Validation of a Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry Method for Determination of All-Trans Retinoic Acid in Human Plasma and Its Application to a Bioequivalence Study

Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Development and Optimization
2.1.1. Optimization of Sample Preparation
2.1.2. Optimization of the LC-MS-MS Condition
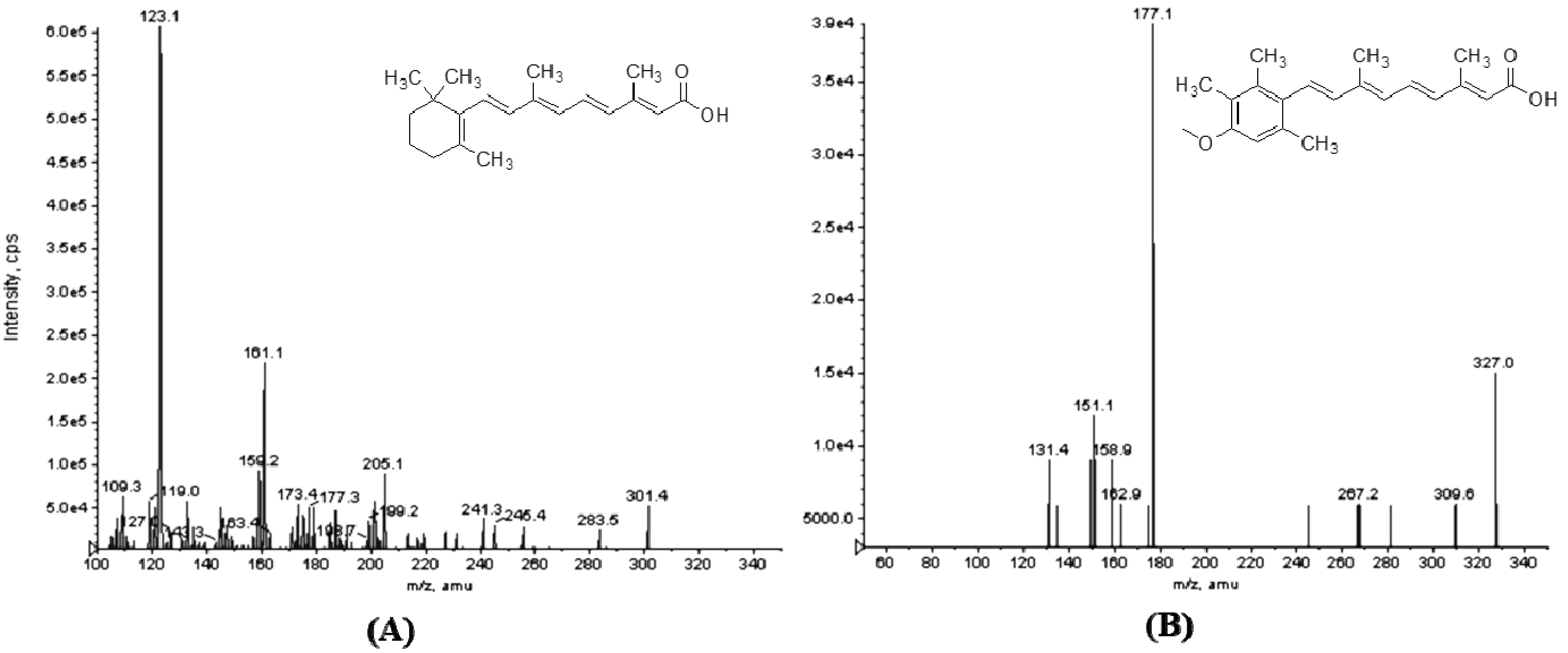
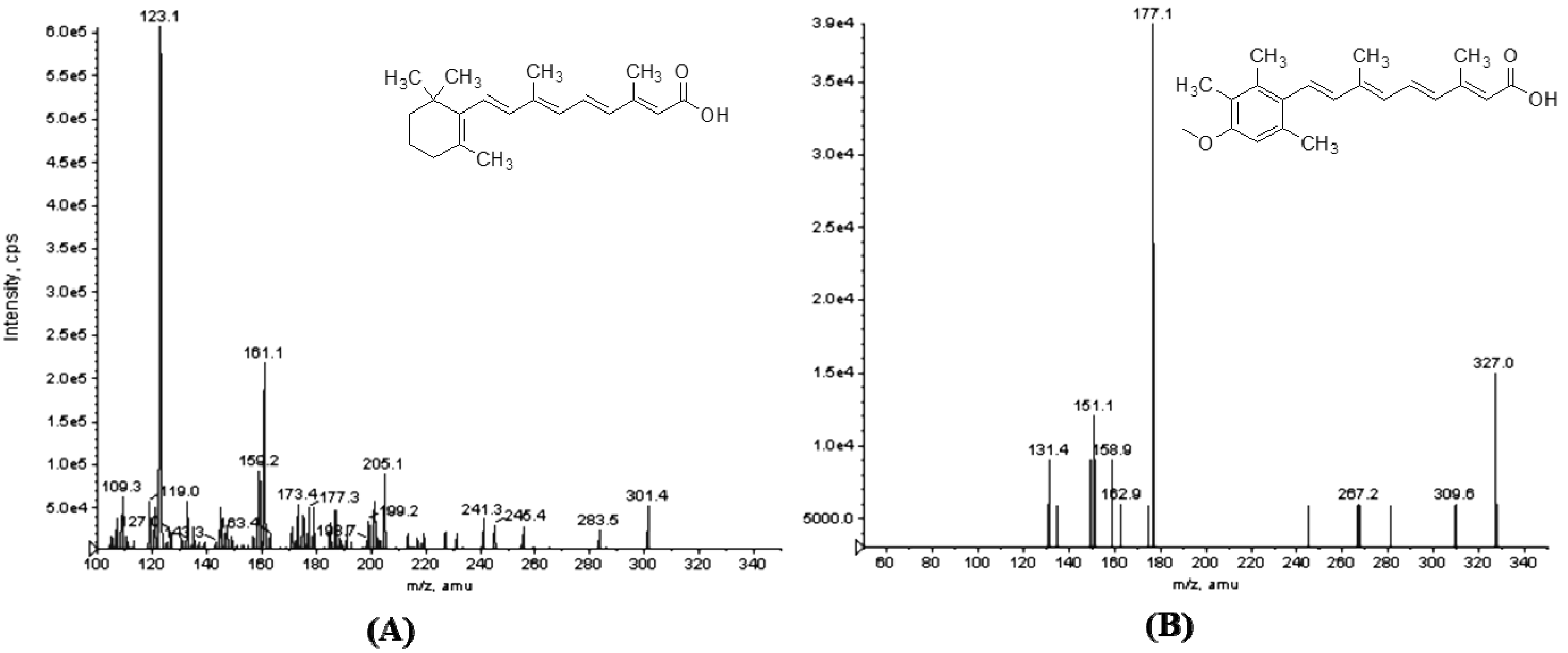
2.2. Method Validation
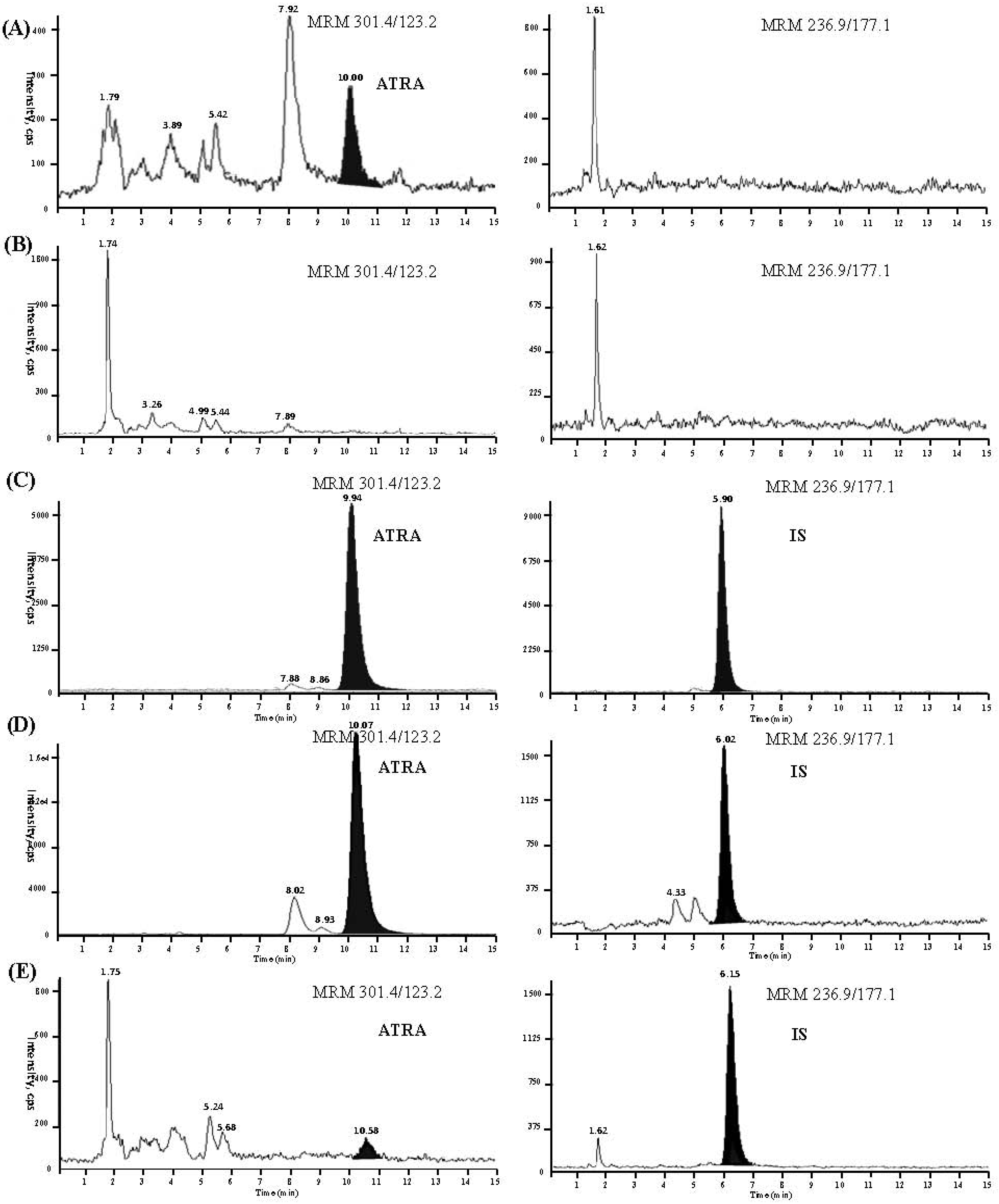
2.2.1. Selectivity
2.2.2. Linearity and Lower Limit of Quantification
2.2.3. Precision and Accuracy
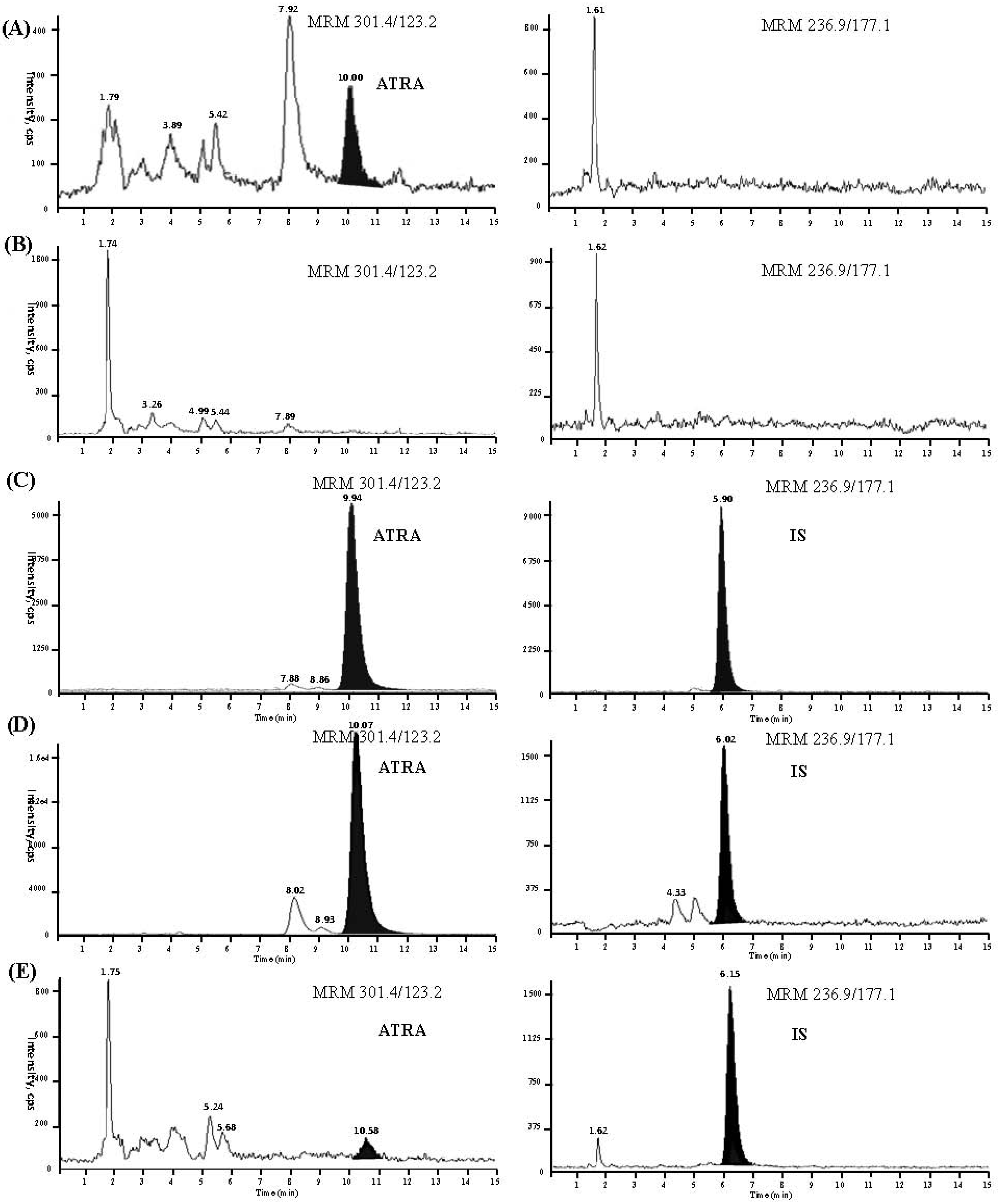

{kind=link}
{kind=link}
{kind=link}
| Theoretical conc. (ng/mL) | Intra-batch (n = 5) | Inter-batch (n = 15) | ||||
|---|---|---|---|---|---|---|
| Detected conc. (mean ± SD) (ng/mL) | Accuracy (%) | Precision (%) | Detected conc. (mean ± SD) (ng/mL) | Accuracy (%) | Precision (%) | |
