRef]
- Penning-Van Beest, F.J.; Koerselman, J.; Herings, R.M. Risk of major bleeding during concomitant use of antibiotic drugs and coumarin anticoagulants. J. Thromb. Haemost. 2008, 6, 284–290. [Google Scholar]
- Kontogiorgis, C.; Hadjipavlou-Litina, D. Biological evaluation of several coumarin derivatives designed possible anti-inflammatory/antioxidant agents. J. EnzymeInhib. Med. Chem. 2003, 18, 63–69. [Google Scholar]
- Liu, Z.Q.; Yu, W.; Liu, Z.L. Antioxidative and prooxidative effects of coumarin derivatives on free radical initiated and photosensitized peroxidation of human low-density lipoprotein. Chem. Phys. Lipids 1999, 103, 125–135. [Google Scholar] [CrossRef]
- Smyth, T.; Ramachandran, V.N.; Smyth, W.F. A study of the antimicrobial activity of selected naturally occurring and synthetic coumarins. Int. J. Antimicrob. Ag. 2009, 33, 421–426. [Google Scholar] [CrossRef]
- Niu, X.; Xing, W.; Li, W.; Fan, T.; Hu, H.; Li, Y. Isofraxidin exhibited anti-inflammatory effects in vivo and inhibited TNF-α production in LPS-induced mouse peritoneal macrophages in vitro via the MAPK pathway. Int. Immunopharmacol. 2012, 14, 164–171. [Google Scholar] [CrossRef]
- Jiménez-Orozco, F.A.; Rosales, A.A.R.; Vega-López, A.; Domínguez-López, M.L.; García-Mondragón, M.J.; Maldonado-Espinoza, A.; Lemini, C.; Mendoza-Patiño, N.; Mandoki, J.J. Differential effects of esculetin and daphnetin on in vitro cell proliferation and in vivo estrogenicity. Eur. J. Pharmacol. 2011, 668, 35–41. [Google Scholar] [CrossRef]
- Liu, H.; Li, Z.; Yu, L.; Zhang, Y. Antitumor activity and mechanisms of scoparone. J. Zhong Guo Yao Li Tong Xun 2005, 22, 40–41. [Google Scholar]
- Liu, W.; Hua, J.; Zhou, J.; Zhang, H.; Zhu, H.; Cheng, Y.; Gust, R. Synthesis and in vitro antitumor activity of novel scopoletin derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 5008–5012. [Google Scholar]
- Rouesac, F.; Leclerc, A. An efficient synthesis of isofraxidin. Synth. Commun. 1993, 23, 1147–1153. [Google Scholar] [CrossRef]
- Chen, W. Total synthesis of isofraxidin. Chin. J. Med. Chem. 1996, 6, 269–271. [Google Scholar]
- Tanaka, H.; Kato, I.; Ito, K. Total synthesis of daphnetin, a coumarin lignold. Chem. Pharm. Bull. 1986, 34, 628–632. [Google Scholar] [CrossRef]
- Ahmed, B.; Khan, S.A.; Alam, T. Synthesis and antihepatotoxic activity of some heterocyclic compounds containing the 1,4-dioxane ring system. Pharmazie 2003, 58, 173–176. [Google Scholar]
- Hwu, J.R.; Singha, R.; Hong, S.C.; Chang, Y.H.; Das, A.R.; Vliegen, I.; de Clercq, E.; Neyts, J. Synthesis of new benzimidazole coumarin conjugates as anti-hepatitis C virus agents. Antivir. Res. 2008, 77, 157–162. [Google Scholar] [CrossRef]
- Neyts, J.; de Clercq, E.; Singha, R.; Chang, Y.H.; Das, A.; Chakraborty, S.; Hong, S.C.; Tsay, S.C.; Hsu, M.H.; Hwu, J.R. Structure–activity relationship of new anti-hepatitis C virus agents: Heterobicycle-coumarin conjugates. J. Med. Chem. 2009, 52, 1486–1490. [Google Scholar] [CrossRef]
- Hwu, J.R.; Lin, S.Y.; Tsay, S.C.; de Clercq, E.; Leyssen, P.; Neyts, J. Coumarin-purine ribofuranoside conjugates as new agents against hepatitis C virus. J. Med. Chem. 2011, 54, 2114–2126. [Google Scholar] [CrossRef]
- Tsay, S.C.; Hwu, J.R.; Singha, R.; Huang, W.C.; Chang, Y.H.; Hsu, M.H.; Shieh, F.K.; Lin, C.C.; Hwang, K.C.; Horng, J.C.; et al. Coumarins hinged directly on benzimidazoles and their ribofuranosides to inhibit hepatitis C virus. Eur. J. Med. Chem. 2013, 63, 290–293. [Google Scholar] [CrossRef]
- Aslam, K.; Khosa, M.K.; Jahan, N.; Nosheen, S. Synthesis and application of coumarin. Pak. J. Pharm. 2010, 23, 449–454. [Google Scholar]
- Mahesh, K.P.; Swapnil, S.M.; Manikrao, M.S. Coumarin synthesises via Pechmann condensation in Lewis acidic chloroaluminate ionic liquid. Tetrahedron Lett. 2001, 42, 9285–9287. [Google Scholar]
- Shaabani, A.; Ghadari, R.; Rahmati, A.; Rezayan, A.H. Coumarin synthesis via knoevenagel condensation reation in 1,1,3,3-N,N,N'N'-tetramethylguanidinium trifluoroacetate ionic liquid. J. Iran Chem. Soc. 2009, 6, 710–714. [Google Scholar] [CrossRef]
