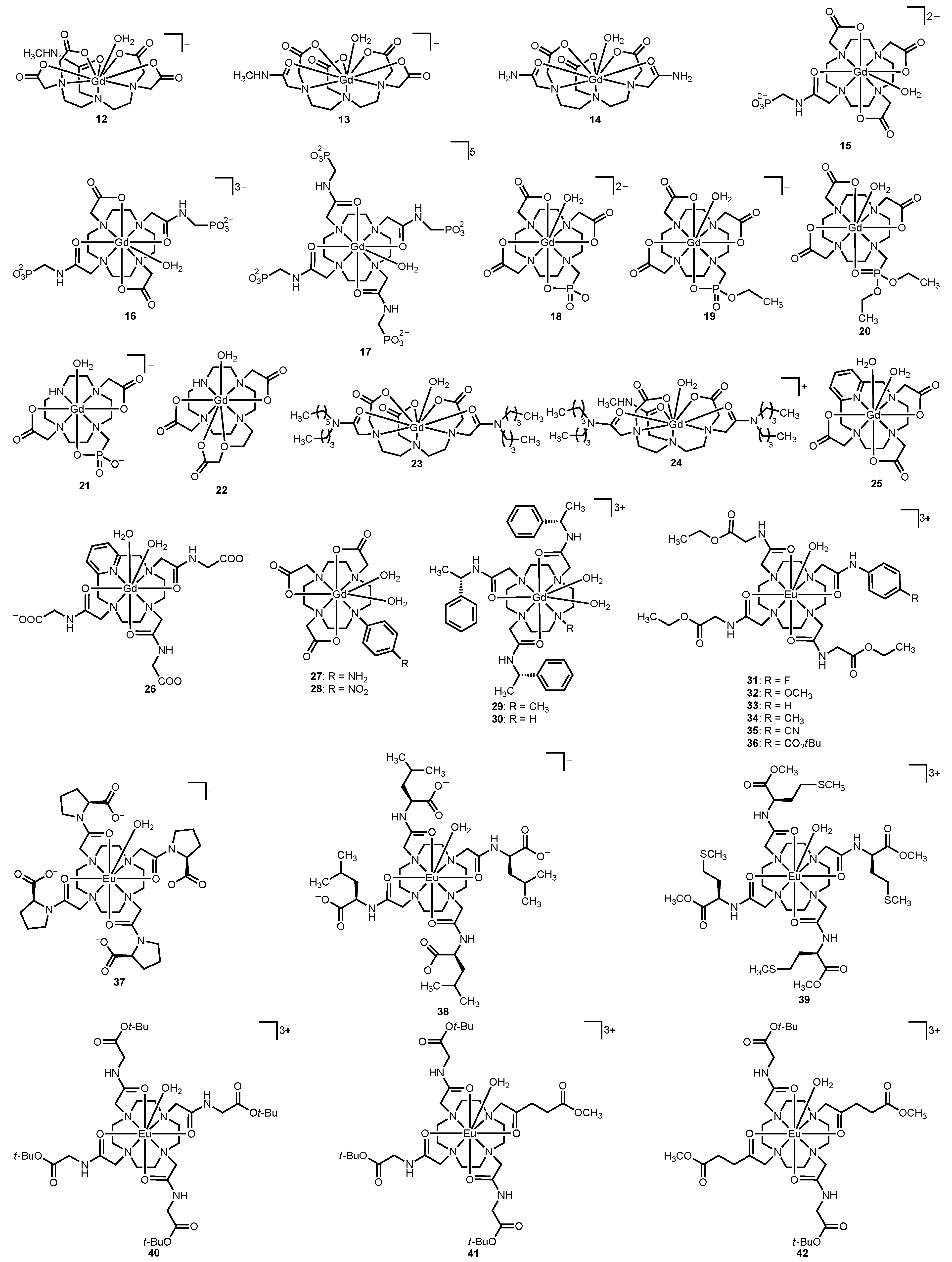Coordination-chemistry-based strategies that have been used to tune the water-exchange rates of Ln
III-containing complexes for
T1-shortening and PARACEST agents include modification of (1) the mechanism of water exchange; (2) the charge of the Ln
III-containing complex; (3) the steric hindrance at the site of water coordination; (4) the ligand side chains; and (5) the ratio of twisted-square-antiprism (TSAP) to square-antiprism (SAP) isomers for 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate (DOTA)-type complexes. In addition to the five strategies listed above, the identity of the Ln
III ion in a complex also influences the magnitude of water-exchange rates [
23,
24,
25,
26,
27]. However, this review focuses on strategies useful to both
T1-shortening and PARACEST agents, and therefore, a description of the influence of Ln
III ion on water-exchange rates is not included because Ln
III ions other than Gd
III are not useful for
T1-shortening agents. The following text is divided into separate sections that describe the five strategies listed above in terms of their implications to
T1-shortening and PARACEST agents. Although these strategies are separated in this text to enable structure–function comparisons to be made, it is important to note that these strategies are interrelated and cannot be completely isolated from each other.
2.1. Modification of Water-Exchange Mechanism
The mechanism of water exchange is an important determinant of the magnitude of water-exchange rates of Ln
III-containing complexes. In this section, the relationship between the mechanism of water exchange and water-exchange rate will be discussed using complexes
1–
11 (
Figure 2 and
Table 1). Water exchange in Ln
III-containing complexes proceeds via dissociative, associative, or interchange mechanisms. In a dissociative mechanism, dissociation of the coordinated water is rate limiting and precedes coordination of the incoming water molecule. In an associative mechanism, coordination of the incoming water molecule is rate limiting and precedes the dissociation of the coordinated water molecule. The interchange mechanisms are where transition states are not observed. Interchange mechanisms are divided in to dissociative interchange and associative interchange mechanisms depending on the relative contributions from bond breaking and forming: higher bond-breaking contributions lead to dissociative interchange and higher bond-forming contributions lead to associative interchange. Ln
III-containing complexes with coordination numbers of nine tend to undergo dissociative or dissociative interchange mechanisms of exchange, and complexes with coordination numbers of eight tend to undergo associative or associative interchange mechanisms of exchange.