| 0.90 | 0.96 ± 0.06 | 106.19 | 5.90 | 0.95 ± 0.07 | 105.48 | 7.46 |
| 12.06 | 11.94 ± 0.35 | 98.98 | 2.95 | 12.35 ± 0.65 | 102.39 | 5.30 |
| 173.60 | 176.50 ± 5.90 | 101.66 | 3.34 | 178.99 ± 8.07 | 103.10 | 4.51 |
2.2.4. Extraction Recovery and Matrix Effect
| Analyte | Concentration (ng/mL) | Recovery (C/B, %) | Matrix effect (B/A, %) |
|---|---|---|---|
| ATRA | 0.90 | 81.61 ± 8.66 | 74.33 ± 4.88 |
| 12.06 | 81.40 ± 7.77 | 77.64 ± 3.39 | |
| 173.60 | 75.63 ± 1.63 | 86.38 ± 6.65 | |
| IS | 114.40 | 76.27 ± 11.41 | 92.24 ± 2.42 |
2.2.5. Stability
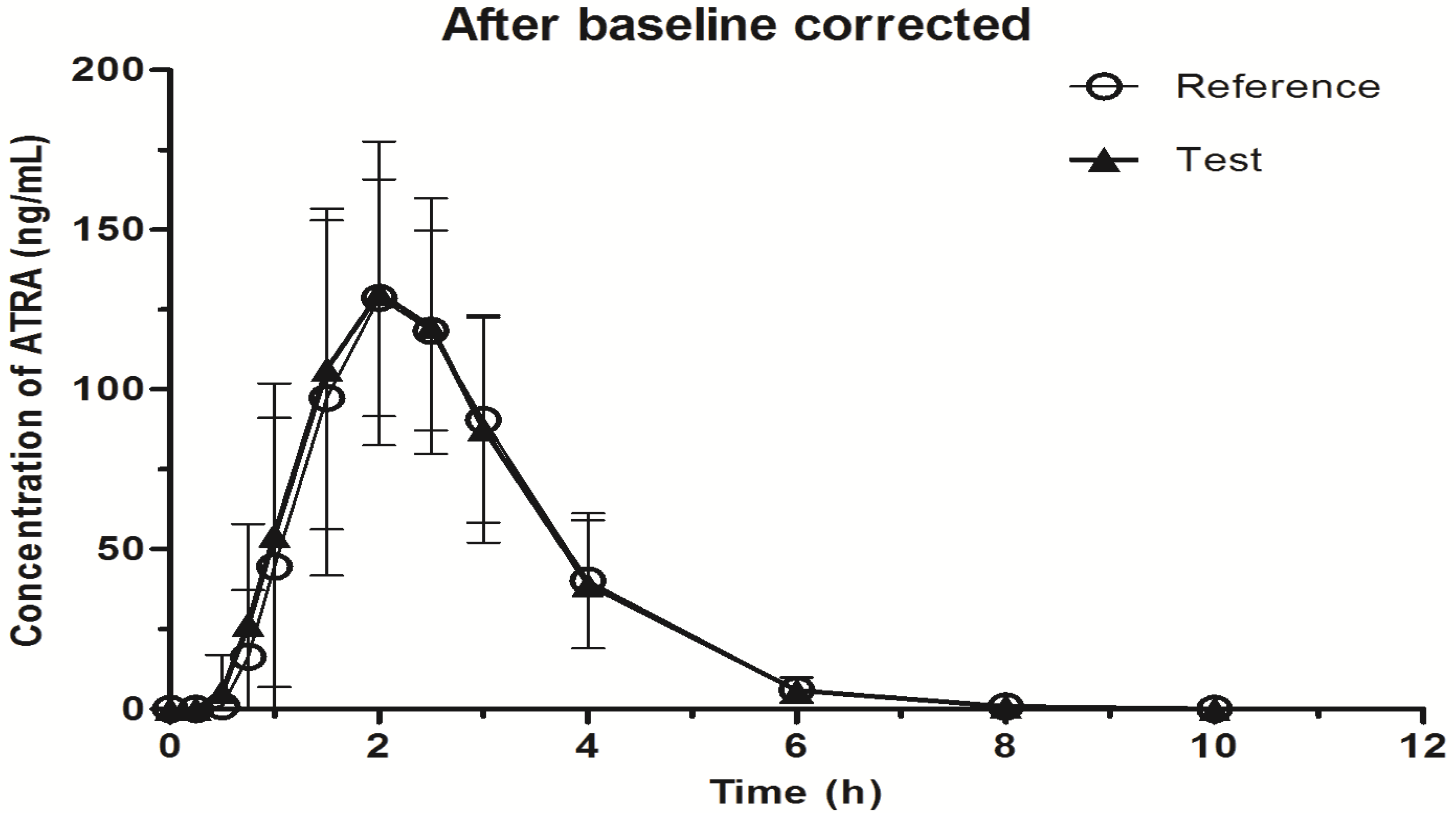
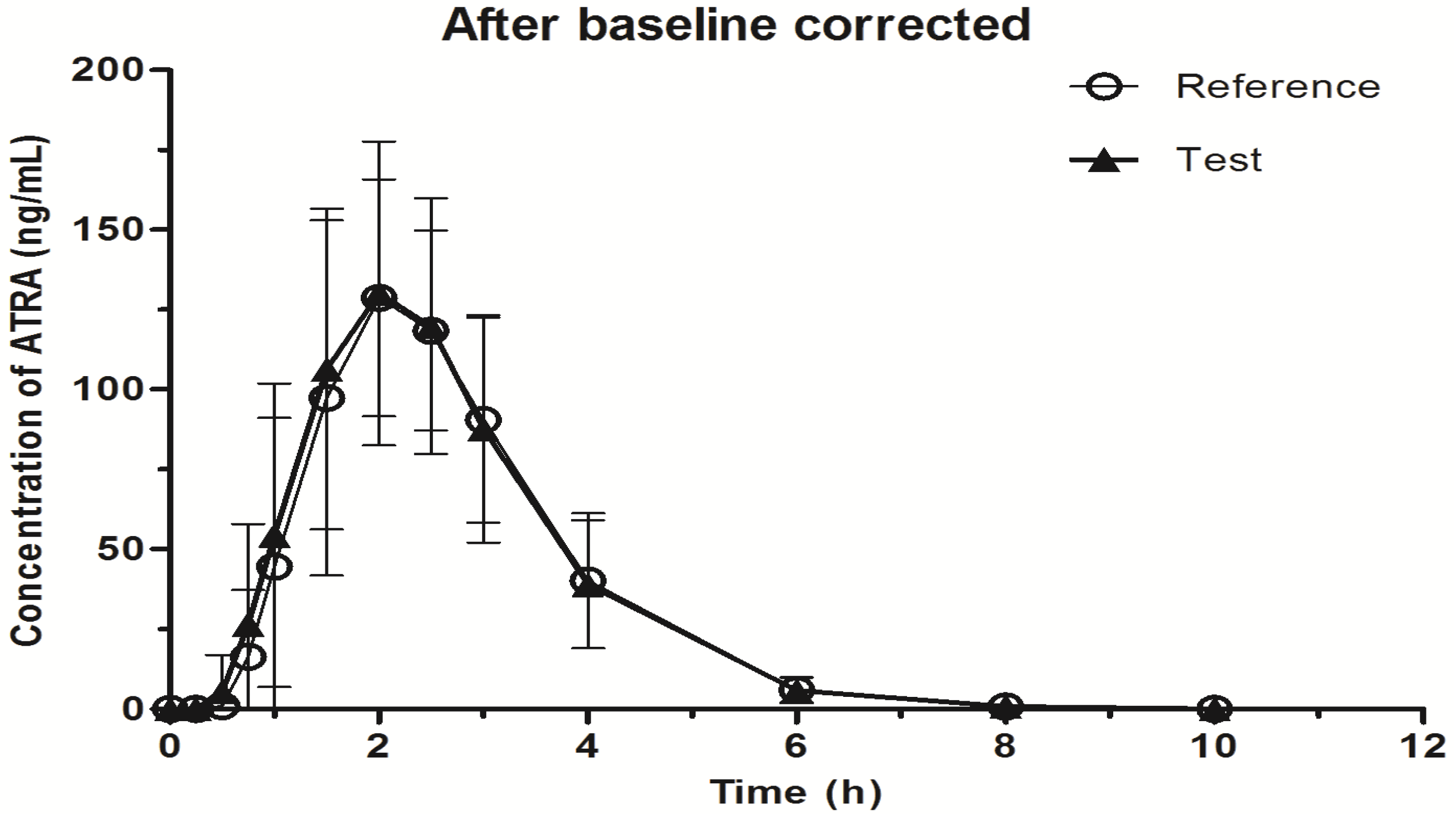
2.3. Application to Biological Samples
| Storage conditions | Theoretical conc. (ng/mL) | Detected conc. (mean ± SD) (ng/mL) | RSD (%) | RE (%) |
|---|---|---|---|---|
| Freeze-thaw (three cycles, −40 °C) | 0.90 | 0.87 ± 0.07 | 8.57 | −3.56 |
