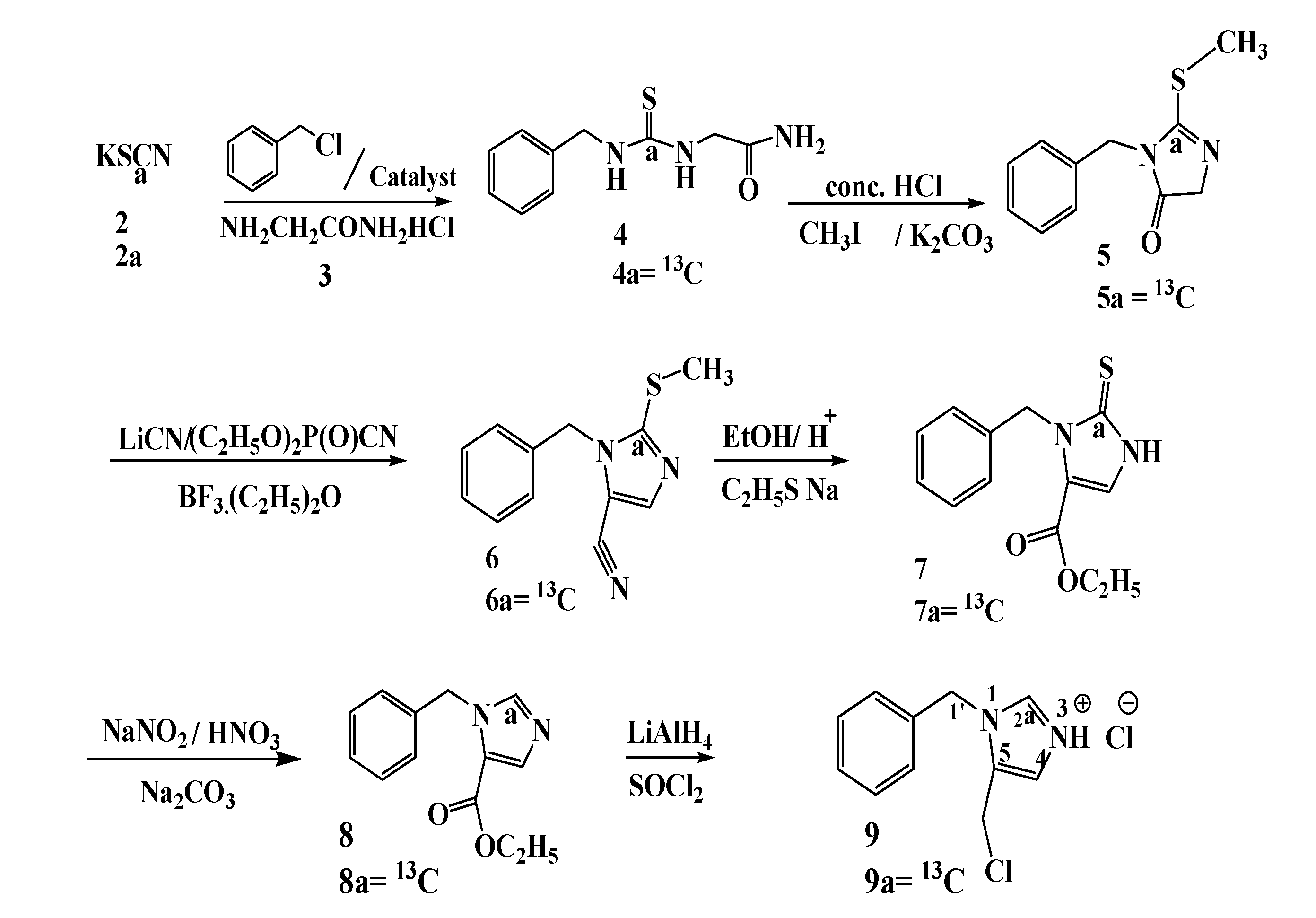
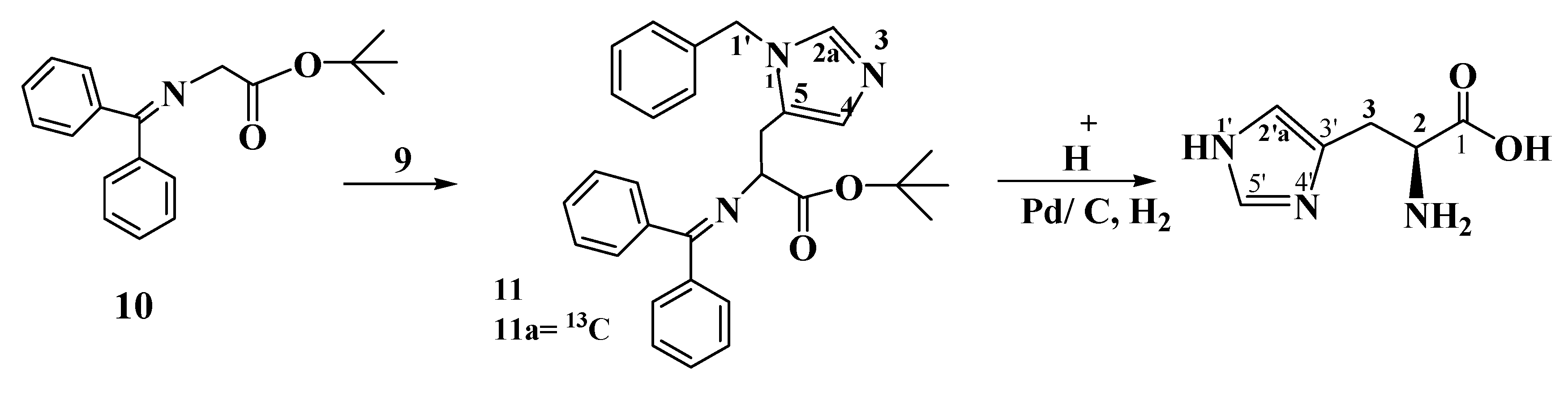
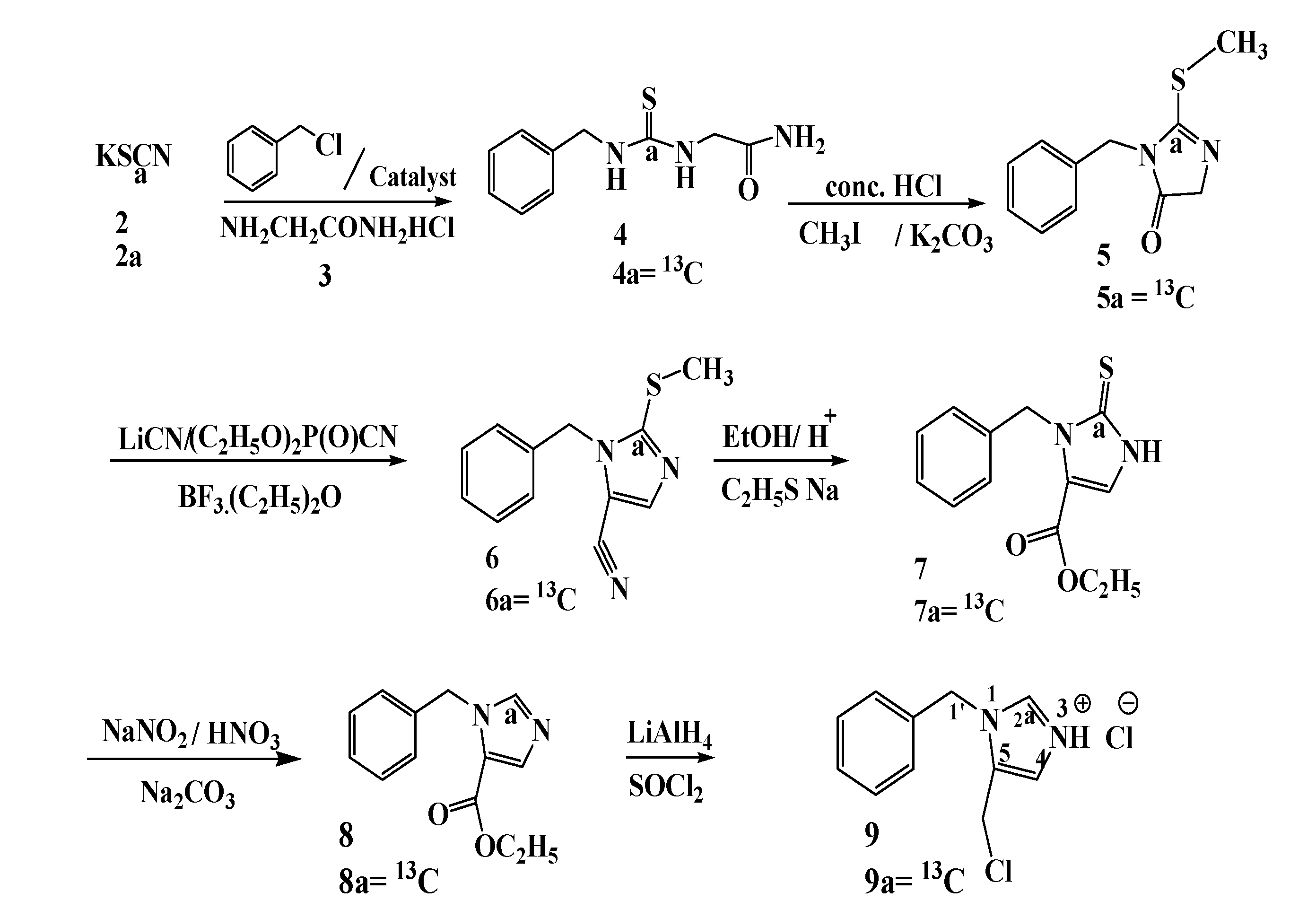
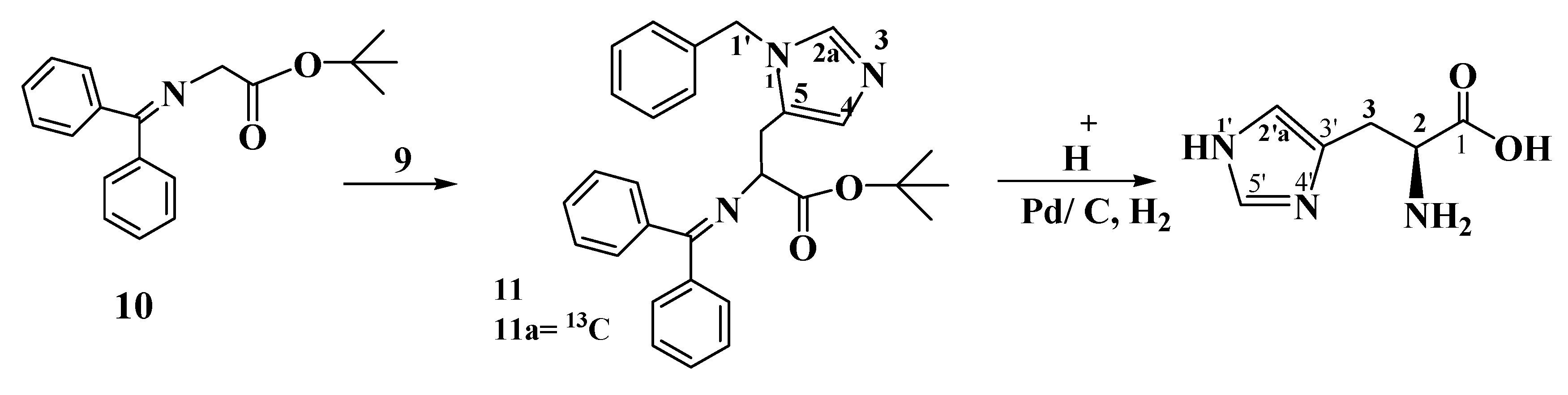
3.2. Preparation of Compounds
N-Benzyl-N'-(2'-acetamido) thiourea (4). First benzyl isothiocyanate is prepared. Benzyl chloride (3.0 g, 23.7 mmol, 2.27 mL) is dissolved in o-dichlorobenzene (10 mL) to which the phase transfer catalyst bis(triphenyl) phosphoranylidene ammonium chloride (0.57 g, 0.5 mmol) and solid potassium thiocyanate (2, 2.5 g, 25.7 