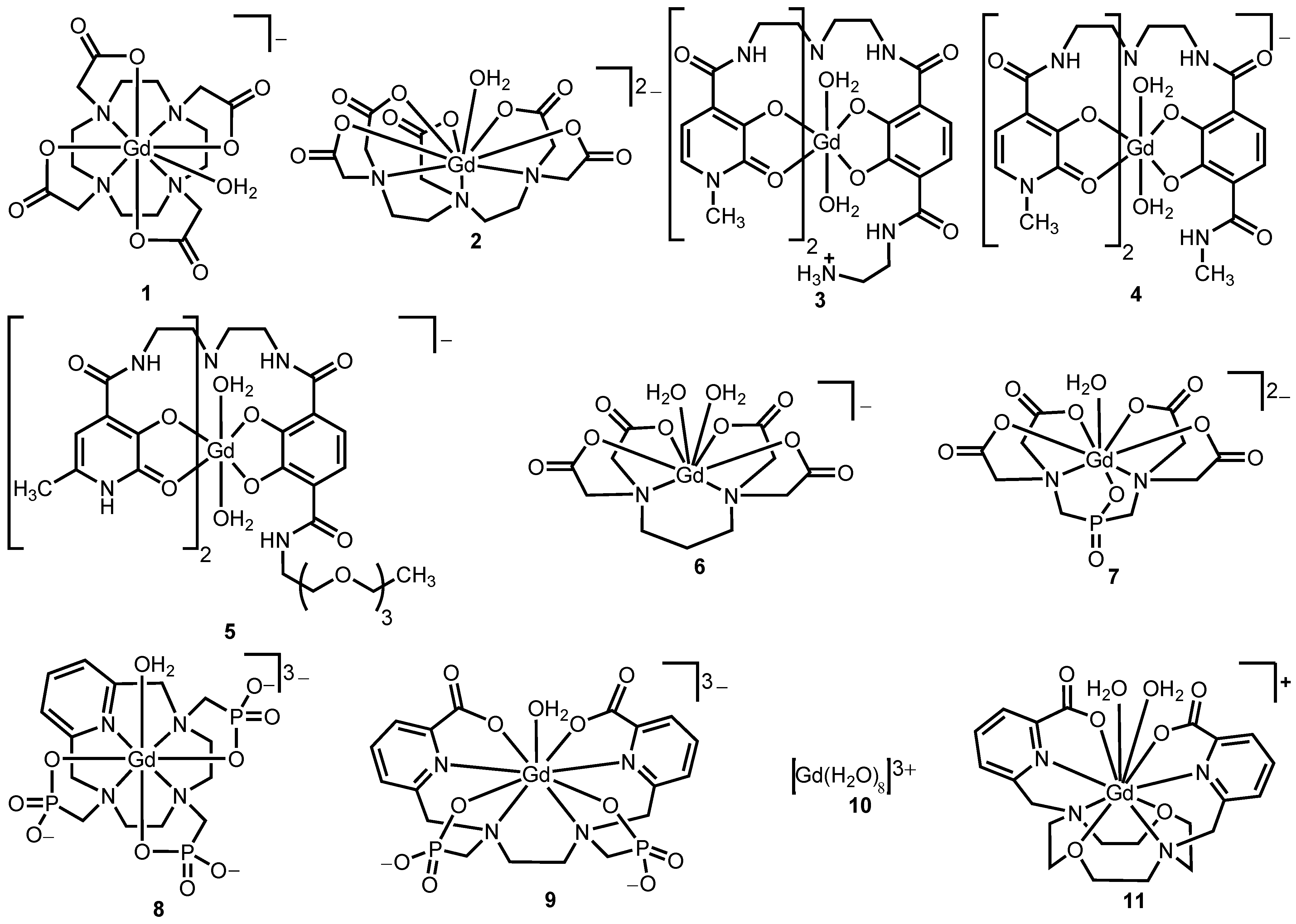
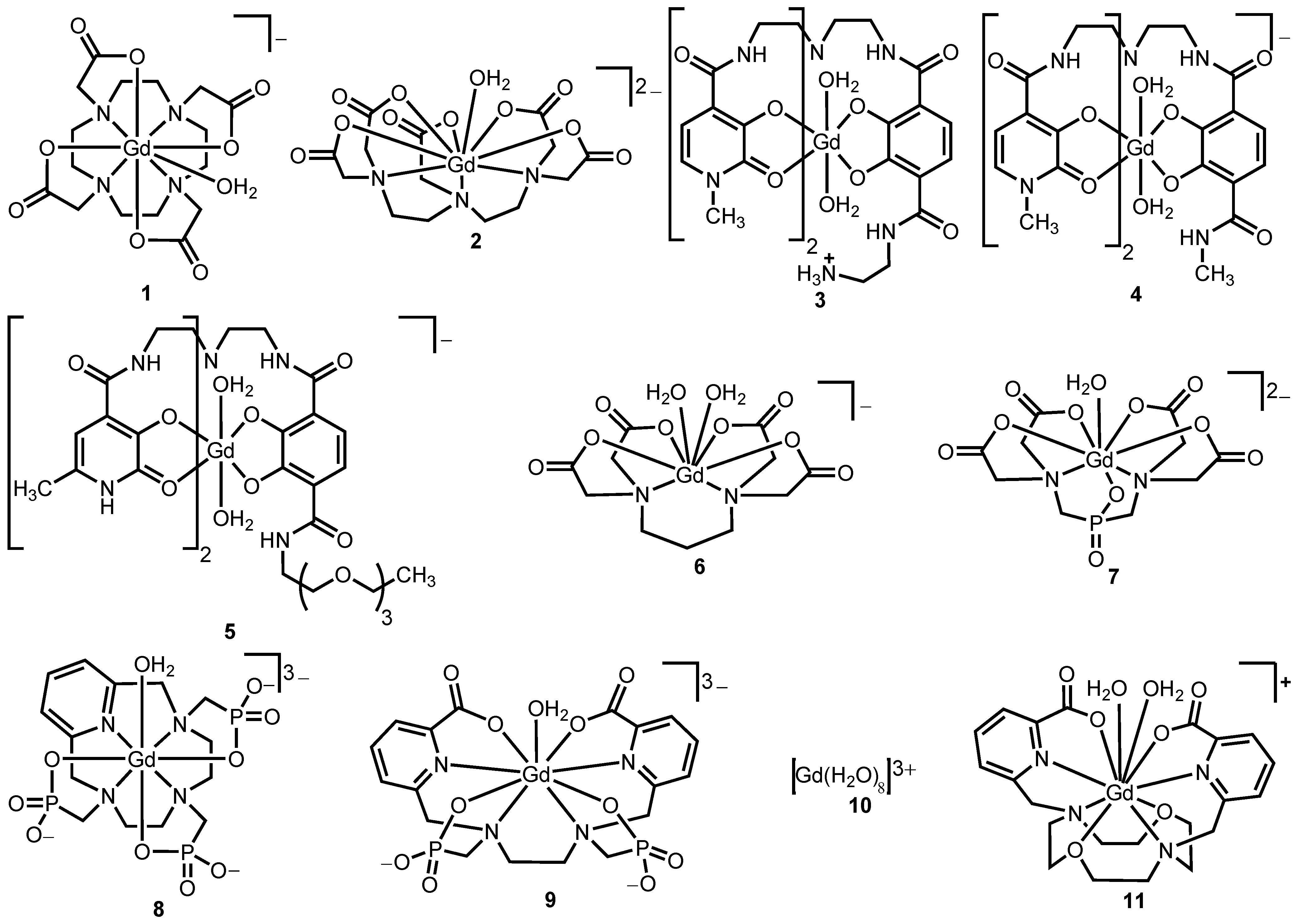
Figure 2.
Representative Gd
III-containing complexes that undergo water exchange via dissociative (
1 and
2), dissociative interchange (
9), associative (
3,
4,
7,
8,
10, and
11), and associative interchange (
5 and
6) processes [
28,
29,
30,
31,
32,
33,
34,
35,
36,
37].
Figure 2.