| 12.06 | 12.14 ± 0.33 | 2.73 | 0.70 | |
| 173.60 | 183.60 ± 13.65 | 7.44 | 5.78 | |
| Auto sampler (48 h, 25 °C) | 0.90 | 0.99 ± 0.03 | 3.23 | 9.17 |
| 12.06 | 12.99 ± 0.55 | 4.25 | 7.71 | |
| 173.60 | 180.10 ± 15.78 | 8.76 | 3.76 | |
| Short-term (4 h, 25 °C) | 0.90 | 1.00 ± 0.03 | 2.97 | 10.72 |
| 12.06 | 11.88 ± 0.17 | 1.42 | −1.53 | |
| 173.60 | 167.10 ± 10.55 | 6.31 | −3.74 | |
| Long-term (65 days, −40 °C) | 0.90 | 0.99 ± 0.04 | 4.56 | 9.21 |
| 12.06 | 11.28 ± 0.39 | 3.43 | −6.50 | |
| 173.60 | 171.20 ± 11.83 | 6.91 | −1.36 |
| Parameter | Before baseline corrected | After baseline corrected | ||
|---|---|---|---|---|
| Reference drug | Test drug | Reference drug | Test drug | |
| T1/2 (h) | 1.62 ± 3.68 | 1.32 ± 2.24 | 0.79 ± 0.47 | 0.71 ± 0.12 |
| Tmax (h) | 2.10 ± 0.40 | 2.10 ± 0.30 | 2.05 ± 0.36 | 2.07 ± 0.29 |
| Cmax (ng/mL) | 141.75 ±31.64 | 142.52 ± 34.09 | 140.79 ± 31.64 | 141.59 ± 34.09 |
