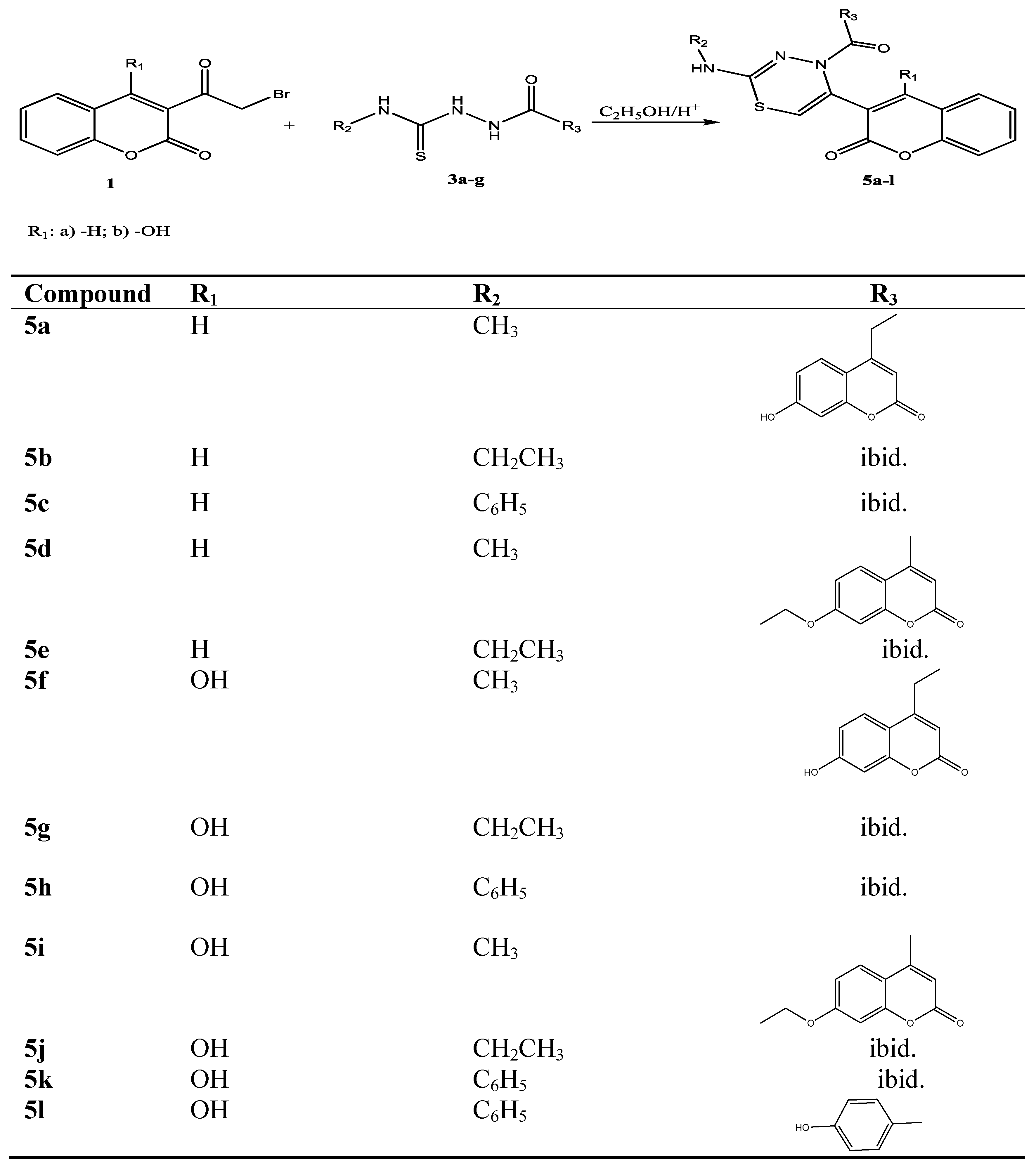
Design and Synthesis of Some New 1,3,4-Thiadiazines with Coumarin Moieties and Their Antioxidative and Antifungal Activity




Abstract
:1. Introduction
2. Results and Discussion
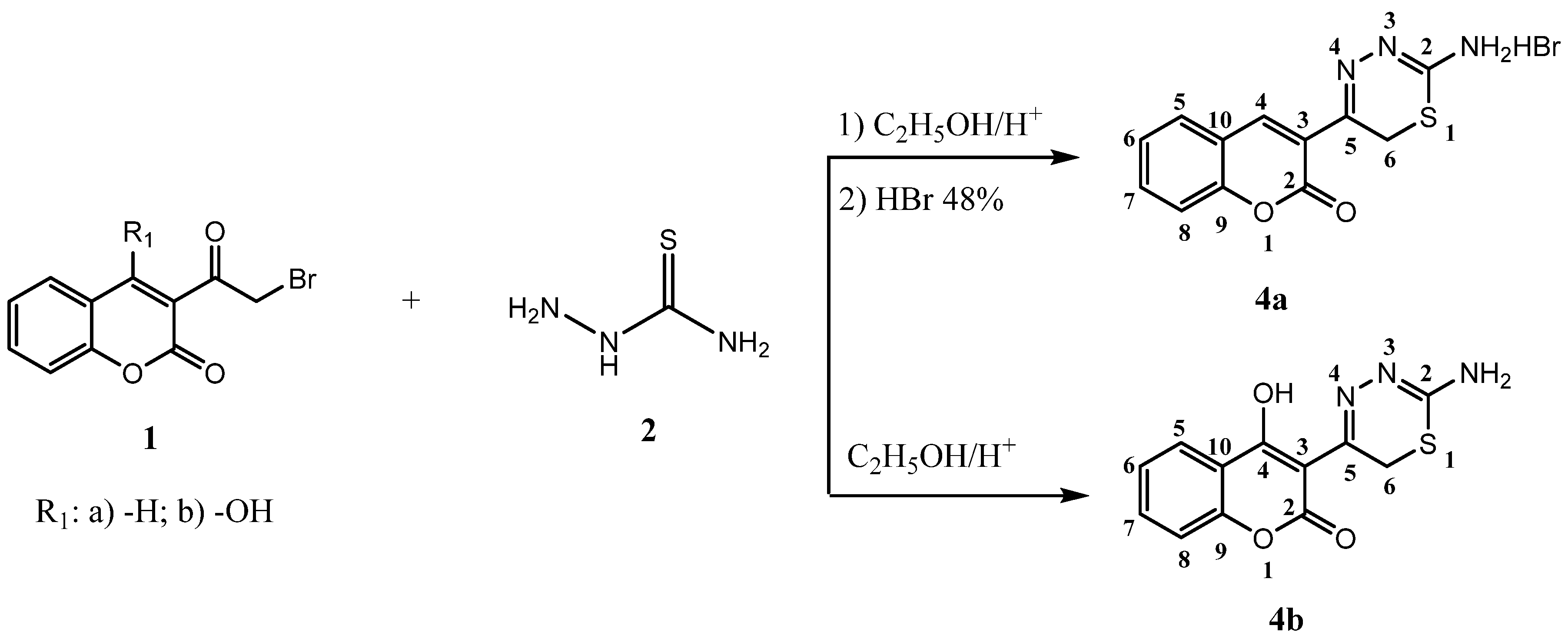
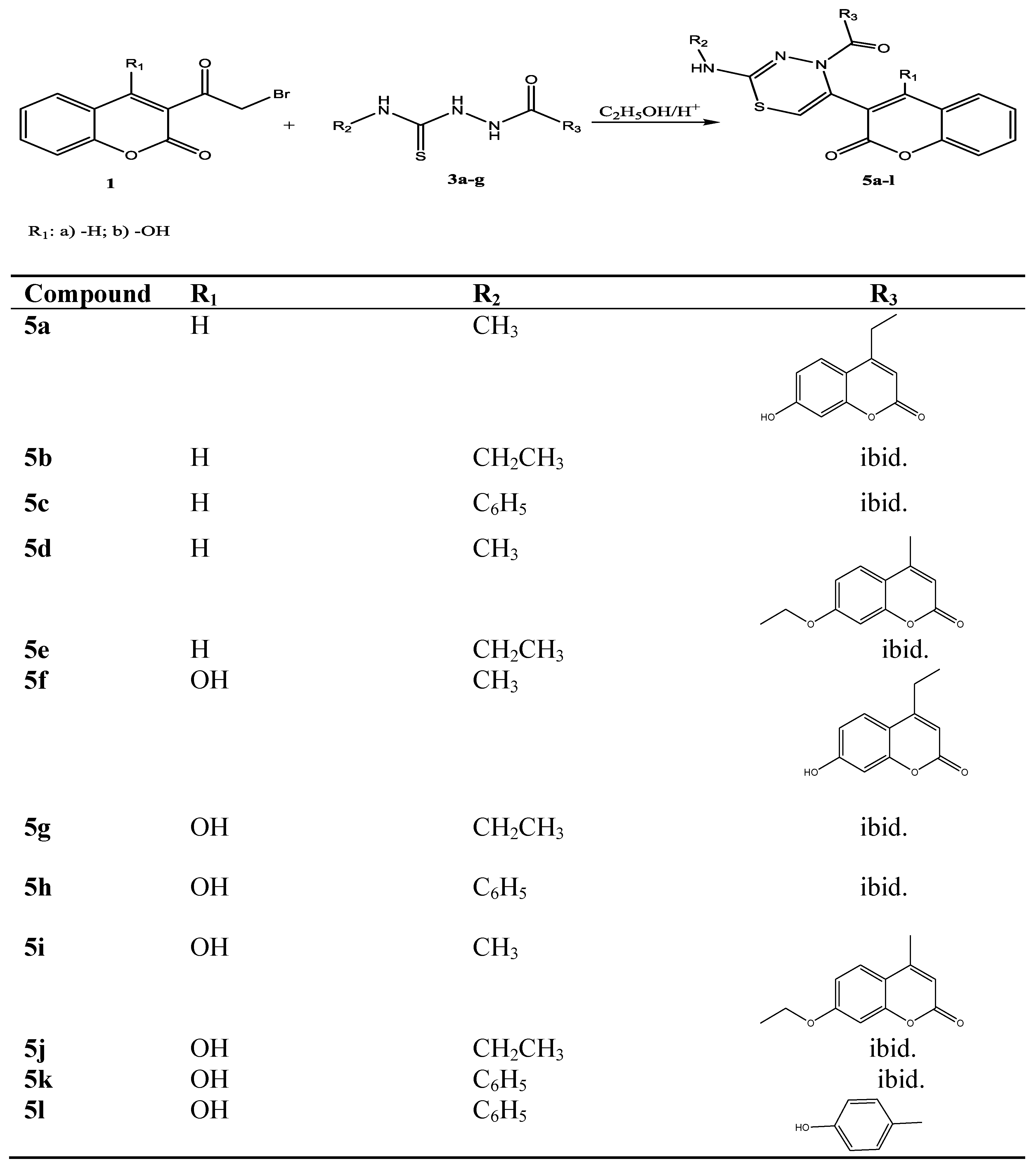
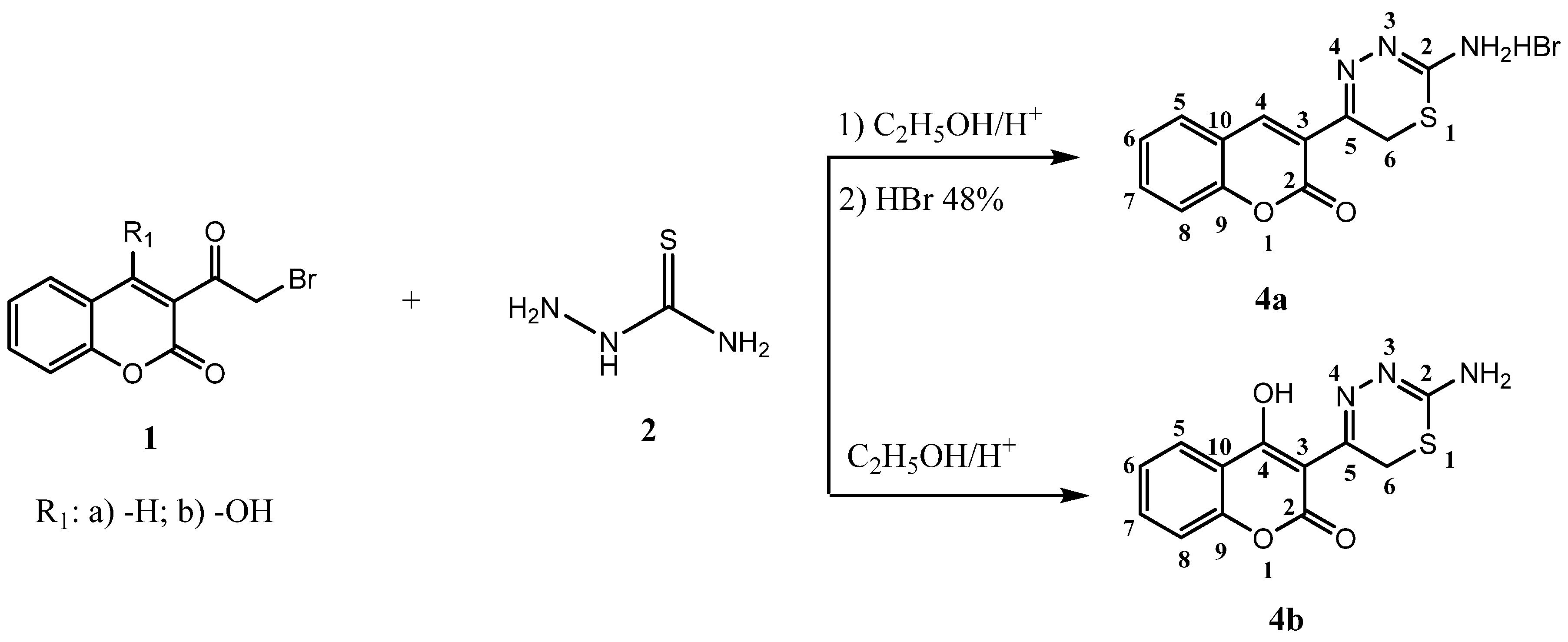
2.1. Chemistry
2.2. DPPH-Scavenging Activity

{kind=link}
{kind=link}
{kind=link}
| Compound | % DPPH scavenging activity | % Chelating activity |
|---|---|---|
| ascorbic acid b | 85.2 ± 6.1 | - |
| EDTA c | - | 90.9 ± 7.22 |
| 4a | 17.2 ± 0.59 | 0.0 |
| 4b | 90.0 ± 1.24 | 15.1 ± 4.27 |
| 5a | 36.0 ± 2.07 | - |
| 5b | 94.4 ± 1.73 | 0 |
| 5c | 17.7 ± 1.26 | 0,0 |
| 5d | 33.3 ± 0.27 | - |
| 5e | 80.7 ± 1.61 | 0.0 |
| 5f | 94.1 ± 1.35 | 0.0 |
| 5g | 82.3 ± 0.70 | 0.0 |
| 5h | 80.6 ± 0.98 | 62.5 ± 2.25 |
| 5i | 79.7 ± 1.29 | 5.3 ± 0.56 |
| 5j | 90.0 ± 1.08 | 53.5 |
| 5k | 77.2 ± 0.93 | 5.18 ± 0.81 |
| 5l | 80.2 ± 0.44 | 10.2 ± 0.42 |
2.3. Iron Chelating Activity
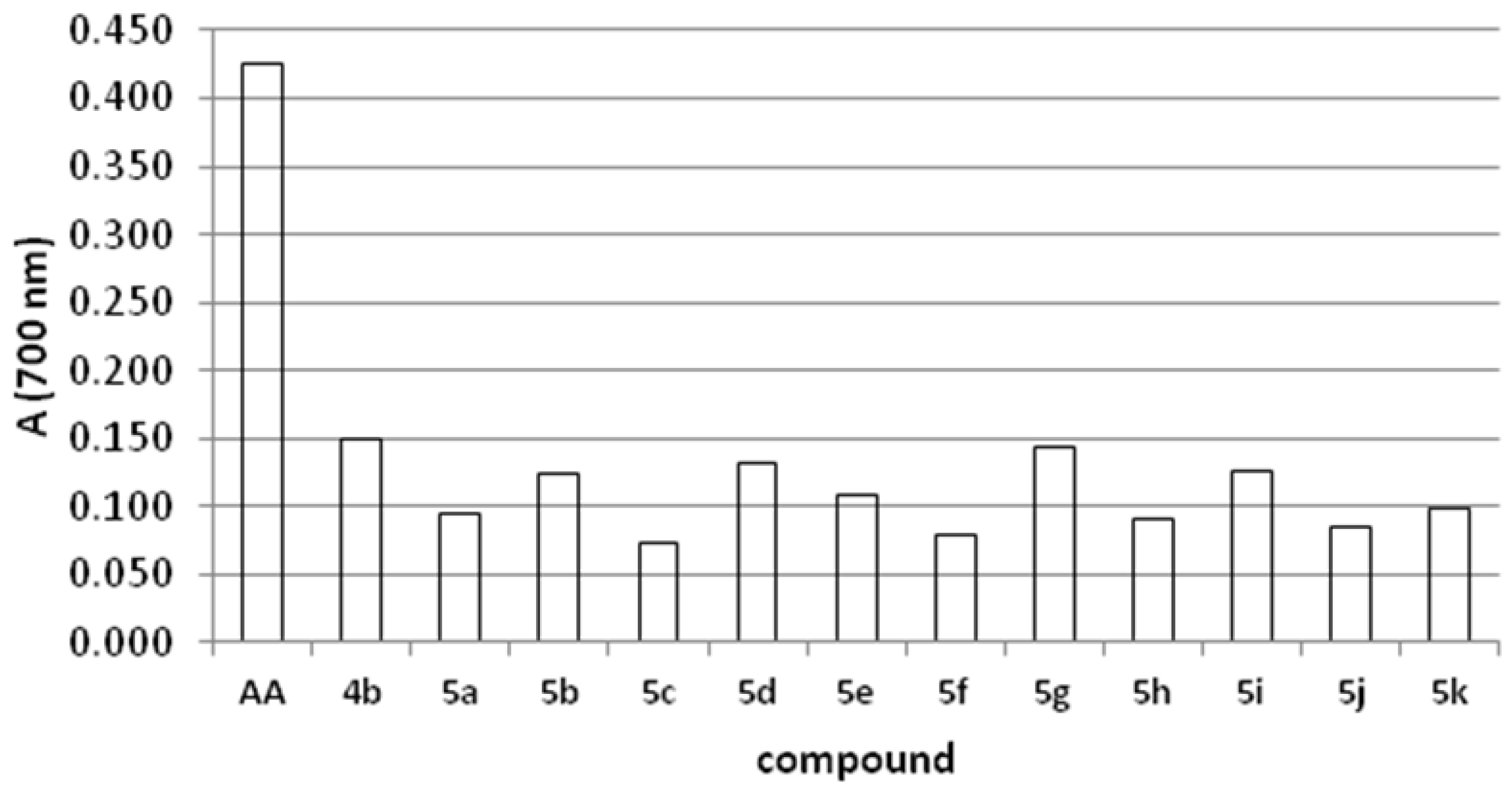
2.4. Reducing Power
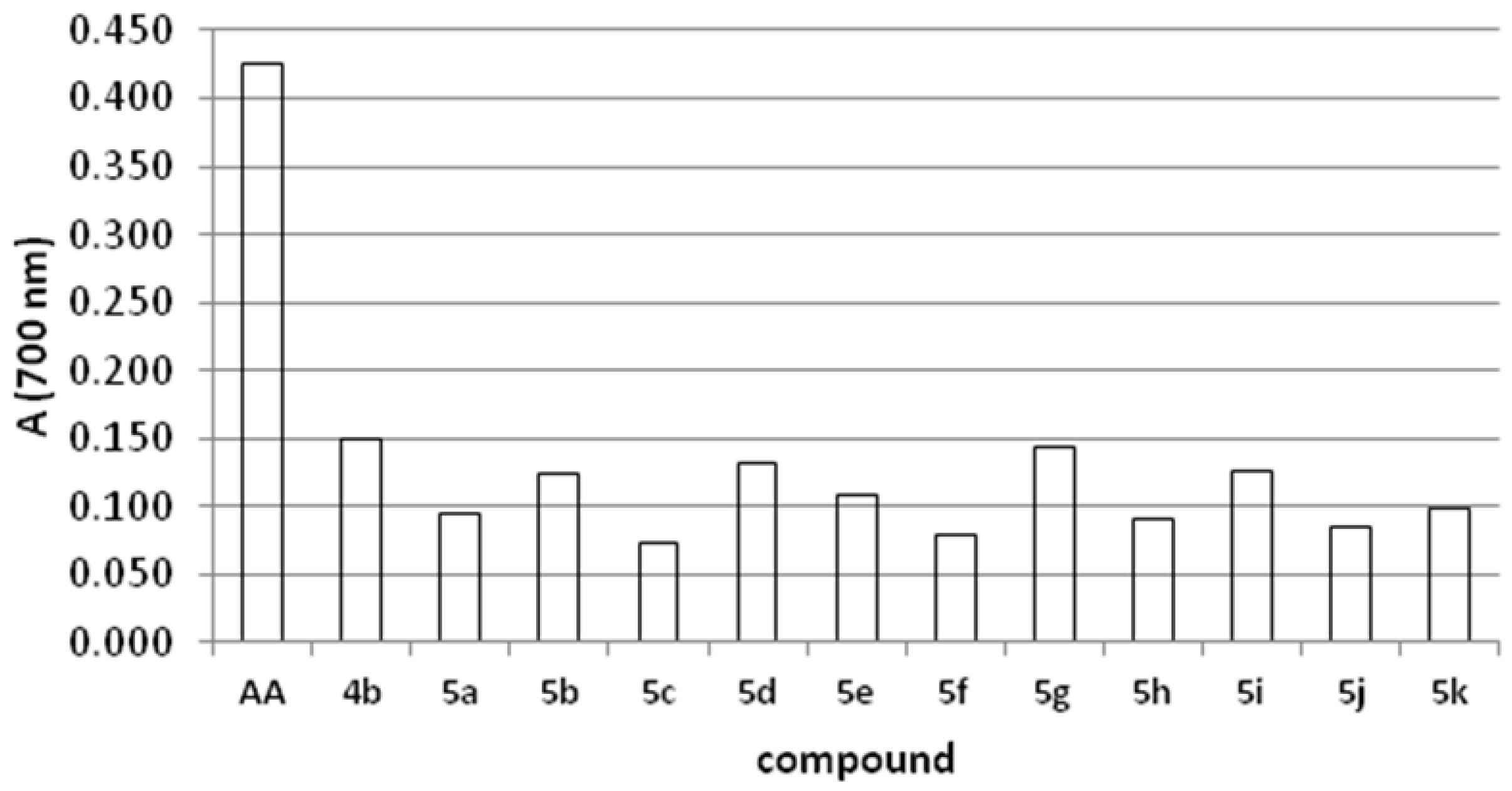
2.5. Antifungal Activity
| MIC50 | ||||
|---|---|---|---|---|
| Compound | A. flavus | A. ochraceus | F. graminearum | F. verticillioides |
| 4a | / | 0.01 | 0.01 | 0.1 |
| 5a | 1 | 0.1 | 0.1 | 0.1 |
| 5b | / | 0.1 | 0.01 | 0.1 |
| 5c | 0.1 | 0.01 | 0.01 | 0.1 |
| 5d | 0.1 | 0.1 | 0.01 | 1 |