mmol) is added. The mixture is heated under reflux for three hours at 180 °C. After cooling to room temperature the solvent is removed under reduced pressure, the residue is extracted with chloroform (3 × 10 mL). The chloroform solution is washed with water and then dried over MgSO4. After drying the solvent is removed under reduced pressure to yield benzyl isothiocyanate as a yellow oil 2.9 g, 19.46 mmol (82.1%). 1H-NMR spectroscopy shows that the product consists of a major compound (benzyl isothiocyanate , 83.0%) and a minor compound (benzyl thiocyanate, 17.0%). Benzyl isothiocyanate: 1H-NMR (300 MHz, DMSO, δ in ppm): 4.91 (2H, benzyl CH2), 7.31–7.42 (5H, aromatic CH); and (2) 13C-NMR (75.5 MHz, DMSO, δ in ppm): 47.9 (benzyl CH2), 127.0 (CHPh), 128.0 (CHPh), 128.5 (CHPh), 129.2 (CS), 134.6 (Cq). Benzyl thiocyanate: 1H-NMR (300 MHz, DMSO, δ in ppm): 4.34 (2H, benzyl CH2), 7.31–7.42 (5H, aromatic CH) and (2) 13C-NMR (75.5 MHz, DMSO, δ in ppm): 37.0 (benzyl CH2), 127.0 (CHPh), 128.0 (CHPh), 128.5 (CHPh), 129.2 (CS), 133.5 (Cq).