| AUC0→t (ng∙h/mL) | 343.95 ± 100.7 | 352.67 ± 121.78 | 334.11 ± 100.32 | 343.19 ± 121.56 |
| AUC0→∞ (ng∙h/mL) | 345.67 ± 98.96 | 353.91 ± 120.71 | 335.09 ± 100.26 | 344.11 ± 121.58 |
3. Experimental
3.1. Chemicals and Reagents
3.2. Liquid Chromatographic Conditions
3.3. Mass Spectrometry Conditions
3.4. Preparation of Standard and Quality Control Samples
3.5. Plasma Sample Preparation
3.6. Method Validation
3.6.1. Selectivity
3.6.2. Linearity and Lower Limit of Quantification
3.6.3. Precision and Accuracy
3.6.4. Extraction Recovery and Matrix Effect
3.6.5. Stability
3.7. Bioequivalence Study
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Chen, S.J.; Chen, Z. Targeting agents alone to cure acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 186–187. [Google Scholar] [CrossRef]
- Barbui, T.; Finazzi, G.; Falanga, A. The impact of all-trans retinoic acid on the coagulopathy of acute promyelocytic leukemia. Blood 1998, 1, 3093–3102. [Google Scholar]
- Muindi, J.R.; Frankel, S.R.; Huselton, C.; DeGrazia, F.; Garland, W.A.; Young, C.W.; Warrell, R.P., Jr. Clinical pharmacology of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res. 1992, 52, 2138–2142. [Google Scholar]
- Quere, R.; Baudet, A.; Cassinat, B.; Bertrand, G.; Marti, J.; Manchon, L.; Piquemal, D.; Chomienne, C.; Commes, T. Pharmacogenomic analysis of acute promyelocytic leukemia cells highlights CYP26 cytochrome metabolism in differential all-trans retinoic acid sensitivity. Blood 2007, 109, 4450–4460. [Google Scholar] [CrossRef]
- Taimi, M.; Helvig, C.; Wisniewski, J.; Ramshaw, H.; White, J.; Amad, M.; Korczak, B.; Petkovich, M. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J. Biol. Chem. 2004, 279, 77–85. [Google Scholar]