Representative Gd
III-containing complexes that undergo water exchange via dissociative (
1 and
2), dissociative interchange (
9), associative (
3,
4,
7,
8,
10, and
11), and associative interchange (
5 and
6) processes [
28,
29,
30,
31,
32,
33,
34,
35,
36,
37].
Table 1.
Water-exchange parameters obtained from 17O-NMR spectroscopy and coordination numbers of complexes 1−11.
Table 1.
Water-exchange parameters obtained from 17O-NMR spectroscopy and coordination numbers of complexes 1−11.
Complex
number | kex
(×106 s−1) | ∆V‡
(cm3 mol−1) | q | Coordination number | Mechanism | Reference |
|---|
| 1 | 4.1 | 10.5 | 1 | 9 | dissociative | [31] |
| 2 | 3.3 | 12.5 | 1 | 9 | dissociative | [31] |
| 2 | 7.0 (37 °C) | nr | 1 | 9 | dissociative | [38] |
| 3 | 380 | nr | 2 | 8 | associative a | [30] |
| 4 | 130 | nr | 2 | 8 | associative a | [30] |
| 5 | 53 | ߝ5 | 2 | 8 | associative interchange | [29] |
| 6 | 102 | –1.5 | 2 | 8 | associative interchange | [33] |
| 7 | 27 | nr | 1 | 8 | associative a | [34] |
| 8 | 170 | nr | 1 | 8 | associative a | [32] |
| 9 | 700 | 8.3 | 0.6 | 8 and 9 | dissociative interchange | [36] |
| 10 | 804 | –3.3 | 8 | 8 | associative | [31] |
| 11 | 220 | nr | 1.4 | 9 and 10 | associative a | [37] |
In aqueous solution, Ln
III aqua ions usually have water-coordination numbers of eight or nine, with large lanthanides (La
III to Nd
III) tending toward water-coordination number nine, medium-sized lanthanides (Sm
III to Gd
III) existing mostly in equilibrium between water-coordination numbers eight and nine, and small lanthanides (Tb
III to Lu
III) likely having water-coordination numbers of eight. Many polyaminopolycarboxylate-based ligands occupy eight coordination sites of Ln
III ions allowing space for the coordination of one water molecule. Because these types of complexes have coordinatively saturated (nine coordinate) ground states, they tend to undergo dissociative water exchange through a relatively unstable eight-coordinate transition state. Dissociative exchange often leads to slow water-exchange rates due to the large energy difference between the nine-coordinate ground state and the eight-coordinate transition state, and the rates of exchange could be increased by stabilizing the eight-coordinate transition state or destabilizing the nine-coordinate ground state. On the other hand, water-exchange rates could be decreased by stabilizing the nine-coordinate ground state or destabilizing the eight-coordinate transition state (
Figure 3).
Different from nine-coordinate complexes, eight-coordinate Ln
III-containing complexes used as contrast agents tend to have six or seven coordination sites occupied by a multidentate ligand, enabling the coordination of one or two water molecules in the inner coordination sphere of the Ln
III ion. Due to the coordinatively unsaturated ground state, eight-coordinate complexes tend to undergo associative water exchange through a nine-coordinate transition state. Because the energy gaps between eight-coordinate ground states and nine-coordinate transition states tend to be small, associative exchange mechanisms often display fast water-exchange rates [
28]. Therefore, with eight-coordinate complexes, stabilization of the nine-coordinate transition state is a potential method to increase water-exchange rates, and stabilization of the eight-coordinate ground state is likely to decrease water-exchange rates.
Nine-coordinate polyaminopolycarboxylate complexes
1 and
2 display water-exchange rates on the order of 10
6 s
−1, and eight-coordinate complexes (compounds
3–
8) display relatively fast water-exchange rates (10
7–10
8 s
−1). These differences in water-exchange rates can be attributed in part to the differences in the mechanism of water exchange. The water-exchange rates of hydroxypyridonate (HOPO)-based complexes
3–
5, reported by Raymond and co-workers, are closer to the optimum water-exchange rate (10
8 s
−1) of
T1-shortening agents at 1.5 T than those of clinically approved polyaminopolycarboxylate-based complexes
1 and
2 [
28,
29,
30,
31]. Similar to HOPO-based complexes, the fast water-exchange rates observed for polyaminopolycarboxylate-based complexes
6 and
7 and phosphonate-containing complex
8 can be attributed in part to an associative exchange mechanism [
32,
33,
34].
Figure 3.
Ground and transition states of the water-exchange process. A. Stabilization and destabilization of the ground state lead to slower and faster water exchange-rates, respectively. B. Stabilization and destabilization of the transition state lead to faster and slower water-exchange rates, respectively.
Figure 3.
Ground and transition states of the water-exchange process. A. Stabilization and destabilization of the ground state lead to slower and faster water exchange-rates, respectively. B. Stabilization and destabilization of the transition state lead to faster and slower water-exchange rates, respectively.
As mentioned earlier, water-exchange rates can be increased by stabilizing the transition state formed during the exchange process. This idea is exemplified by the 3-fold faster water-exchange rate of complex
3 relative to complex
4 [
30]. This difference in water-exchange rate is likely due to the accommodation of a third water molecule in the inner-coordination sphere through intra-molecular hydrogen bonding between the primary amine of complex
3 and a third water molecule. Stabilization of the nine-coordinate transition state through intra-molecular hydrogen bonding is likely the cause of the faster water-exchange rate for complex
3 [
30]. In another example, linear complex
9, despite undergoing dissociative exchange, displays a water-exchange rate that is 210-times greater than the rate for linear complex
2 and is nearly as fast as the Gd
III-aqua complex,
10. This extremely fast water-exchange rate is likely due to the existence of both eight- and nine-coordinate species in solution as observed by variable temperature UV–vis spectroscopy of the Eu
III analog of complex
9, implying that the eight-coordinate transition state is stabilized [
35,
36]. In a similar study, but on a macrocyclic complex, Tóth and co-workers demonstrated that macrocyclic complex
11 displayed a water-exchange rate that is 54-times faster than the rate of macrocyclic complex
1, because of an equilibrium between two hydration states (
q = 1 and
q = 2) with coordination-numbers nine and ten based on variable temperature UV-vis spectroscopy. Complex
11 likely undergoes associative exchange based on the large negative activation entropy (–35 J mol
−1 K
−1); however, the authors did not report the volume of activation for complex
11 [
37].
