Two New Secondary Metabolites from Xylaria sp. cfcc 87468


 ,
,
Abstract
:1. Introduction
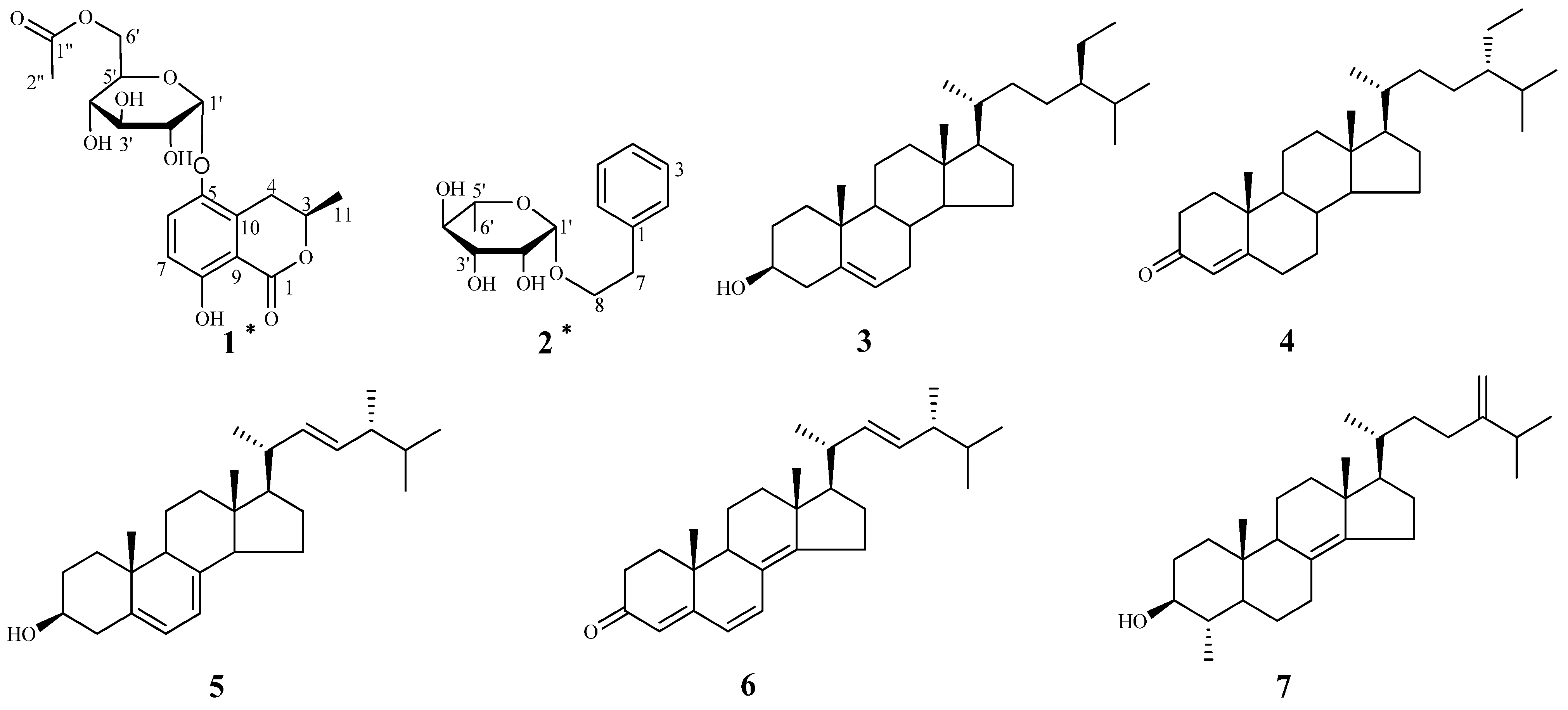
2. Results and Discussion
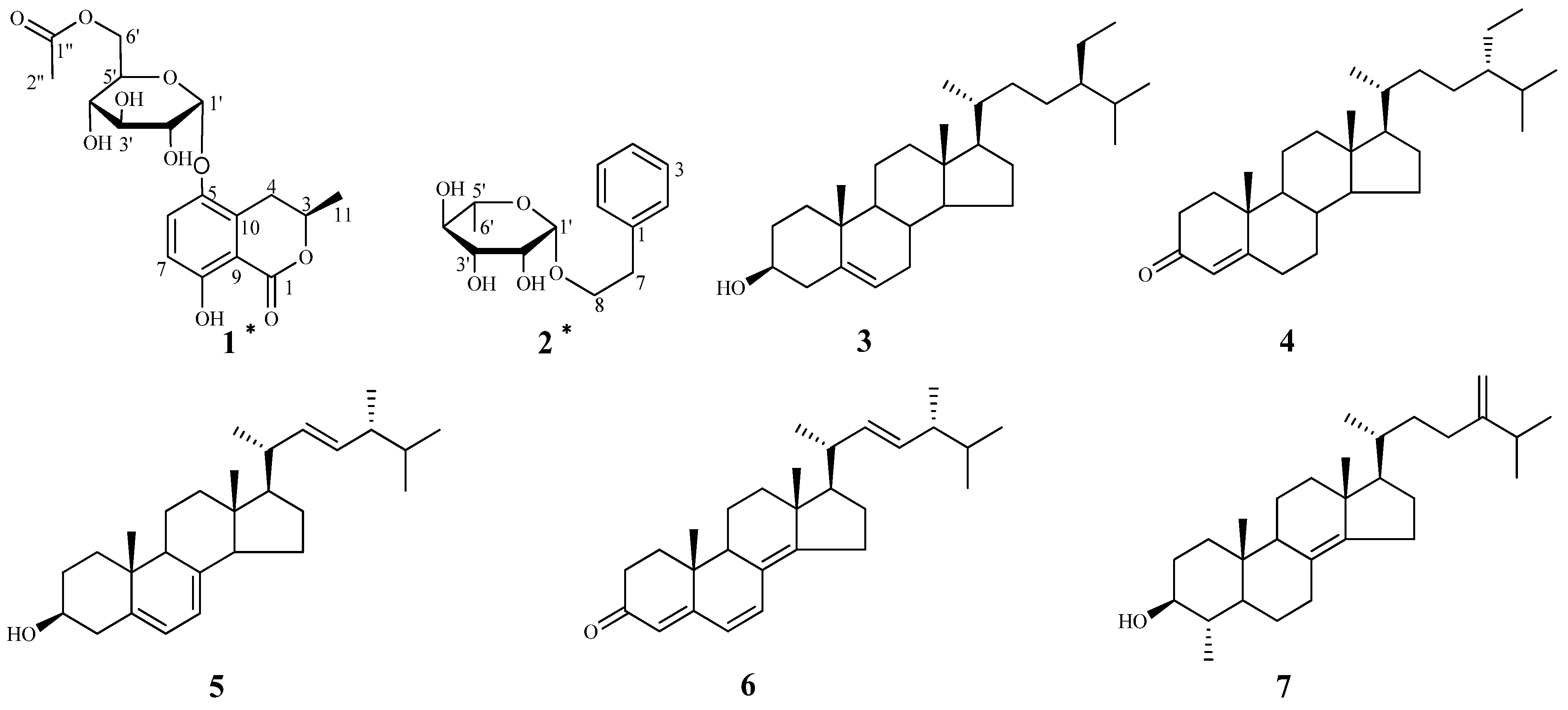
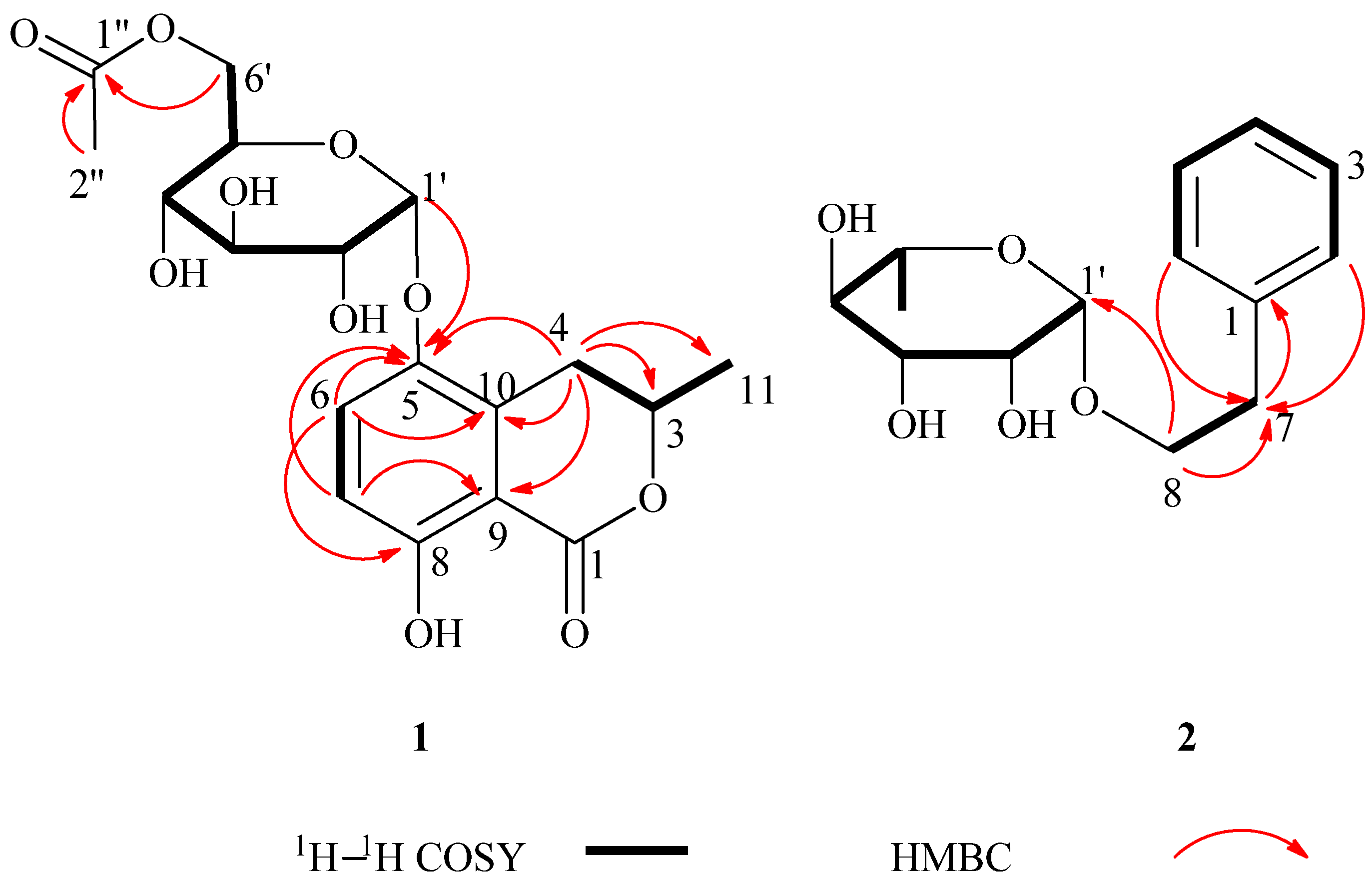
 +60 (c = 1.33, CH3OH). A molecular formula of C18H22O10 was assigned based on the interpretation of HRESIMS peak at m/z 421.1097 [M+Na]+ (calcd. 421.1105). Its IR spectrum showed characteristic hydroxyl group (3397 cm–1), and two ester carbonyl group (1676 and 1737 cm–1) absorptions. The 1H-NMR data of 1 (Table 1) showed two aromatic proton signals at δH 6.83 (d, J = 9.2 Hz, 1H) and 7.45 (d, J = 9.2 Hz, 1H), two methyls at δH 1.98 (s, 3H) and 1.52 (d, J = 6.3 Hz, 3H), and an oxygenated proton at δH 5.35 (d, J = 3.7 Hz, 1H). The 13C-NMR and DEPT spectra of compound 1 displayed 18 carbon signals, including two methyls, two methylenes (one oxygenated methylene), six methines, six aromatic carbons (four quaternary carbons), and two ester carbonyl groups. One set of proton signals at δH 3.3–4.3, 5.35, and their corresponding carbons resonating at δC 64.9, 71.9, 72.1, 73.2, 74.8, and 100.5, suggested the presence of a hexosyl sugar moiety in the molecule. The 1H-NMR spectrum exhibited protons signals at δH 2.69 (dd, J = 17.0, 11.7 Hz, H-4a), 3.46 (dd, J = 17.0, 3.3 Hz, H-4b), and 4.72 (m, H-3).