| 5e | 0.01 | 0.1 | 0.01 | 0.1 |
| 5f | 0.1 | 0.1 | 0.01 | 0.1 |
| 5g | 1 | 0.1 | 0.01 | 0.01 |
| 5i | 1 | 0.1 | 0.01 | 1 |
| 5j | 1 | 0.1 | 0.01 | 0.1 |
3. Experimental
3.1. General
3.2. Synthesis
3.2.1. General Procedure for the Preparation of 1,4-Disubstituted Thiosemicarbazides 3 [21]
3.2.2. Synthesis of 5-(2-Oxo-2H-chromen-3-yl)-6H-1,3,4-thiadizin-2-aminium Bromide (4a)
3.2.3. Synthesis of 3-(2-Amino-6H-1,3,4-thiadiazin-5-yl)-4-hydroxy-2H-chromen-2-one (4b)
3.2.4. General Procedure for Preparation 2,4,5-Trisubstituted 4H-1,3,4- thiadiazines 5a–l
3.3. DPPH-Scavenging Activity
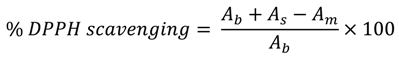
3.4. Iron Chelating Activity
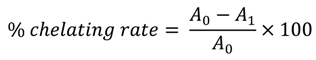
3.5. Reducing Power
3.6. Antifungal Activity
3.6.1. General
3.6.2. Tested Fungi
3.6.3. Preparation of Inoculum
3.6.4. Medium
3.6.5. Drug Dilutions
3.6.6. Incubation and MIC Determination
4. Conclusions
Conflicts of Interest
References
- El-Agrody, A.; Abd El-Latif, M.; El-Hady, N.; Fakery, A.; Bedair, A. Hetero aromatization with 4-hydroxycoumarin Part II: Synthesis of some new pyrano[2,3-d]pyrimidines, [1,2,4]triazolo[1,5-c]pyrimidines and pyrimido[1,6-b][1,2,4]triazine derivatives. Molecules 2001, 6, 519–527. [Google Scholar] [CrossRef]
- Rositca, D.N.; Vayssilov, G.N.; Rodios, N.; Bojilova, A. Regio- and stereoselective [2+2] photodimerization of 3-substituted 2-alkoxy-2-oxo-2H-1,2-benzoxaphosphorines. Molecules 2002, 7, 420–432. [Google Scholar] [CrossRef]