The fast water-exchange rates (107–108 s−1) of complexes 3–9 and 11 are desirable starting points for the design of T1-shortening agents. Designing eight-coordinate complexes with the ability to undergo associative exchange and complexes in which eight- and nine-coordinate species are in equilibrium with one another is desirable to attain fast water-exchange rates for T1-shortening agents. In addition to complexes that undergo associative exchange, complexes that undergo dissociative exchange, but with stable eight-coordinate transition states are likely to display fast water-exchange rates desirable for T1-shortening agents. However, stabilizing the eight-coordinate transition state can be challenging for most polyaminopolycarboxylate-based ligands because these ligands tend to favor complexes with coordination number nine. On the other hand, the fast water-exchange rates of complexes 3–9 and 11 and the relatively slower exchange rates (106 s−1) of nine-coordinate polyaminopolycarboxylate-based complexes 1 and 2 are orders of magnitude too fast to be useful for PARACEST agents. Therefore, designing coordinatively saturated nine-coordinate complexes that undergo dissociative exchange is a useful starting point for the development of PARACEST agents. Moreover, stabilizing the nine-coordinate ground state, for example through hydrogen-bond interactions, would lead to a larger energy gap between the ground and transition states, potentially leading to slow water-exchange rates. Stabilization of the nine-coordinate ground state leads to slow water-exchange rates, which at first glance seems opposite to the fast water-exchange rate observed for complex 3 when the nine-coordinate transition state was stabilized. However, the opposite trends in water-exchange rates can be rationalized based on the difference in the mechanism of exchange: dissociative mechanisms have slow water-exchange rates with stable nine-coordinate ground states, and associative mechanisms have fast water-exchange rates with stable nine-coordinate transition states.
2.2. Modification of the Charge of LnIII-Containing Complex
The charge of Ln
III-containing complexes is another important determinant of the magnitude of water-exchange rate, and this section describes the influence of complex charge and electron density of the coordinating atoms on water-exchange rate using complexes
12–
42 as examples (
Figure 4 and
Table 2). It is important to note that modification of charge toward positively charged complexes is expected to lead to increased proton-exchange rates [
11]; however, discussion of proton-exchange rates is beyond the scope of this review. Also, the differences in water-exchange rates described in this section are explained in terms of charge density only, although the charge density at the Ln
III center closely correlates with Lewis acidity of the Ln
III ion. The influence of the charge of a complex on water-exchange rates is exemplified by the 2- to 8-times slower water-exchange rate that is observed for monoamide complexes
12 and
13 and bisamide complex
14 compared to the all carboxylate complex
2 [
38,
39]. Not only is the charge of a Ln
III-containing complex important for tuning water-exchange rates, but the charge density at the Ln
III center is equally important. The atoms directly coordinating to a Ln
III ion tend to have the greatest impact on the charge density at the Ln
III center. Modifying the coordinating atoms can change the charge density, thereby altering the water-exchange rate. For example, a slowing of water-exchange rates is observed in macrocyclic amide derivatives
15–
17 relative to the all-carboxylate macrocyclic complex
1 [
40]. This slowing of water-exchange rates occurs due to the higher positive charge density at the Gd
III center, regardless of the increasing negative charge of the phosphonate side chains in the series
15–
17. The slowing of water-exchange rates in amide-containing complexes is proportional to the number of amide groups that coordinate to the Ln
III ion.
Figure 4.
Representative Gd
III- and Eu
III-containing complexes that relate the influence of charge (compounds
12–
26) and electron density of coordinating atoms (compounds
27–
42) to water-exchange rate [
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49].
Figure 4.
Representative Gd
III- and Eu
III-containing complexes that relate the influence of charge (compounds
12–
26) and electron density of coordinating atoms (compounds
27–
42) to water-exchange rate [
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49].
Table 2.
Water-exchange rates of complexes
12–
42 discussed in
section 2.2, determined from
17O-NMR spectroscopy and CEST spectra for Gd
III- and Eu
III-containing complexes, respectively.
Table 2.
Water-exchange rates of complexes 12–42 discussed in section 2.2, determined from 17O-NMR spectroscopy and CEST spectra for GdIII- and EuIII-containing complexes, respectively.
| Complex | kex (×106 s−1) | LnIII ion | Reference |
|---|
| 12 | 1.9 | GdIII | [39] |
| 13 | 1.3 | GdIII | [39] |
| 14 | 0.85 (37 °C) | GdIII | [38] |
| 15 | 0.77 | GdIII | [40] |
| 16 | 0.16 | GdIII | [40] |
| 17 | 0.038 | GdIII | [40] |
| 18 | 80 | GdIII | [41] |
| 19 | 20 | GdIII | [41] |
| 20 | 4.4 | GdIII | [41] |
| 21 | 78.7 | GdIII | [42] |
| 22 | 0.457 | GdIII | [42] |
| 23 | 0.98 | GdIII | [43] |
| 24 | 0.60 | GdIII | [43] |
| 25 | 14.1 | GdIII | [44] |
| 26 | 6.29 | GdIII | [44] |
| 27 | 17.6 | GdIII | [45] |
| 28 | 7.4 | GdIII | [45] |
| 29 | 1.7 | GdIII | [46] |
| 30 | 0.66 | GdIII | [46] |
| 31 | 0.0069 | EuIII | [47] |
| 32 | 0.0051 | EuIII | [47] |
| 33 | 0.0037 | EuIII | [47] |
| 34 | 0.0033 | EuIII | [47] |
| 35 | 0.0031 | EuIII | [47] |
| 36 | 0.0028 | EuIII | [47] |
| 37 | 0.018 | EuIII | [48] |
| 38 | 0.012 | EuIII | [48] |
| 39 | 0.005 | EuIII | [48] |
| 40 | 0.00290 | EuIII | [49] |
| 41 | 0.00253 | EuIII | [49] |
| 42 | 0.00211 | EuIII | [49] |
In general, faster water-exchange rates are observed in complexes with less positive charges. For example, an 18- and 4.5-fold difference in water-exchange rate was observed between negatively charged complexes
18 and
19 and structurally similar, but neutral complex
20 [
41]. Another example demonstrated that the water-exchange rate of negatively charged Gd
III-containing 1,4,7,10-tetraazacyclododecane-1,4,7-triacetate (DO3A) derivative
21 with a monodentate phosphonate group displayed a water-exchange rate that is 170-times faster than that of the neutral DO3A derivative
22 with a bidentate ethoxyacetate moiety [
42]. The difference in water-exchange rate between negatively charged and neutral complexes is possibly due to the difference in charge between the two complexes but is likely also due to the change in the functional groups (discussed in
Section 2.3,
Section 2.4). Based on the correlation between fast water-exchange rates and positive charges, complexes with less positive charge at the Gd
III center are desirable for designing Gd
III-containing
T1-shortening agents with fast water-exchange rates.
On the other hand, reducing the water-exchange rate is desirable when designing PARACEST agents, and complexes with high positive charges on the Ln
III ion tend to lead to slow water-exchange rates. As mentioned earlier, the observed 8.2-fold difference in water-exchange rate between negatively charged linear complex
2 and the analogous neutral linear complex
14 at 37 °C is an example of slowing of water-exchange rates with higher overall positive charge and higher positive charge on the Ln
III ion [
38]. Similarly, a 1.6-fold difference in water-exchange rate was observed between neutral, linear complex
23 and the analogous positively charged complex
24 [
43]. Complexes
23 and
24 are structurally similar but have different charges; consequently, the slowing of water-exchange rate can be attributed at least partially to the difference in charge between
23 and
24. In another example, a 2.2-fold difference in water-exchange rate was observed between neutral, macrocyclic Gd
III-containing complex
25 and neutral glycine derivative
26 [
44]. Although complexes
25 and
26 are both neutral, the charge distribution on the complexes is different. Complex
26 has the negative ligand-based charges farther away from the Gd
III ion than in complex
25, resulting in a lower density of positive charge close to the Gd
III ion in complex
25 relative to
26. This difference in charge density at the Gd
III center likely is responsible for the differences in water-exchange rates between complexes
25 and
26. Observations that slow water-exchange rates occur with more positive charges suggest that complexes with more positive charges are potentially useful in designing PARACEST agents with slow water-exchange rates.