+60 (c = 1.33, CH3OH). A molecular formula of C18H22O10 was assigned based on the interpretation of HRESIMS peak at m/z 421.1097 [M+Na]+ (calcd. 421.1105). Its IR spectrum showed characteristic hydroxyl group (3397 cm–1), and two ester carbonyl group (1676 and 1737 cm–1) absorptions. The 1H-NMR data of 1 (Table 1) showed two aromatic proton signals at δH 6.83 (d, J = 9.2 Hz, 1H) and 7.45 (d, J = 9.2 Hz, 1H), two methyls at δH 1.98 (s, 3H) and 1.52 (d, J = 6.3 Hz, 3H), and an oxygenated proton at δH 5.35 (d, J = 3.7 Hz, 1H). The 13C-NMR and DEPT spectra of compound 1 displayed 18 carbon signals, including two methyls, two methylenes (one oxygenated methylene), six methines, six aromatic carbons (four quaternary carbons), and two ester carbonyl groups. One set of proton signals at δH 3.3–4.3, 5.35, and their corresponding carbons resonating at δC 64.9, 71.9, 72.1, 73.2, 74.8, and 100.5, suggested the presence of a hexosyl sugar moiety in the molecule. The 1H-NMR spectrum exhibited protons signals at δH 2.69 (dd, J = 17.0, 11.7 Hz, H-4a), 3.46 (dd, J = 17.0, 3.3 Hz, H-4b), and 4.72 (m, H-3).
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 171.4, s | 140.4, s | ||
| 2 | 7.26 (overlap, 1H) | 129.3, d | ||
| 3 | 4.72 (m, 1H) | 77.8, d | 7.24 (overlap, 1H) | 129.9, d |
| 4 | 3.46 (dd, J = 17.0, 3.3 Hz, 1H) | 29.4, t | 7.19 (overlap, 1H) | 127.2, d |
| 2.69 (dd, J = 17.0, 11.7 Hz, 1H) | ||||
| 5 | 146.7, s | 7.24 (overlap, 1H) | 129.9, d | |
| 6 | 6.83 (d, J = 9.2 Hz, 1H) | 116.7, d | 7.26 (overlap, 1H) | 129.3, d |
| 7 | 7.45 (d, J = 9.2 Hz, 1H) | 126.7, d | 2.86 (t, J = 6.7 Hz, 2H) | 37.1, t |
| 8 | 158.5, s | 3.85 (dt, J = 9.7, 6.9 Hz, 1H) | 69.4, t | |
| 3.63 (dt, J = 9.7, 6.7 Hz, 1H) | ||||
| 9 | 130.6, s | |||
| 10 | 109.5, s | |||
| 11 | 1.52 (d, J = 6.3 Hz, 3H) | 21.1, q | ||
| 1'' | 172.6, s | |||
| 2'' | 1.98 (s, 3H) | 20.7, q | ||
| 1' | 5.35 (d, J = 3.7 Hz, 1H) | 100.5, d | 4.65 (d, J = 1.5 Hz, 1H) | 101.5, d |
| 2' | 3.60 (dd, J = 9.7, 3.7 Hz, 1H) | 73.2, d | 3.77 (dd, J = 3.3, 1.7 Hz, 1H) | 72.2, d |
| 3' | 3.82 (m, 1H) | 74.8, d | 3.60 (dd, J = 5.9, 3.2 Hz, 1H) | 72.4, d |