Glycinamide hydrochloride (3, 4.0 g, 36.1 mmol) is dissolved in water (24 mL). To this mixture a 50% solution of KOH (35.7 mmol, 2 g in 3.0 mL of water) is added to adjust the pH to 8.5, and then ethanol (72.0 mL) is added. The solution is cooled in ice bath to 0 °C, and then treated dropwise with the previously prepared mixture containing 83.0% benzyl isothiocyanate and 17.0% benzyl thiocyanate (5.39 g, 4.7 mL, 36.1 mmol). The reaction mixture is stirred for 3 h, and frozen at −30 °C overnight. The mixture is allowed to reach room temperature, the solvent is decanted from the solid compound, and the solid compound is dried in vacuo to remove the remaining ethanol and water. The product is recrystallized from acetone to afford 7.0 g (31.4 mmol, 86.98%) of N-benzyl-N'-(2'-acetylamido) thiourea as pale yellow crystals. 1H-NMR (300 MHz, DMSO, δ in ppm): 4.04 (2H, C-5), 4.66 (2H, C-1), 7.32 (5H, aromatic CH), 8.30 (1 H, N-2), 7.54 (1H, N-4), 7.11 (2H, N-7); 13C-NMR (75.5 MHz, DMSO, δ in ppm): 42.3 (C-5), 45.1 (C-1), 126.8, 127.2, 128.2, 134.19 (Cq), 170.5 (C6, Cq), 175.0 (C3, Cq).
[13C] N-Benzyl-N'-(2'-acetamido) thiourea (4a). First 13C benzyl isothiocyanate (1.2 g, 8.0 mmol) is prepared (80.0% yield) as described above from KS13CN (1.0 g, 10.3 mmol). 1H-NMR: the signal at 4.91 ppm was split into a doublet (3JC-H1 = 2.6 Hz).13C-NMR: enhanced signal at 129.2 ppm. Then [13C]-N-benzyl-N'-(2'-acetamido)thiourea (4a, 1.5 g, 6.5 mmol) is prepared in 89.04% yield from 13C-benzyl isothiocyanate (1.1 g, 7.3 mmol) as described for 4. 1H-NMR: the signals at 4.11 and 4.65 were split into doublets (3JC3H5 = 4.8 Hz, 3JC3H1 = 5.6 Hz); 13C-NMR: enhanced signal at 175.0 ppm.