To further probe the influence of charge density near a Ln
III ion on water-exchange rates, the electronic effects of coordinating atoms have been studied. As an example, Aime and co-workers demonstrated that the water-exchange rates of neutral DO3A-type complexes can be varied by altering the electronic properties of coordinating macrocyclic nitrogen atoms [
45]. In this example, complexes
27 and
28 differ in the substituent on the
para-position of the phenyl group attached to a coordinating macrocyclic nitrogen atom. A 2.4-fold faster water-exchange rate was observed with complex
27, with an electron-donating amino group, relative to complex
28, with an electron-withdrawing nitro group [
45]. Moreover, the influence of electron-donating methyl groups on water-exchange rates of triamide derivatives of DO3A-type complexes has also been investigated. Complex
29 that contains an electron-donating methyl substituent displayed a 2.6-fold faster water-exchange rate than non-methyl-containing analog
30 [
46]. The relatively fast water-exchange rates in complexes
27 and
29 compared to
28 and
30 can be attributed to the electron density on the macrocyclic nitrogen atoms. This density can neutralize some of the positive charge on the Ln
III ion resulting in weak interactions with water and facilitate the dissociation of coordinated water from the Ln
III ion. In addition to electron density, steric hindrance at the water-coordination site caused by the methyl group in complex
29 (described in
Section 2.3) is likely another contributing factor to the faster water-exchange rate of complex
29 relative to complex
30.
The influence of the electron density of macrocyclic nitrogen donors on water-exchange rates is similar to the influence of electron density on arm donors. In one study, Sherry and co-workers investigated the series of complexes
31–
36 containing substituents with different electronic properties, and demonstrated that water-exchange rate can be tuned by varying the electron density on the coordinating amide group. In this study, mesomeric electron-donating groups (OCH
3) led to fast exchange rates, and mesomeric electron-withdrawing groups (CO
2t-Bu and CN) led to slow exchange. However, the opposite trend was reported for inductively donating (CH
3) and withdrawing (F) groups [
47]. In another example, a series of three complexes (compounds
37–
39) was investigated, where complexes
37 and
38 displayed 3.6- and 2.4-times faster water-exchange rates, respectively, relative to complex
39. This observation was expected based on the calculated Mulliken charges on the coordinating carbonyl oxygen atoms. In this series of complexes, the negative charge on the coordinating carbonyl oxygen atoms ranges from most negative in complex
37 to least negative in complex
39 [
48]. The idea that lower charge density on the coordinating atom leads to slower water-exchange rates was expanded to investigate the suitability of ketones to achieve slow water-exchange rates desirable for PARACEST agents [
49]. In this study of complexes
40–
42, water-exchange rates were 1.1- to 1.4-times faster in complex
40 relative to complexes
41 and
42, respectively. These observations are consistent with water-exchange rate being dependent on the number of poorly electron donating ketone-donor arms in the complex.
Based on the studies described in this section, negatively charged complexes with carboxylate and phosphonate donors lead to fast water-exchange rates compared to positively charged complexes with amide donors. Moreover, water-exchange rates become faster as a function of the negative charge of the complex, low positive charge density at the LnIII center, or both. These features that lead to fast water-exchange rates are desirable for the design of T1-shortening agents. The opposite is true for PARACEST agents, where positively charged complexes with amide and ketone donors tend to be useful as PARACEST agents because complexes with high positive charges favor slow water-exchange rates. However, complexes with poorly donating amide and ketone groups tend to display lower stability relative to negatively charged complexes with strong carboxylate donors, and stability of these complexes is critical in designing contrast agents.
2.3. Modification of Steric Hindrance at the Site of Water Coordination
Another parameter that influences water-exchange rates is the degree of steric hindrance at the water-coordination site. This parameter is related to mechanism of exchange and isomer ratio (described in
Section 2.5); increased steric hindrance leads to faster water-exchange rates in complexes that undergo dissociative water exchange because crowding the water-coordination site favors dissociation of the coordinated water molecule, which is the rate-limiting step. The influence of steric hindrance at the site of water coordination on water-exchange rate is described in this section using complexes
43–
65 (
Figure 5 and
Table 3).
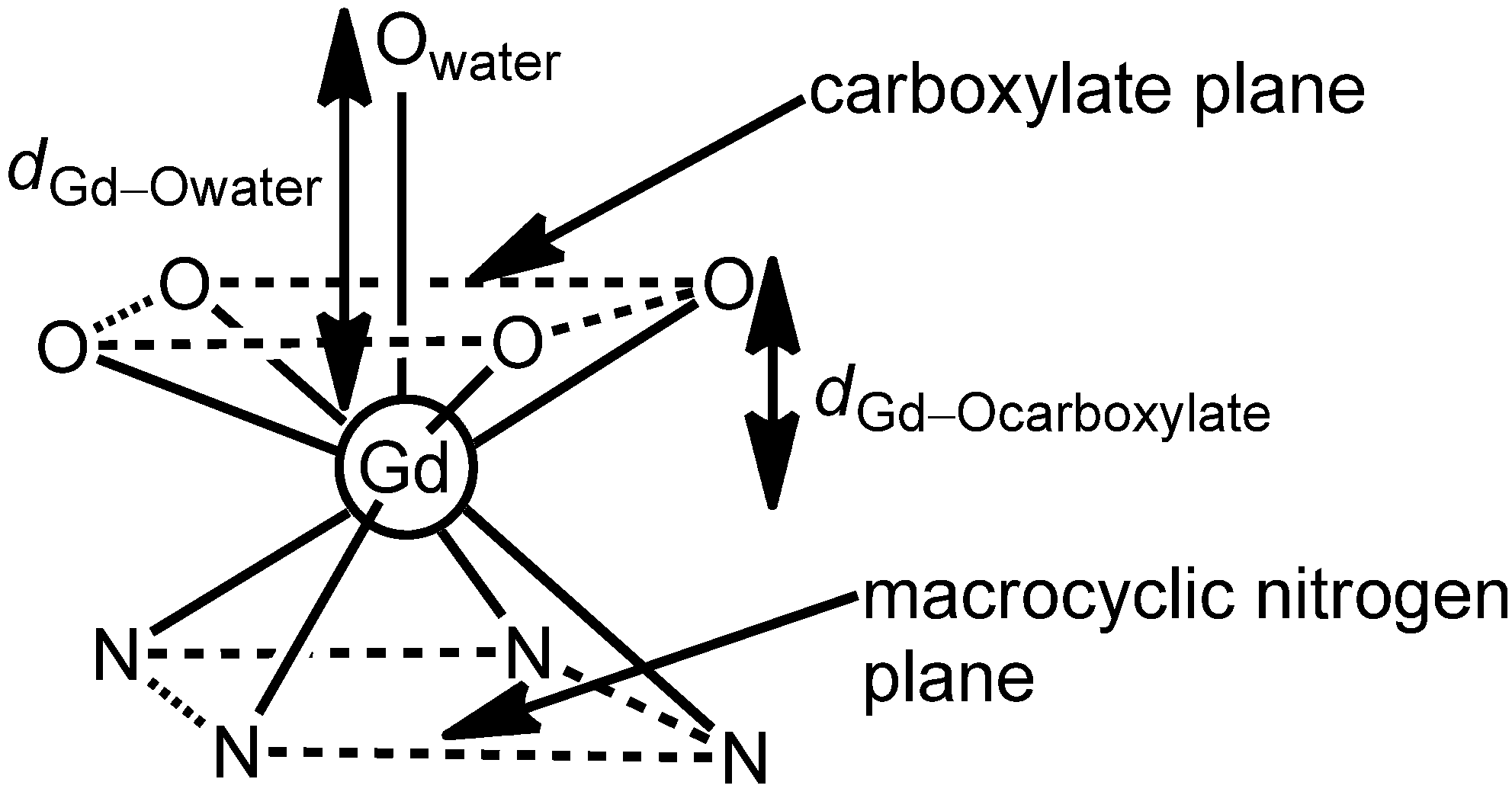
Merbach and coworkers demonstrated that complex
43, an analog of macrocyclic complex
1 with an extended macrocyclic backbone that is one carbon longer than the macrocyclic backbone of complex
1, has a 66-fold faster water-exchange rate relative to complex
1. The difference in water-exchange rates was attributed to the increased steric encumbrance at the site of water coordination that results from the difference between the five-membered and six-membered rings formed between the macrocycles and the Ln
III ions [