- Flašík, R.; Stankovičová, H.; Gáplovský, A.; Donovalová, J. Synthesis and study of novel coumarin derivatives potentially utilizable as memory media. Molecules 2009, 14, 4838–4848. [Google Scholar] [CrossRef]
- Kovalenko, S.; Bylov, I.; Sytnik, K.; Chernykh, V.; Bilokin, Y. A new pathway to 3-hetaryl-2-oxo-2H-chromenes: On the proposed mechanisms for the reaction of 3-carbamoyl-2-iminochromenes with dinucleophiles. Molecules 2000, 5, 1146–1165. [Google Scholar] [CrossRef]
- El-Saghier, A.; Khodairy, A.; Khodiyar, A. New synthetic approaches to condensed and spiro coumarins: Coumarin-3-thiocarboxamide as building block for the the synthesis of condense and spiro coumarins. Phosphorus Sulfur. 2000, 160, 105–119. [Google Scholar] [CrossRef]
- Al-Amiery, A.A.; Al-Bayati, R.; Saour, K.; Radi, M. Cytotoxicity, antioxidant and antimicrobial activities of novel 2-quinolone derivatives derived from coumarins. Res. Chem. Intermediat. 2012, 38, 559–569. [Google Scholar] [CrossRef]
- Azizian, J.; Mohammadi, A.; Bidar, I.; Mirazaei, P. KAl(SO4)2∙12H2O (alum) a reusable catalyst for the synthesis of some 4-substituted coumarins via Pechmann reaction under solvent-free conditions. Montash. Chem. 2008, 139, 805–808. [Google Scholar] [CrossRef]