1-Benzyl-2-(methylthio)-imidazol-5-ketone (5). Benzyl thiourea (4, 7.0 g, 31.3 mmol) is dissolved in acetone (25 mL) and treated carefully with concentrated HCl (3 mL for each gram of compound) and the mixture is then heated overnight in an oil bath at 40 °C (the reaction is followed with TLC until the starting compound has disappeared). The acetone is removed in vacuo and the residue is treated with sat. aq. NaHCO3 to neutralize the excess acid and the mixture is extracted three times with CH2Cl2. Then the combined organic layers are washed with water and brine (100 mL), the dichloromethane layer is dried with MgSO4, filtered and concentrated in vacuo. The product is purified on a column of silica gel (dichloromethane/methanol = 95/5) to give a yellow crystalline compound (5.05 g, 78.1%). This compound (3-benzyl-2-thioimidazolidin-4-one) is dissolved in acetonitrile (30 mL). To this mixture iodomethane (5.24 g, 36.9 mmol) and potassium carbonate (1.7 g, 12.3 mmol) are added. The mixture is stirred at 40 °C for 14 h. The reaction mixture is cooled to room temperature and then filtered. The solvents are removed in vacuo. The residue is dissolved in a mixture of dichloromethane and hexane (1:1, 50 mL), filtered and the solvents are removed in vacuo. The product is purified on a column of silica gel (dichloromethane/methanol = 97/3) yielding 1-benzyl-2-(methylthio)-imidazol-5-ketone as a green crystalline compound (5.15 g, 23.4 mmol, 75%). 1H-NMR (300 MHz, DMSO, δ in ppm): 2.22 (3H, methyl thiogroup CH3), 2.99 (2H, 5-CH2), 4.65 (2H, benzyl CH2), 7.21–7.33 (5H, aromatic CH); 13C-NMR (75.5 MHz, DMSO, δ in ppm): 12.0 (CH3), 43.0 (C-5), 58.3 (benzyl CH2), 127.0, 127.6, 128.5, 136.1 (Cq), 162.1 (C2), 179.8 (C4, Cq).
[13C] 1-Benzyl-2-(methylthio)-imidazol-5-ketone (5a). 13C-1-benzyl-2-(methylthio)-imidazol-5-ketone (5a, 1.25 g, 5.6 mmol, 96.5% yield) is prepared as described for 5 from 13C-benzylthiourea (1.3 g, 5.8 mmol). 1H-NMR: the signals at 2.22 and 4.65 ppm were split into doublets (3JC2H5 = 4.8 Hz, 3JC2H3' = 5.6 Hz); 13C-NMR: enhanced signal at 162.1 ppm.
1-Benzyl-2-(methylthio)-5-imidazolcarbonitrile (6). 1-Benzyl-2-(methylthio)-imidazol-5- ketone (5, 3 g, 13.6 mmol) is dissolved in THF (130 mL), and to this mixture diethyl phosphorocyanidate (DEPC, 6.67 g, 40.9 mmol) and lithium cyanide (1.3 g, 39.3 mmol) are added gradually [the lithium cyanide is prepared by using a 250 mL round bottomed flask equipped with a magnetic stirrer, nitrogen inlet and a 60 mL addition funnel; lithium hydride (1.0 g, 126 mmol) is dissolved in anhydrous tetrahydrofuran (100 mL). The stirred suspension is cooled in an ice bath and acetone cyanohydrin (9.14 g, 9.14 mL, 100 mmol) is added dropwise over 15 min. After the addition is completed, the ice bath is removed and the mixture stirred for 2 h at room temperature. The magnetic stirring bar is removed and the solvent evaporated in vacuo to yield lithium cyanide as a white powder (4.07 g, 123.4 mmol, 99.2%)].
After the addition of LiCN is completed the reaction mixture is stirred at room temperature for 10 min. After removal of THF by evaporation the residue is dissolved in water (130 mL) and a mixture of benzene/ethyl acetate (1/1, 400 mL). The organic layer is separated and washed with water (2 × 100 mL) and saturated aqueous sodium chloride (1 × 100 mL). The organic layer is separated and then dried over MgSO4, followed by concentration to give green oil, which is dissolved directly in benzene (20 mL) followed by addition of boron trifluoride etherate (5.8 g, 40.9 mmol). The reaction mixture is stirred at room temperature for 2 h under nitrogen. After 2 h benzene (200 mL) and water (40 mL) are added to the reaction mixture, the organic layer is separated, and washed with water (2 × 100 mL) and saturated aqueous sodium chloride (150 mL). The organic layer is separated, dried over MgSO4 followed by concentration giving a brown oil of the nitrile, which is purified on a silica gel column (CH2Cl2/MeOH = 95/5). The result is 2.3 g (10.04 mmol, 73.8%) of the desired compound 6 as a brown oil. 1H-NMR (300 MHz, DMSO, δ in ppm): 2.49 (3H, CH3), 5.15 (2H, benzyl CH2), 6.92 (H, C4), 7.16 (2H, aromatic CH), 7.41 (3H, aromatic CH); 13C-NMR (75.5 MHz, DMSO, δ in ppm): 15.9 (methyl thio group CH3), 46.1 (benzyl CH2), 104.1 (C5), 111.9 (CN), 126.5, 127.1, 128.6, 136.1 (Cq), 136.4 (C4, Cq), 138.0 (C2).