10]. The argument of increased steric hindrance is supported by the differences in the Gd
III–O
carboxylate distances and the O
carboxylate–Gd
III–O
carboxylate angles (
Figure 6). Although complexes
1 and
43 have similar Gd
III–O
water distances of 2.45 and 2.48 Å, respectively, they have markedly different Gd
III–O
carboxylate distances of 0.70 and 0.83 Å, respectively. These data indicate that the negatively charged carboxylate plane is closer to the axially coordinated water molecule in complex
43 than in complex
1. This argument is further supported by the differences in the O
carboxylate–Gd
III–O
carboxylate angles (136.7 and 142.7° for complex
43 vs 146° for
1) implying that the carboxylate plane around the Gd
III ion is more compact in complex
43 compared to complex
1 [
10].
Figure 5.
Representative Gd
III-containing complexes
43–
65 that relate the influence of steric hindrance at the water-coordination site to water-exchange rate [
10,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62].
Figure 5.
Representative Gd
III-containing complexes
43–
65 that relate the influence of steric hindrance at the water-coordination site to water-exchange rate [
10,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62].
The steric difference between five and six-membered rings in chelation also can be observed in the arms of the complexes. Complex
44, with an extended carboxylate arm that is one carbon longer than the arms in complex
1, displayed a 15-fold faster water-exchange rate than complex
1. This difference is likely due to the increased steric hindrance at the site of water coordination, similar to the system with an extended macrocycle [
50]. Based on the water-exchange rates of
43 and
44, extending the macrocyclic backbone has a more pronounced effect in increasing the water-exchange rates of DOTA-type complexes compared to extending one of the arms.
Table 3.
Water-exchange rates of Gd
III-containing complexes
43–
65 discussed in
section 2.3 from
17O-NMR spectroscopy.
Table 3.
Water-exchange rates of GdIII-containing complexes 43–65 discussed in section 2.3 from 17O-NMR spectroscopy.
| Complex | kex (×106 s−1) | Reference |
|---|
| 43 | 270 | [10] |
| 44 | 61 | [50] |
| 45 | 110 | [51] |
| 46 | 1.6 | [52] |
| 47 | 81.2 | [53] |
| 48 | 1.1 | [53] |
| 49 | 86 | [54] |
| 50 | 0.6 | [54] |
| 51 | 330 | [55] |
| 52 | 31 | [56] |
| 53 | 80 | [50] |
| 54 | 31 | [50] |
| 55 | 18 (37 °C) | [57] |
| 56 | 12 (37 °C) | [57] |
| 57 | 11 (37 °C) | [57] |
| 58 | 10 (37 °C) | [57] |
| 59 | 9.26 (37 °C) | [57] |
| 60 | 43 | [58] |
| 61 | 11 | [59] |
| 62 | 34 | [54] |
| 63 | 59 | [60] |
| 64 | 29 | [61] |
| 65 | 63 | [62] |
Figure 6.
Coordination polyhedron of Gd
III-containing complex
43 [
10].
Figure 6.
Coordination polyhedron of Gd
III-containing complex
43 [
10].
In another example, DOTA-monoamide derivative
45, with an amide group separated by an ethylene bridge from the macrocyclic nitrogen, displayed a water-exchange rate that is 69-times faster than the analogous DOTA-monoamide complex
46 [
51,
52]. The fast water-exchange rate of
45 is likely due to the steric hindrance at the water-coordination site, imposed by the seven membered chelate formed by the ethylene bridge between the macrocyclic nitrogen and the amide oxygen compared to the five membered chelate in DOTA-monoamide complex
46. A similar trend in water-exchange rates was observed between DOTA-monoamide complexes
47 and
48 where complex
47 displayed a water-exchange rate that is 74-times faster than that of complex
48 [
53]. The faster water-exchange rate of complex
47 relative to complex
48 is possibly due to the steric constraints at the site of water-coordination caused by the six membered chelate in
47 relative to the five membered chelate in
48. Similar to the fast water-exchange rates observed for DOTA-monoamide derivatives
45 and
47, a 143-fold difference in water-exchange rate was observed between triaza-macrocyclic complex
49 with a propionate arm and the analogous triaza-macrocyclic complex
50 with an acetate arm. The difference in water-exchange rate between complexes
49 and
50 was reportedly due to the increased steric hindrance at the site of water coordination caused by the extended propionate arm [
54]. These studies suggest that the steric environment caused by ring size is generalizable to different sizes of macrocyclic ligand backbones.
The work on elongation of the macrocyclic backbones was extended into linear diethylene triamine pentaacetate (DTPA)-type systems by Merbach and coworkers. In one study, complex
51, which is structurally similar to complex
2 but with an extra carbon in the nitrogen backbone, displayed a 100-fold faster water-exchange rate relative to that of complex
2. The faster water-exchange rate of
51 relative to
2 was reported to be due to the steric hindrance caused by the extra carbon in the backbone of
51 [
55]. In another example, complex
52 with an ether-based oxy-ethylene bridge in the backbone led to a water-exchange rate that is 9.4-times faster than that of complex
2. The difference in water-exchange rate between
2 and
52 can be attributed to the 3% longer Gd–O
water distance in
52 compared to
2, possibly due to steric hindrance caused by the ethylene bridge on the backbone of
52, which facilitates the dissociation of the coordinated water molecule [
56]. However, because there are multiple structural differences between complexes
2 and
52, it is likely that no single difference is responsible for the entire change in water-exchange rate.