- Satyanarayan, V.S.; Sreevani, P.; Sivakumar, A. Synthesis and antimicrobial activity of new Schiff bases containing coumarin moiety and their spectral characterization. Arkivoc 2008, 17, 221–233. [Google Scholar]
- Garazd, M.M.; Muzychka, O.V.; Voyk, A.I.; Nagorichna, I.V.; Ogorodniichuk, A.S. Modified coumarins. 27. Synthesis and antioxidant activity of 3-substituted 5,7-dihydroxy-4-methylcoumarins. Chem. Nat. Compd. 2007, 43, 19–23. [Google Scholar] [CrossRef]
- Smitha, G.; Sanjeeva, R. ZrCl4-catalyzed Pechmann reaction: Synthesis of coumarins under solvent-free conditions. Synth. Commun. 2004, 34, 3997–4003. [Google Scholar] [CrossRef]
- Kotali, A.; Lafazanis, I.; Harris, P. Synthesis of 6,7-diacylcoumarins via the transformation of a hydroxy into a carbonyl group. Synth. Commun. 2008, 38, 3996–4006. [Google Scholar] [CrossRef]
- Nofal, Z.M.; El-Zahar, M.; Abd El-Karim, S. Novel coumarin derivatives with expected biological activity. Molecules 2000, 5, 99–113. [Google Scholar] [CrossRef]
- Kennedy, R.O.; Thornes, R.D. Coumarins: Biology, Applications and Mode of Action; John Wiley and Sons: Chichester, UK, 1997. [Google Scholar]
- Zabradnik, M. The Production and Application of Fluorescent Brightening Agents; John Wiley and Sons: New York, NY, USA, 1992. [Google Scholar]
- Heravi, M.; Sadjadi, S.; Oskooie, H.; Shoar, R.; Bamoharram, F. The synthesis of coumarin-3-carboxylic acids and 3-acetyl-coumarin derivatives using heteropolyacids as heterogeneous and recyclable catalysts. Catal. Commun. 2008, 9, 470–474. [Google Scholar] [CrossRef]