- Conley, B.A.; Egorin, M.J.; Sridhara, R.; Finley, R.; Hemady, R.; Wu, S.; Tait, N.S.; van Echo, D.A. Phase I clinical trial of all-trans-retinoic acid with correlation of its pharmacokinetics and pharmacodynamics. Cancer Chemother. Pharmacol. 1997, 39, 291–299. [Google Scholar] [CrossRef]
- Thudi, N.R.; Shrivastav, V.K.; Monif, T.; Gurule, S.; Partani, P.O.; Tandon, M.; Mathur, R. Pharmacokinetic and bioequivalence study of endogenous compound tretinoin 10 mg capsules in healthy volunteers by base line correction approach. Clin. Res. Regul. Aff. 2011, 28, 68–73. [Google Scholar] [CrossRef]
- Lanvers, C.; Hempel, G.; Blaschke, G.; Boos, J. Simultaneous determination of all-trans-, 13-cis- and 9-cis-retinoic acid, their 4-oxo metabolites and all-trans-retinol in human plasma by high-performance liquid chromatography. J. Chromatogr. B Biomed. Appl. 1996, 685, 233–240. [Google Scholar] [CrossRef]
- Napoli, J.L. Quantification of physiological levels of retinoic acid. Methods Enzymol. 1986, 123, 112–124. [Google Scholar]