In addition to extending the linear backbone, DTPA-type complexes with extended arms have been synthesized to study the influence of arm extensions on water-exchange rates. For example, a 24-fold difference in water-exchange rate was observed between DTPA analog
53 with an extended carboxylate arm and the parent complex
2 [
50]. Furthermore, with DTPA-type complexes, the position of the extended carboxylate arm (whether terminal or central) also influences the water-exchange rate. Complex
53, with a terminal extended carboxylate arm, has a water-exchange rate that is 2.6-times faster than that of complex
54 with a central extended carboxylate arm [
50]. This observation implies that the extension of terminal carboxylates causes more steric hindrance at the site of water coordination and leads to faster water-exchange rates compared to extension of the central carboxylate arm. Unlike with macrocyclic complexes, extension of the nitrogen backbone with linear complexes generally has a higher impact on water-exchange rates than extending one of the carboxylate arms.
In addition to extending multidentate ligand backbones and arms, the influence of steric crowding caused by backbone substitution on the water-exchange rates was studied using the series of linear complexes
55–
59 [
57]. In this series, the complexes differ from one another in the alkyl-group substitution on the ethylene carbons. The dialkyl substituted complex
55 has a water-exchange rate that is 2.6-times faster than complex
2 at 37 °C, and monoalkyl substituted complexes
56–
59 display water-exchange rates that are 1.3- to 1.7-times faster than the parent unsubstituted complex
2 at 37 °C. The faster water-exchange rates of the alkyl substituted complexes relative to parent complex
2 are likely due to the increased steric hindrance in the inner-coordination sphere [
57]. A similar study demonstrated that dimethyl-substituted complex
60 had a 1.3-fold faster water-exchange rate than parent complex
6 [
58]. A confounding point with complex
6 is that it has two different reported water-exchange rates (102 × 10
6 s
−1 [
33] and 33 × 10
6 s
−1 [
58]), and the 1.3-fold difference in rates is not observed if the wrong value is used when comparing complexes
6 and
60. As with the other systems, this result is likely due to the increased steric hindrance from the methyl substitution.
In addition to using alkyl groups to investigate the influence of steric crowding on water-exchange rates, complexes containing bulky phosphonate arms display faster water-exchange rates relative to complexes with no phosphonates. This difference in rates is likely at least partially due to the steric hindrance at the site of water-coordination caused by the large size of the phosphonate groups. For example, phosphonate-containing complex
61 displayed a water-exchange rate that is 3.5-times faster than that of the parent complex
2 [
59]. In another example, phosphonate-containing triaza-macrocyclic complex
62 displayed a 57-fold faster water-exchange rate relative to the non-phosphonate-containing analog
50 [
54].
Rigidifying the backbone of polyaminopolycarboxylate-based ligands is another strategy that often results in faster water-exchange rates, possibly due to enhanced steric crowding caused by substituents used to rigidify the ligand backbone. For example, backbone rigidification in complex
63 using two piperidine moieties led to a 1.9-fold faster water-exchange rate than non-rigid complex
52 [
60]. In another example, complex
64 with a cyclohexylene-bridge-containing rigidified macrocycle led to a water-exchange rate that was 2.4-fold faster than that of non-rigid
25 [
61]. Another example demonstrated that a rigid macrocyclic ligand framework based on diazapyridinophane (complex
65) displayed a 15-fold faster water-exchange rate than complex
1 [
62]. The faster water-exchange rates of rigidified complexes
63–
65 with respect to non-rigid analogs
52,
25, and
1 are likely due to a variety of differences, but one of the influences is the increased steric hindrance brought about by the cyclic functionalities that enhance the rigidity of the backbone.
Increasing steric hindrance at the site of water coordination—using strategies including extension of ligand backbone or arms, incorporating bulky phosphonates, introducing bulky alkyl groups on the ligand backbone, and rigidification of the ligand backbone—leads to complexes with fast water-exchange rates; therefore, these strategies are desirable in T1-shortening agents. However, it is important to note that extension of ligand backbone and arms often leads to less stable complexes relative to analogous complexes without backbone and arm extensions, and stability is a critical consideration in the design of contrast agents. On the other hand, releasing steric encumbrance at the site of water coordination using less sterically demanding coordinating groups leads to complexes with slow water-exchange rates that might be desirable for use as PARACEST agents.
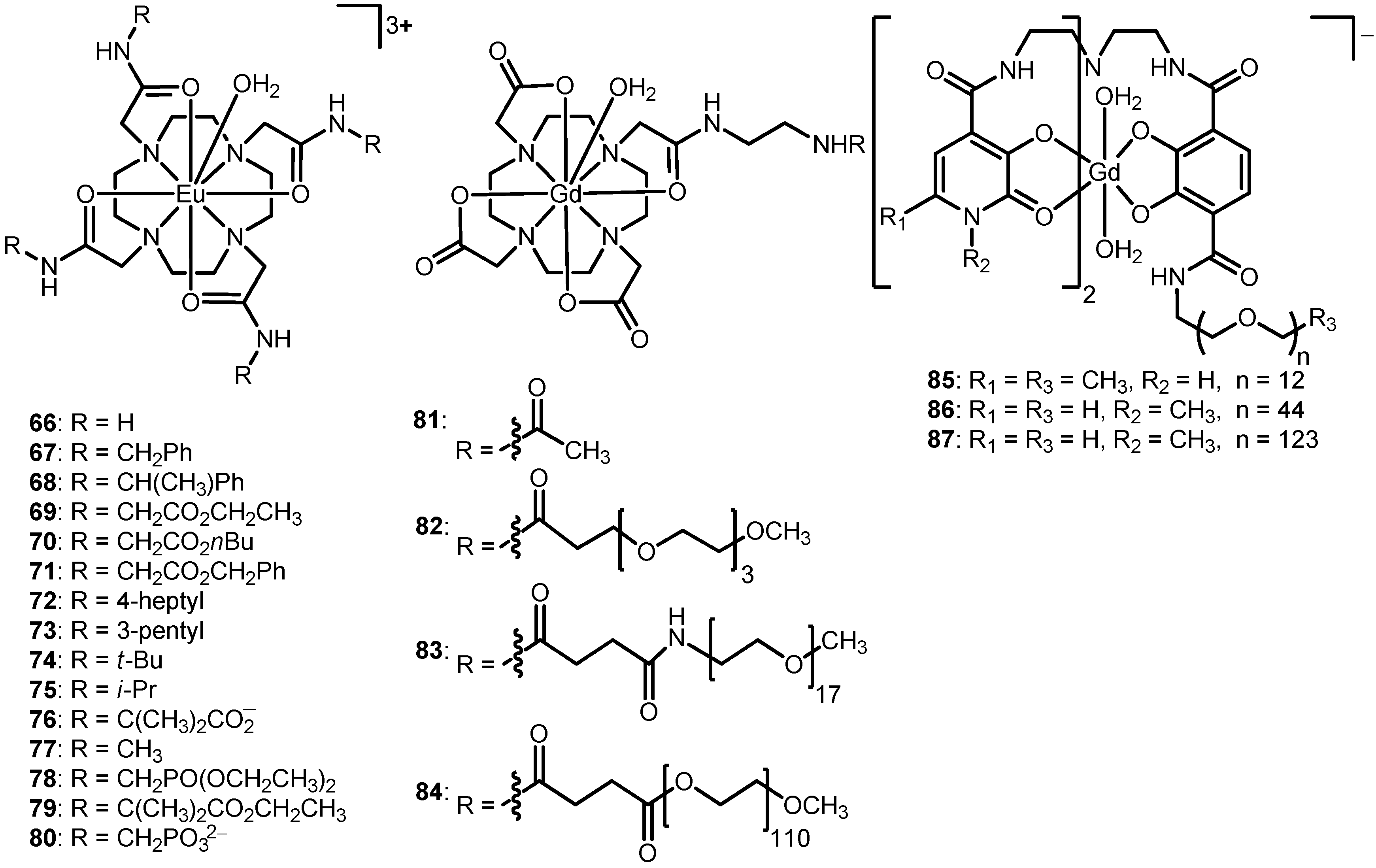
2.4. Modification of the Ligand Side Chains
The water-exchange rates of Ln
III-containing complexes tend to depend on the chemical nature of ligand side chains including bulkiness, polarity, and charge as has been described to some extent in the previous sections. This section focuses on the influence of ligand side chains on water-exchange rate using complexes