- arkanj, B.; Molnar, M.; Cacic, M.; Gille, L. 4-Methyl-7-hydroxycoumarin antifungal and antioxidant activity enhancement by substitution with thiosemicarbazide and thiazolidinone moieties. Food Chem. 2013, 139, 488–495. [Google Scholar] [CrossRef]
- Novikova, A.P.; Perova, N.M.; Chupakhin, O.N. Synthesis and properties of functional derivatives of 1,3,4-thiadiazines and condensed systems based on these compounds (review). Khim. Geterot. Soedin. 1991, 11, 1443–1457. [Google Scholar]
- Bose, P.K. Thiodiazines. Part 1. Condensation of thiosemicarbazide with ω-bromo-acetophenone. J. Indian. Chem. Soc. 1924, 1, 51–62. [Google Scholar]
- Pfeiffer, W.D. Methods of Organic Chemistry (Houben-Weyl), 4th ed.; Büchel, K.H., Falbe, J., Hagemann, H., Hanack, M., Klamann, D., Kreher, R., Kropf, H., Regitz, M., Schaumann, E., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 1998; pp. 483–529. [Google Scholar]
- Klosa, J. Preparation of 4-hydroxycoumarin ketones with the help of phosphoroxychloride. Arch. Pharm. Ber. Dtsch. Pharmaz. Ges. 1955, 288, 56–61. [Google Scholar] [CrossRef]
- Gurosi, A.; Karali, N. Synthesis, characterization and primary antituberculosis activity evaluation of 4-(3-Coumarinyl)-3-benzyl-4-thiazolin-2-one benzylidenehydrazone. Tur. J. Chem. 2003, 27, 545–551. [Google Scholar]