[13C] 1-Benzyl-2-(methylthio)-5-imidazol carbonitrile (6a). [13C]-1-benzyl-2-(methylthio)-5-imidazolcarbonitrile (6a, 1.05 g, 3.8 mmol, 70.4% yield) is prepared as described for 6 from 13C-1-benzyl-2-(methylthio)-imidazol-5-ketone (1.2 g, 5.4 mmol). 1H-NMR: the signals at 2.49 and 5.15 ppm were split into doublets (3JC2H5 = 4.6 Hz, 3JC2H3' = 5.6 Hz); 13C-NMR: enhanced signal at 138.0 ppm.
Ethyl 1-benzyl-2,3-dihydro-2-thioxo-5-imidazole-carboxylate (7). 1-Benzyl-2-(methylthio)-5-imidazol-carbonitrile (6, 1.5 g, 6.5 mmol) is dissolved in ethanol (10 mL), the mixture is cooled to 0 °C in an ice bath and 0.53 mL of concentrated sulfuric acid (9.825 mmol) is added dropwise and carefully. After the addition is completed the ice bath is removed and the mixture is heated under reflux for 24 h. The mixture is cooled to room temperature and poured into a flask containing ice water (50 mL); the pH is adjusted to 3 with an aqueous solution of KOH. The mixture is filtered and the filtrate is concentrated in vacuo to yield a yellow solid of the desired compound, the solid is recrystallized from dichloromethane to give brown needles (1.7 g, 5.86 mmol, 89.9%).
The latter is added to a suspension of sodium ethanethiolate (10.6 mmol, 0.85 g) in DMF (8 mL). The mixture is heated at reflux overnight; the DMF is removed in vacuo to afford a crude solid. The solid is dissolved in a minimal amount of ethanol (12 mL), and then silica gel (5 g) is added to this mixture. The mixture is filtered carefully and washed with ethanol (5 mL). The residue is treated with a mixture of CH2Cl2/EtOH = 95/5 to yield a yellow oil (1.29 g, 4.94 mmol, 76.0%) which is used directly in the next step.
[13C] Ethyl 1-benzyl-2,3-dihydro-2-thioxo-5-imidazolecarboxylate (7a). 13C Ethyl 1-benzyl-2,3-dihydro-2-thioxo-5-imidazole-carboxylate (7a, 1.08 g, 4.1 mmol, 95.3% yield) is prepared as described for 7 from 13C 1-benzyl-2-(methylthio)-5-imidazolcarbonitrile (1.0 g, 4.3 mmol).
1-Benzyl 5-carbo-ethoxy imidazole (8). The yellow compound 7 is dissolved in a NaNO2 solution (1.0 mL, 0.5 g NaNO2 in 237 mL of H2O) and then nitric acid (65%, 0.35 mL) is added with continuous stirring. The mixture is stirred for 30 min. and then boiled with a solution of sodium carbonate (40% in water, 8 mL). The product is extracted with ether, dried over MgSO4 and concentrated. Purification is effected by column chromatography (CH2Cl2/EtOH = 97/3) to yield a brown oil (950 mg, 4.13 mmol, 83.6%). 1H-NMR (400 MHz, DMSO, δ in ppm): 1.19 (3H, ethyl ester group CH3), 3.94 (2H, ethyl ester group CH2), 4.45 (2H, benzyl CH2), 6.75 (1H, C5), 7.19–7.35 (5H, aromatic CH), 8.13 (1H, C2); 13C-NMR (100 MHz, DMSO, δ in ppm): 16.1 (CH3), 42.9 (CH2), 43.4 (benzyl CH2), 119.2 (C5), 126.2, 126.7, 127.9, 138.8 (Cq), 134.5 (C4, Cq), 141.2 (C2), 158.2 (CO).
[13C] 1-Benzyl 5-carboethoxy imidazole (8a). 13C 1-Benzyl-5-carboethoxy imidazole (8a, 750 mg, 3.2 mmol, 88.9% yield) is prepared as described for 8 from 13C ethyl 1-benzyl-2,3-dihydro-2-thioxo-5-imidazolecarboxylate (950 mg, 3.6 mmol). 1H-NMR: the signals at 4.45, 6.75 and 8.13 ppm were split into doublets (3JC2H5 = 4.8 Hz, 3JC2H3' = 5.6 Hz, 1JC2H2 = 219 Hz); 13C-NMR: enhanced signal at 141.2 ppm.