- Harayama, T. Synthetic studies on aromatic heterocyclic compounds. Pharm. Soc. Jpn. 2006, 126, 543–564. [Google Scholar]
- Kraus, G.A.; Cui, W.; Seo, Y.H. The reaction of aryl triflates and aryl pivalates with electrophiles. The triflate as a meta-directing group. Tetrahedron Lett. 2002, 43, 7077–7078. [Google Scholar] [CrossRef]
- Castanet, A.S.; Colobert, F.; Broutin, P.E. Mild and regioselective iodination of electron-rich aromatics with N-iodosuccinimide and catalytic trifluoroacetic acid. Tetrahedron Lett. 2002, 43, 5047–5048. [Google Scholar] [CrossRef]
- Zabradnik, M. The Production and Application of Fluorescent Brightening Agents; John Wiley and Sons: New York, NY, USA, 1992. [Google Scholar]
- Heravi, M.; Sadjadi, S.; Oskooie, H.; Shoar, R.; Bamoharram, F. The synthesis of coumarin-3-carboxylic acids and 3-acetyl-coumarin derivatives using heteropolyacids as heterogeneous and recyclable catalysts. Catal. Commun. 2008, 9, 470–474. [Google Scholar] [CrossRef]
- Chen, W.A. Convenient total synthesis of isofraxidin, a natural coumarin. Indian J. Chem. Sect. BOrg. Chem. Incl. Med. Chem. 1996, 35, 1085–1087. [Google Scholar]
- Demyttenaere, J.; Syngel, K.V.; Markusse, A.P.; Vervisch, S.; Debenedetti, S.; de Kimpe, N. Synthesis of 6-methoxy-4H-1-benzopyran-7-ol, a character donating component of the fragrance of Wisteria sinensis. Tetrahedron 2002, 58, 2163–2166. [Google Scholar] [CrossRef]
- Crosby, D.G. Improved synthesis of scopoletin. J. Org. Chem. 1961, 26, 1215–1217. [Google Scholar] [CrossRef]
- Hauer, H.; Ritter, T.; Grotemeier, G. An improved and large scale synthesis of the natural coumarin scopoletin. Arch. Pharm. 1995, 328, 737–738. [Google Scholar] [CrossRef]
- Hao, S.; Zong, G.; Wei, Y.; Meng, Z. Synthesis and pesticidal activity of 6,7-dimethoxy coumarin. J. Qingdao Agric. Univ. 2007, 24, 241–244. [Google Scholar]
- Zhang, S.; Ma, J.; Chen, S.; Li, H.; Xin, J. Improved synthesis techniques of 6,7-dimethoxy coumarin. J. Hebei Univ. Sci. Technol. 2007, 28, 24–28. [Google Scholar]
- Manoharan, A.; Sugumar, M.; Kumar, A.; Jose, H.; Mathai, D.; Khilnani, G.C.; Kapil, A.; Francis, G.; Radhakrishnan, K.; Dutta, T.K; et al. Phenotypic & molecular characterization of AmpC β-lactamases among Escherichia coli, Klebsiella spp. & Enterobacter spp. from five Indian Medical Centers. Indian J. Med. Res. 2012, 135, 359–364. [Google Scholar]
- Scorzoni, L.; de Lucas, M.P.; Mesa-Arango, A.C.; Fusco-Almeida, A.M.; Lozano, E.; Cuenca-Estrella, M.; Mendes-Giannini, M.J.; Zaragoza, O. Antifungal efficacy during Candida krusei infection in non-conventional models correlates with the yeast in vitro susceptibility profile. PLoS One 2013, 8, e60047. [Google Scholar] [CrossRef]
- Grings, M.; Moura, A.P.; Parmeggiani, B.; Marcowich, G.F.; Amaral, A.U.; de Souza Wyse, A.T.; Wajner, M.; Leipnitz, G. Disturbance of brain energy and redox homeostasis provoked by sulfite and thiosulfate: Potential pathomechanisms involved in the neuropathology of sulfite oxidase deficiency. Gene 2013, 531, 191–198. [Google Scholar] [CrossRef]
- Ravi, K.J.; Ganga, R.B. Evaluation of in vitro antioxidant activity of Spathodea campnulata leaves. Int. J. Biol. Pharm. Res. 2013, 4, 328–332. [Google Scholar]
- Sample Availability: Samples of the compounds a1, a2 and 2–5 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Ayed, A.S.; Hamdi, N. A New and Efficient Method for the Synthesis of Novel 3-Acetyl Coumarins Oxadiazoles Derivatives with Expected Biological Activity. Molecules 2014, 19, 911-924. https://doi.org/10.3390/molecules19010911
Al-Ayed AS, Hamdi N. A New and Efficient Method for the Synthesis of Novel 3-Acetyl Coumarins Oxadiazoles Derivatives with Expected Biological Activity. Molecules. 2014; 19(1):911-924. https://doi.org/10.3390/molecules19010911
Chicago/Turabian StyleAl-Ayed, Abdullah Sulaiman, and Naceur Hamdi. 2014. "A New and Efficient Method for the Synthesis of Novel 3-Acetyl Coumarins Oxadiazoles Derivatives with Expected Biological Activity" Molecules 19, no. 1: 911-924. https://doi.org/10.3390/molecules19010911