- Cacic, M.; Trkovnik, M.; Cacic, F.; Has-Schön, E. Synthesis and antibacterial acivity of some derivatives of (7-hydroxy-2-oxo-2H-chromen-4-yl) acetic acid hydrazide. Molecules 2006, 1, 134–147. [Google Scholar]
- Saundaneand, A.R.; Walmik, P. Synthesis, antioxidant, antimicrobial, antimycobacterial, and cytotoxic activities of azetidinone and thiazolidinone moieties linked to indole nucleus. J. Chem. 2013, 2013, 543815:1–543815:9. [Google Scholar]
- Singhal, M.; Paul, A.; Singh, H.P.; Dubey, S.K.; Gaur, K. Evaluation of reducing power assay of chalcone semicarbazones. J. Chem. Pharm. Res. 2011, 3, 639–645. [Google Scholar]
- Jayanthi, P.; Lalitha, P. Reducing power of the solvent extracts of Eichhornia. crassipes (mart.) solms. Int. J. Pharm. Pharm. Sci. 2011, 3, 126–128. [Google Scholar]
- Krstanović, V.; Klapec, T.; Velić, N.; Milaković, Z. Contamination of malt barley and wheat by Fusarium graminearum and Fusarium culmorum from the crop years 2001–2003 in eastern Croatia. Microbiol. Res. 2005, 160, 353–359. [Google Scholar] [CrossRef]
- Manojkumar, P.; Ravi, T.K.; Subbuchettiar, G. Synthesis of coumarin heterocyclic derivatives with antioxidant activity and in vitro cytotoxic activity against tumor cells. Acta Pharm. 2009, 59, 159–170. [Google Scholar] [CrossRef]
- Wu, C.R.; Huang, M.Y.; Lin, Y.T.; Ju, H.Y.; Ching, H. Antioxidant properties of Cortex fraxini and its simple coumarins. Food Chem. 2007, 104, 1464–1471. [Google Scholar] [CrossRef]