1-Benzyl-5-chloromethylimidazolium chloride (9). In a 100 mL three-necked flask lithium aluminum hydride (0.16 g, 4.4 mmol) is suspended in THF (20 mL) under a dry argon atmosphere. To this suspension a solution of 1-benzyl 5carboethoxy imidazole (8, 930 mg, 3.5 mmol) in THF (15 mL) is added dropwise with continuous stirring (15 min). The reaction is followed by TLC until the latter has disappeared (2.5 h, TLC: CH2Cl2/EtOH = 95/5). The mixture is carefully diluted with water (12 mL) and subsequently with 6 M HCl (12 mL). The pH is adjusted to 9 using a 6 M NaOH solution and the aqueous layer is extracted with dichloromethane. The organic layers are combined, washed with saturated aqueous NaCl and dried over anhydrous MgSO4. After filtration the solvent is removed in vacuo, yielding a yellow oil (660 mg, 3.19 mmol, 86.8%). A portion (500 mg) of this compound is placed in a 100 mL round bottomed flask fitted with a reflux condenser to which thionyl chloride (1.23 g, 10.0 mmol) is added. The mixture is refluxed for three h, and then allowed to cool to room temperature. The volatiles are evaporated in vacuo. The result is a brown oil (560 mg, 2.45 mmol, 70.0%) of the desired compound. 1H-NMR (400 MHz, CDCl3, δ in ppm): 4.36 (2H, CH2Cl), 4.69 (2H, benzyl CH2), 6.90 (1H, C5), 7.31–7.40 (5H, aromatic CH), 7.91 (1H, C2); 13C-NMR (100 MHz, CDCl3, δ in ppm): 44.6 (C-4'), 52.2 (benzyl CH2), 111.7 (C5), 127.5, 127.7, 129.0, 131.9 (C2), 135.6 (Cq), 144.0 (C4, Cq).
[13C] 1-Benzyl-5-chloromethylimidazolium chloride (9a). 13C 1-benzyl-5-chloromethylimidazolium chloride (9a, 540 mg, 2.21 mmol, 73.7% yield) is prepared as described for 9 from 13C 1-benzyl-5-carboethoxyimidazole (700 mg, 3.0 mmol). 1H-NMR: the signals at 4.69, 6.90 and 7.91 were split into doublets (3JC2H5 = 4.6 Hz, 3JC2H3' = 5.6 Hz, 1JC2H2 = 221 Hz). 13C-NMR: enhanced signal at 131.9 ppm.
Tert-butyl-3-(1-benzyl-1H-imidazol-5-yl)-2(diphenylmethyleneamino) propanoate (11). N-(Diphenyl-methylene) glycine tert-butyl ester (10, 570 mg, 1.92 mmol) and the chiral catalyst 2,7-bis [O-9-allyl hydrocinchonidinium N-methyl]naphthalene dibromide (19 mg, 0.0019 mmol) are dissolved in toluene (10 mL). To this mixture 1-benzyl-5-chloromethylimidazolium chloride (9, 500 mg, 2.05 mmol) is added. The reaction mixture is then cooled to 0 °C, and 50% aqueous KOH (0.28 mL) is added, the mixture is stirred at 0 °C until the starting material has been consumed (8 h). The ice bath is removed and the mixture is diluted with ether (25 mL), washed with water (2 × 15 mL), the ether layer is separated, dried over MgSO4, filtered and concentrated to yield a brown crystalline compound (780 mg, 1.67 mmol, 87.5% yield). 1H-NMR (300 MHz, DMSO, δ in ppm): 1.39 (9H, CH3), 4.03 (2H, benzyl CH2), 4.48 (2H, CH2), 4.94 (1H, C3'), 6.90 (1H, C5), 7.1–7.55 (15H, aromatic CH), 7.90 (1H, C2); 13C-NMR (100 MHz, DMSO, δ in ppm): 27.6 (3C, CH3), 53.3 (C-4'), 55.7 (benzyl CH2), 67.2(C, C3'), 75.6 (C, OMe3), 125.2 (C5), 126.9–128.8 (15 C, CHPh), 135.0 (C4, Cq), 135.0 (C2), 137.3–138.6 (Cq), 168.9 (C=N), 170.0 (CO).