| 4' | 3.36 (dd, J = 10.0, 8.9 Hz, 1H) | 71.9, d | 3.35 (d, J = 9.2 Hz, 1H) | 73.8, d |
| 5' | 3.87 (m, 1H) | 72.1, d | 3.40 (dd, J = 9.4, 6.0 Hz, 1H) | 69.7, d |
| 6' | 4.36 (dd, J = 11.8, 2.1 Hz, 1H) | 64.9, t | 1.19 (d, J = 6.0 Hz, 3H) | 17.9, q |
| 4.18 (dd, J = 11.9, 6.7 Hz, 1H) | ||||
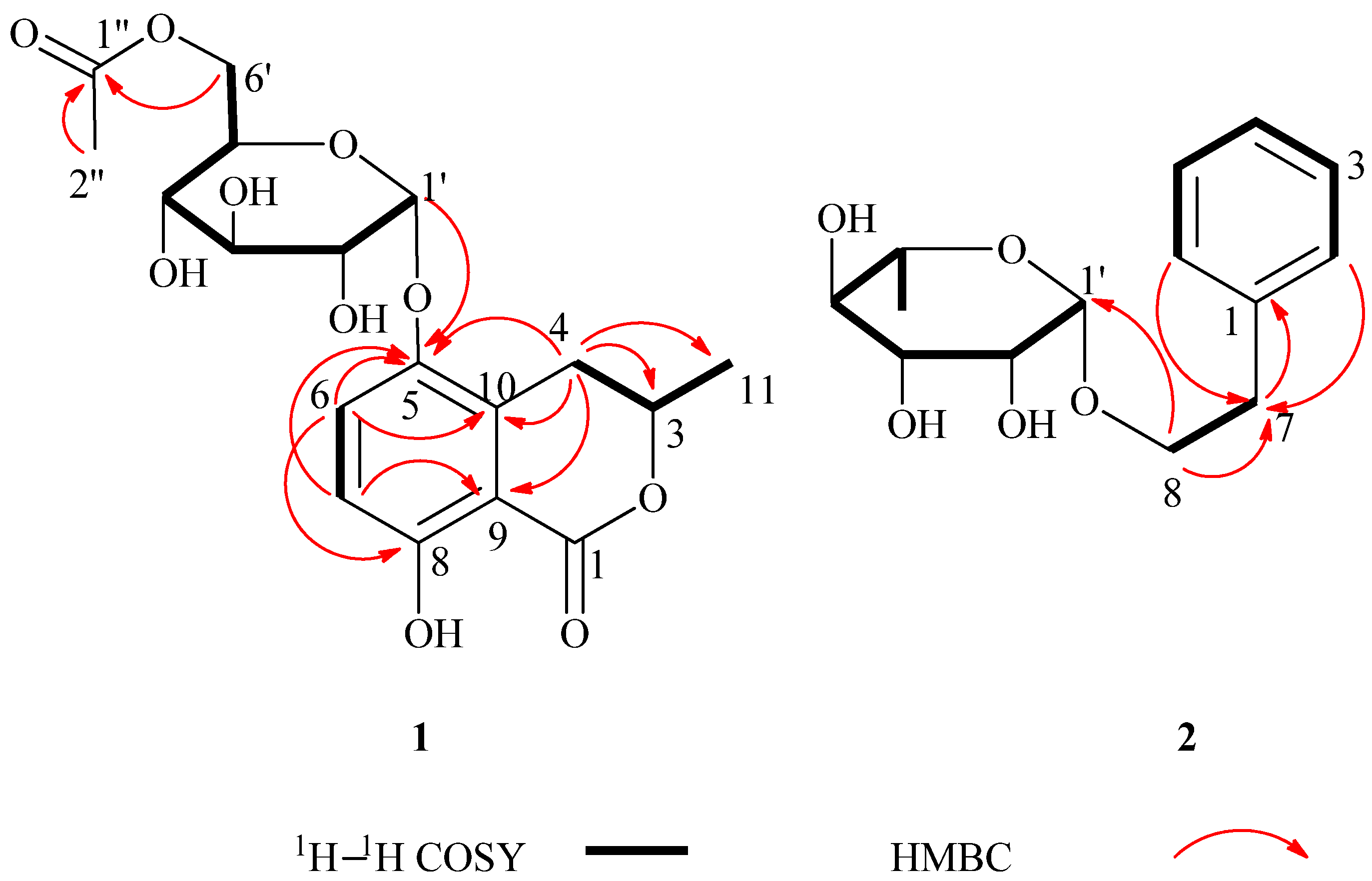 −46 (c = 4.83, CH3OH). The IR spectrum of 2 showed absorptions due to hydroxyl (3398 cm−1) and aromatic (1452 and 1497 cm−1) functionalities. The 1H-NMR data of 2 (Table 1) showed a set of monosubstituted aromatic ring signals at δH 7.15–7.28 (5H, overlapped), and a methyl doublet at δH 1.19 (d, J = 6.0 Hz, 3H), and oxygenated proton at δH 4.65 (d, J = 1.5 Hz, 1H). The 13C-NMR and DEPT showed six aromatic carbons (one quaternary carbon), two methylenes (one oxygenated), five methine carbons (three oxygenated and one anomeric), and one methyl group. According to the signals of six aromatic carbons and 1H–1H COSY signal at H-8 (δH 3.63, 3.85) with H-7 (δH 2.86) indicated compound 2 was a phenylethanol derivative. A series of proton signals in the range of δH 3.3–3.8, 4.65 (d, J = 1.5 Hz, 1H), and 1.19 (d, J = 6.0 Hz, 3H), and their corresponding carbons at δC 69.7, 72.2, 72.4, 73.8, 101.5, and 17.9 were observed in the 1H- and 13C-NMR spectra, which indicated the existence of a α-rhamnose moiety. After acid hydrolysis of 2, the sugar moiety was identified to be α-l-rhamnose according to the GC analysis. In the HMBC spectrum, the correlation of H-8 (δH 3.63, 3.85) with C-1' (δC 101.5) showed that the α-rhamnose was attached to C-8 (Figure 2), hence compound 2 was identified as (−)-phenylethyl-8-O-α-l-rhamnopyranoside.
−46 (c = 4.83, CH3OH). The IR spectrum of 2 showed absorptions due to hydroxyl (3398 cm−1) and aromatic (1452 and 1497 cm−1) functionalities. The 1H-NMR data of 2 (Table 1) showed a set of monosubstituted aromatic ring signals at δH 7.15–7.28 (5H, overlapped), and a methyl doublet at δH 1.19 (d, J = 6.0 Hz, 3H), and oxygenated proton at δH 4.65 (d, J = 1.5 Hz, 1H). The 13C-NMR and DEPT showed six aromatic carbons (one quaternary carbon), two methylenes (one oxygenated), five methine carbons (three oxygenated and one anomeric), and one