- Taskova, R.; Mitova, M.; Mikhova, B.; Duddeck, H. Bioactive phenolics from Carthamus. lanatus, L. Z. Naturforsch. C 2003, 58c, 704–707. [Google Scholar]
- Zhao, G.-R.; Xiang, Z.-J.; Ye, T.-X.; Yuan, Y.-J.; Guo, Z.-X. Antioxidant activities of Salvia miltiorrhiza and Panax notoginseng. Food Chem. 2006, 99, 767–774. [Google Scholar] [CrossRef]
- Igbinosa, O.O.; Igbinosa, I.H.; Chigor, V.N.; Uzunuigbe, O.E.; Oyedemi, S.O.; Odjadjare, E.E.; Okoh, A.I.; Igbinosa, E.O. Polyphenolic contents and antioxidant potential of stem bark extracts from Jatropha. curcas (Linn). Int. J. Mol. Sci. 2011, 12, 2958–2971. [Google Scholar] [CrossRef]
- National Committee for Clinical Laboratory Standards. Reference Method For Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi; In Approved standard, NCCLS document M38-A; CLSI: Wayne, PA, USA, 2002. [Google Scholar]
- Hussein, H.S.; Brasel, J.M. Toxicity, metabolism, and impact of mycotoxins on humans and animals. Toxicology 2001, 167, 101–134. [Google Scholar] [CrossRef]
- Clement, M.; Tremblay, J.; Lange, M.; Thibodeau, J.; Belhumeur, P. Purification and identification of bovine cheese whey fatty acids exhibiting in vitro antifungal activity. J. Dairy Sci. 2008, 91, 2535–2544. [Google Scholar] [CrossRef]