- McCaffery, P.; Evans, J.; Koul, O.; Volpert, A.; Reid, K.; Ullman, M.D. Retinoid quantification by HPLC/MSn. J. Lipid Res. 2002, 43, 1143–1149. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, W.Y.; Prins, G.S.; van Breemen, R.B. Simultaneous determination of all-trans, 9-cis, 13-cis retinoic acid and retinol in rat prostate using liquid chromatography-mass spectrometry. J. Mass Spectrom. 2001, 36, 882–888. [Google Scholar] [CrossRef]
- Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry, Bioanalytical Method Validation; U.S. Department of Health and Human Services: Rockville, MD, USA, 2001. [Google Scholar]
- Sample Availability: Samples of the compounds and materials are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peng, J.-B.; Luo, C.-H.; Wang, Y.-C.; Huang, W.-H.; Chen, Y.; Zhou, H.-H.; Tan, Z.-R. Validation of a Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry Method for Determination of All-Trans Retinoic Acid in Human Plasma and Its Application to a Bioequivalence Study. Molecules 2014, 19, 1189-1200. https://doi.org/10.3390/molecules19011189
Peng J-B, Luo C-H, Wang Y-C, Huang W-H, Chen Y, Zhou H-H, Tan Z-R. Validation of a Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry Method for Determination of All-Trans Retinoic Acid in Human Plasma and Its Application to a Bioequivalence Study. Molecules. 2014; 19(1):1189-1200. https://doi.org/10.3390/molecules19011189
Chicago/Turabian StylePeng, Jing-Bo, Chen-Hui Luo, Yi-Cheng Wang, Wei-Hua Huang, Yao Chen, Hong-Hao Zhou, and Zhi-Rong Tan. 2014. "Validation of a Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry Method for Determination of All-Trans Retinoic Acid in Human Plasma and Its Application to a Bioequivalence Study" Molecules 19, no. 1: 1189-1200. https://doi.org/10.3390/molecules19011189