methyl group. According to the signals of six aromatic carbons and 1H–1H COSY signal at H-8 (δH 3.63, 3.85) with H-7 (δH 2.86) indicated compound 2 was a phenylethanol derivative. A series of proton signals in the range of δH 3.3–3.8, 4.65 (d, J = 1.5 Hz, 1H), and 1.19 (d, J = 6.0 Hz, 3H), and their corresponding carbons at δC 69.7, 72.2, 72.4, 73.8, 101.5, and 17.9 were observed in the 1H- and 13C-NMR spectra, which indicated the existence of a α-rhamnose moiety. After acid hydrolysis of 2, the sugar moiety was identified to be α-l-rhamnose according to the GC analysis. In the HMBC spectrum, the correlation of H-8 (δH 3.63, 3.85) with C-1' (δC 101.5) showed that the α-rhamnose was attached to C-8 (Figure 2), hence compound 2 was identified as (−)-phenylethyl-8-O-α-l-rhamnopyranoside.3. Experimental
3.1. General Procedures
3.2. Fungal Material and Aphylogenetic Analysis of ITS 1–4 Gene Sequence
3.3. Fermentation and Isolation
3.4. Hydrolysis and Determination of the Absolute Configuration of the Sugar Moiety
3.5. Spectroscopic Data
+60 (c = 1.33, CH3OH); CD (MeOH): λmax nm (Δε)= 205.0 (+2.33), 209.0 (+2.24), 226.6 (−2.02), 255.0 (−3.75); UV (MeOH) λmax (lg ε) 213 (4.47), 258 (3.70) nm; IR (film) vmax 3397, 2921, 1737, 1676, 1476, 1390, 1248, 1128, 1050, 910, 872 cm−1; 1H and 13C-NMR data, see Table 1; HRESIMS m/z 421.1097 [M + Na]+ (calcd for C18H22O10Na, 421.1105). −46 (c = 4.83, CH3OH); UV (MeOH) λmax (lg ε) 218 (2.85), 333 (2.13) nm; IR (film) νmax 3398, 2930, 1497, 1542, 1130, 1092, 1053, 981, 806, 749, 699 cm−1; 1H and 13C-NMR data, see Table 1; HRESIMS m/z 291.1200 [M+Na]+ (calcd for C14H20O5Na, 291.1203).4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Strobel, G.; Daisy, B. Bioprospecting for microbial endophytes and their natural products. Mol. Biol. Rev. 2003, 67, 491–502. [Google Scholar] [CrossRef]
- Strobel, G.; Daisy, B.; Castillo, U.; Harper, J. Natural products from endophytic microorganisms. J. Nat. Prod. 2004, 67, 257–268. [Google Scholar] [CrossRef]
- Stadler, M.; Hellwig, V.; Pandalai, S.G. Chemotaxonomy of the Xylariaceae and remarkable bioactive compounds from Xylariales and their associated asexual stages. Recent Res. Dev. Phytochem. 2005, 9, 41–93. [Google Scholar]