- Santos, M.M.M.; Faria, N.; Iley, J.; Coles, S.J.; Hurrsthouse, M.B.; Martins, M.L. Reaction of naphthoquinones with substituted nitromethanes. Facile synthesis and antifungal activity of naphtho[2,3-d]isoxazole-4,9-diones. Bioorg. Med. Chem. Lett. 2010, 20, 193–195. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 5a–l are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Čačić, M.; Pavić, V.; Molnar, M.; Šarkanj, B.; Has-Schön, E. Design and Synthesis of Some New 1,3,4-Thiadiazines with Coumarin Moieties and Their Antioxidative and Antifungal Activity. Molecules 2014, 19, 1163-1177. https://doi.org/10.3390/molecules19011163
Čačić M, Pavić V, Molnar M, Šarkanj B, Has-Schön E. Design and Synthesis of Some New 1,3,4-Thiadiazines with Coumarin Moieties and Their Antioxidative and Antifungal Activity. Molecules. 2014; 19(1):1163-1177. https://doi.org/10.3390/molecules19011163
Chicago/Turabian StyleČačić, Milan, Valentina Pavić, Maja Molnar, Bojan Šarkanj, and Elizabeta Has-Schön. 2014. "Design and Synthesis of Some New 1,3,4-Thiadiazines with Coumarin Moieties and Their Antioxidative and Antifungal Activity" Molecules 19, no. 1: 1163-1177. https://doi.org/10.3390/molecules19011163
APA StyleČačić, M., Pavić, V., Molnar, M., Šarkanj, B., & Has-Schön, E. (2014). Design and Synthesis of Some New 1,3,4-Thiadiazines with Coumarin Moieties and Their Antioxidative and Antifungal Activity. Molecules, 19(1), 1163-1177. https://doi.org/10.3390/molecules19011163