[13C] Tert-butyl-3-(1-benzyl-1H-imidazol-5-yl)-2(diphenylmethyleneamino) propanoate (11a). 13C tert-Butyl-3-(1-benzyl-1H-imidazol-5-yl)-2(diphenylmethyleneamino) propanoate (11a, 1.1 g, 2.36 mmol, 95.9% yield) is prepared as described for 11 from 13C 1-benzyl-5-chloromethylimidazolium chloride (600 mg, 2.46 mmol). 1H-NMR: the signals at 4.03, 6.90 and 7.90 were split into doublets (3JC2H5 = 4.8 Hz, 3JC2H1' = 5.6 Hz, 1JC2H2 = 220 Hz). 13C-NMR: enhanced signal at 135.0 ppm.
L-Histidine dihydrochloride (
1).
tert-Butyl-3-(1-benzyl-1
H-imidazol-5-yl)-2-(diphenylmethylene-amino) propanoate (
11, 400 mg, 0.86 mmol) is dissolved in THF (10 mL) and then stirred with 10% citric acid solution (5 mL) overnight. TLC shows complete disappearance of the starting material. The mixture is extracted twice with ether (20 mL). The water layer is then brought to pH 12 with a K
2CO
3 solution and extracted with ethyl acetate (10 mL). The combined organic layers are dried over MgSO
4 and concentrated
in vacuo to give brown crystals (250 mg, 0.828 mmol, 96.3%). This compound is then treated with 1 N HCl (5 mL), and the mixture is refluxed overnight. The reaction is followed by TLC until the starting material has disappeared (dichloromethane/ethanol = 99/1). The mixture is allowed to reach room temperature, diluted with a small amount of dichloromethane, filtered and concentrated
in vacuo. The product is a yellow oil (230 mg, 0.709 mmol, 85.7%). The latter is dissolved in a mixture of cyclohexene/methanol (1/1). Palladium black (100 mg) is added to the mixture. The mixture is refluxed until the conversion is complete, which takes 5 days. On the second day a freshly prepared Pd pellet [
6] (50 mg in 10 mL of the same solvent mixture) is added to the mixture. The suspension is cooled down to room temperature and filtered over Celite. The solvent is evaporated
in vacuo; the compound is redissolved in methanol and then concentrated to yield a yellow solid (165 mg, 0.727 mmol, 84.5%).
1H-NMR (400 MHz, D
2O/DCl, δ in ppm): 3.46 (1H, H3a), 3.62 (1H, H3b), 4.42 (1H, H2), 7.49 (1H, H5'), 8.74 (1H, H2');
13C-NMR (100 MHz, D
2O/DCl, δ in ppm): 27.9 (C3),54.7 (C2), 118.5 (C5'), 130.0 (C4'), 135.8 (C2'), 174.0 (C1).
2'-[13C]-L-Histidine dihydrochloride (
1a). 2'-
13C-
L-Histidine dihydrochloride (
1a, 390 mg, 1.7 mmol, 80.9% yield) is prepared as described for
1 from
13C tert-butyl-3-(1-benzyl-1H-imidazol-5-yl)-2-(diphenylmethyleneamino) propanoate (1.0 g, 2.1 mmol).
1H-NMR: the signals at 7.49 and 8.74 ppm were split into doublets (
3J
C2H5' = 6.5 Hz,
1J
C2'H2' = 221 Hz).
13C-NMR: enhanced signal at 135.8 ppm. Both chemical and optical purity is 99% as found by an HPLC method, in agreement with the literature [
5]. The more volatile n-butyl ester of 2'-
13C-L-histidine was investigated by double focus chemical ionization mass spectroscopy (ionization gas CH
4). The signal at
m/z = 213.1439 corresponds with
12C
913C
1H
1814N
316O
2 (calculated mass: 213.1433). In the single focus EI/70 eV mass spectrum the fragment at
m/z = 82 is formed by McLafferty rearrangement involving the imidazole ring. From the base peak and the M-1, M+1 and M+2 peaks 99% enrichment with
13C at the C2' could be established within experimental error and in agreement with the
13C incorporation in
13C thiocyanate. No isotope dilution has taken place during the reaction sequence.

{kind=link}
{kind=link}