- Whalley, A.; Edwards, R.L. Secondary metabolites and systematic arrangement within the xylariaceae. Can. J. Bot. 1995, 73, s802–s810. [Google Scholar] [CrossRef]
- Lutz-Kutschera, G.; Engelmeier, D.; Hadacek, F.; Werner, A.; Greger, H.; Hofer, O. Synthesis of side chain substituted 3-butylisocoumarins and absolute configurations of natural isocoumarins from Artemisia dracunculus. Monatshefte Chem. 2003, 134, 1195–1206. [Google Scholar] [CrossRef]
- Rukachaisirikul, V.; Buadam, S.; Sukpondma, Y.; Phongpaichit, S.; Sakayaroj, J.; Hutadilok-Towatana, N. Indanone and mellein derivatives from the Garcinia-derived fungus Xylaria sp. PSU–G12. Phytochem. Lett. 2013, 6, 135–138. [Google Scholar] [CrossRef]
- Lu, C.; Lin, X.; Shen, Y. A new dihydroisocoumarin from the strain Aspergillus sp. CMM, an endophytic fungus of Cephalotaxus mannii. Chem. Nat. Compd. 2008, 44, 569–571. [Google Scholar] [CrossRef]
- Venkatasubbaiah, P.; Chilton, W.S. Phytotoxins of Botryosphaeria obtusa. J. Nat. Prod. 1990, 53, 1628–1630. [Google Scholar] [CrossRef]
- Lai, Y.; Ding, S.; Qian, H.; Zhang, J.; Xue, Y.; Luo, Z.; Yao, G.; Zhang, Y. A new lignan glucoside from the whole plants of Salvia Scapiformis. Molecules 2013, 18, 11377–11383. [Google Scholar] [CrossRef]
- Yang, M.H.; Luo, J.G.; Huang, X.F.; Kong, L.Y. Flavonol glycosides with α-d-aldohexoses from Rhododendron irroratum. Nat. Prod. Res. 2010, 24, 920–925. [Google Scholar] [CrossRef]
- Li, W.H.; Chang, S.T.; Chang, S.C.; Chang, H.T. Isolation of antibacterial diterpenoids from Cryptomeria japonica bark. Nat. Prod. Res. 2008, 22, 1085–1093. [Google Scholar] [CrossRef]
- Lee, I.K.; Kim, M.A.; Lee, S.Y.; Hong, J.K.; Lee, J.H.; Lee, K.R. Phytochemical constituents of Schizonepeta tenuifolia briquet. Nat. Prod. Sci. 2008, 14, 100–106. [Google Scholar]
- Shirane, N.; Takenaka, H.; Ueda, K.; Hashimoto, Y.; Katoh, K.; Ishii, H. Sterol analysis of DMI-resistant and-sensitive strains of venturia inaequalis. Phytochemistry 1996, 41, 1301–1308. [Google Scholar] [CrossRef]
- Ciminiello, P.; Fattorusso, E.; Magno, S.; Mangoni, A.; Pansini, M. A novel conjugated ketosteroid from the marine sponge Dictyonella incisa. J. Nat. Prod. 1989, 52, 1331–1333. [Google Scholar] [CrossRef]
- Giner, J.; Zhao, H.; Beach, D.H.; Parish, E.J.; Jayasimhulu, K.; Kaneshiro, E.S. Comprehensive and definitive structural identities of Pneumocystis carinii sterols. J. Lipid. Res. 2002, 43, 1114–1124. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, Y.; Castlebury, L.A.; Cerniglia, C.E. A method for the large scale isolation of high transformation efficiency fungal genomic DNA. FEMS Microbiol. Lett. 1996, 145, 261–265. [Google Scholar] [CrossRef]
- Hsieh, H.M.; Lin, C.R.; Fang, M.J.; Rogers, J.D.; Fournier, J.; Lechat, C.; Ju, Y.M. Phylogenetic status of Xylaria subgenus Pseudoxylaria among taxa of the subfamily Xylarioideae (Xylariaceae) and phylogeny of the taxa involved in the subfamily. Mol. Phylogenet. Evol. 2010, 54, 957–969. [Google Scholar] [CrossRef]
- Stadler, M.; Kuhnert, E.; Peršoh, D.; Fournier, J. The Xylariaceae as model example for a unified nomenclature following the “One Fungus–One Name” (1F1N) Concept. Mycol. Int. J. Fungal. Biol. 2013, 4, 5–21. [Google Scholar]
- Sample Availability: Samples of the compounds 1–7 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, F.; Han, S.; Hu, S.; Xue, Y.; Wang, J.; Xu, H.; Chen, L.; Zhang, G.; Zhang, Y. Two New Secondary Metabolites from Xylaria sp. cfcc 87468. Molecules 2014, 19, 1250-1257. https://doi.org/10.3390/molecules19011250
Wang F, Han S, Hu S, Xue Y, Wang J, Xu H, Chen L, Zhang G, Zhang Y. Two New Secondary Metabolites from Xylaria sp. cfcc 87468. Molecules. 2014; 19(1):1250-1257. https://doi.org/10.3390/molecules19011250
Chicago/Turabian StyleWang, Fuqian, Shishi Han, Song Hu, Yongbo Xue, Jianping Wang, Hongfeng Xu, Lu Chen, Geng Zhang, and Yonghui Zhang. 2014. "Two New Secondary Metabolites from Xylaria sp. cfcc 87468" Molecules 19, no. 1: 1250-1257. https://doi.org/10.3390/molecules19011250
APA StyleWang, F., Han, S., Hu, S., Xue, Y., Wang, J., Xu, H., Chen, L., Zhang, G., & Zhang, Y. (2014). Two New Secondary Metabolites from Xylaria sp. cfcc 87468. Molecules, 19(1), 1250-1257. https://doi.org/10.3390/molecules19011250