4 (
Figure 2),
40 (
Figure 4) and
66–
87 (
Figure 7 and
Table 4,
Table 5). For example, a series of Eu
III-tetraamide-based complexes (
40 and
66–
71) was synthesized to investigate the influence of ligand side chains in terms of water-accessible surface area (calculated from molecular modeling) on water-exchange rates [
63]. The complexes in the series likely undergo dissociative water exchange based on their nine coordinate ground state structures. Based on the findings of this study, the water-exchange rate decreased with decreasing water-accessible surface area (
66 >
40 >
67 >
68 >
69 >
70 =
71) [
63]. The rationale used to explain these data is that large water-accessible surfaces give rise to a large amount of second-sphere hydration, subsequently decreasing the activation energy for the water-exchange process. Moreover, any substituent that facilitates the access of incoming water molecules through second-sphere hydration was reported to lead to fast water-exchange rates. On the other hand, substituents on the ligand side chains that hinder the access of incoming water molecules lead to slow water-exchange rates [
63].
Figure 7.
Representative complexes
66–
87 that relate the influence of bulkiness and polarity of ligand side chains and water-exchange rates [
15,
27,
63,
64,
65,
66,
67,
68,
69].
Figure 7.
Representative complexes
66–
87 that relate the influence of bulkiness and polarity of ligand side chains and water-exchange rates [
15,
27,
63,
64,
65,
66,
67,
68,
69].
Table 4.
Water-exchange rates of Eu
III-containing DOTA-tetraamide complexes
40 and
66–
80 discussed in
Section 2.4.
Table 4.
Water-exchange rates of EuIII-containing DOTA-tetraamide complexes 40 and 66–80 discussed in Section 2.4.
| Complex | kex (×106 s−1) | Side chain | Method | Reference |
|---|
| 40 | 0.007 | CH2CO2t-Bu | NMR spectroscopyc | [63] |
| 66 | 0.01 | H | NMR spectroscopyc | [63] |
| 66 | 0.0083a | H | 1H-NMR spectroscopy | [65] |
| 67 | 0.005 | CH2Ph | NMR spectroscopyc | [63] |
| 68 | 0.004 | CH(CH3)Ph | NMR spectroscopyc | [63] |
| 69 | 0.003 | CH2CO2CH2CH3 | NMR spectroscopyc | [63] |
| 69 | 0.0013a | CH2CO2CH2CH3 | 1H-NMR spectroscopy | [27] |
| 70 | 0.002 | CH2CO2nBu | NMR spectroscopyc | [63] |
| 71 | 0.002 | CH2CO2CH2Ph | NMR spectroscopyc | [63] |
| 72 | >1 a | 4-heptyl | CEST | [15] |
| 73 | 0.059 a | 3-pentyl | CEST | [15] |
| 74 | 0.10 a | t-Bu | CEST | [15] |
| 75 | 0.027 a | i-Pr | CEST | [15] |
| 76 | 0.0096 a | C(CH3)2CO2− | CEST | [15] |
| 77 | 0.0064 a | CH3 | 1H-NMR spectroscopy | [65] |
| 78 | 0.00077 a | CH2PO(OCH2CH3)2 | 1H-NMR spectroscopy | [66] |
| 79 | 0.0048 a | C(CH3)2CO2CH2CH3 | CEST | [15] |
| 80 | 0.015 b | CH2PO32− | 1H-NMR spectroscopy | [27] |
Table 5.
Water-exchange rates and water-coordination numbers of Gd
III-containing polyaminopolycarboxylate-type (
81–
84) and HOPO-based (
4 and
85–
87) PEG conjugates discussed in
section 2.4.
Table 5.
Water-exchange rates and water-coordination numbers of GdIII-containing polyaminopolycarboxylate-type (81–84) and HOPO-based (4 and 85–87) PEG conjugates discussed in section 2.4.
| Complex | kex (×106 s−1) | q | Reference |
|---|
| 4 | 130 | 2 | [30] |
| 81 | 2.7 | 0.9 | [67] |
| 82 | 1.5 | 0.9 | [67] |
| 83 | 0.83 | 1.0 | [67] |
| 84 | 0.67 | 0.8 | [67] |
| 85 | 77 | 1 | [68] |
| 86 | 53 | 1 | [69] |
| 87 | 32 | 1 | [69] |
In addition to studying the influence of water-accessible surface area on water-exchange rates, a recent study correlated the steric bulk of ligand side chains and water-exchange rates using complexes
72–
75. Based on this study, large amounts of steric bulk on ligand side chains resulted in fast water-exchange rates [
15]. However, this trend cannot be generalized to any bulky group because there are other factors involved including polarity of the side chains. The relationship between steric bulk of ligand side chains and water-exchange rate is similar to that observed in section 2.3, where water-exchange rate increased with steric hindrance imposed at the site of water-coordination. Furthermore, at least a 17-times faster water-exchange rate was observed for more bulky heptyl-containing complex
72 relative to less bulky pentyl-containing complex
73, and a 3.7-fold faster water-exchange rate was observed for complex
74 (with more bulky
tert-butyl substituents) relative to
75 (with less bulky isopropyl substituents) [
15]. To explain these observations, it was suggested that side chains with bulky substituents interact with one another through steric interactions making the coordinated water exposed to bulk water [
15]. The exposure of coordinated water to bulk water facilitates water exchange leading to fast water-exchange rates.
Because the steric bulk and polarity of side chains are intertwined, complexes
74 and
76 were used to isolate the effects of steric bulk and polarity. Despite being similar in terms of steric bulk, complexes
74 and
76 display different water-exchange rates (
Table 4), suggesting that the polarity of the side chains influences the magnitude of water-exchange rates. Complex
74 displayed a 10-fold faster water-exchange rate than complex
76 with polar carboxylate groups. This difference in water-exchange rate can be attributed to the ability of polar groups to sustain second-sphere hydration via hydrogen-bond interactions, thereby stabilizing the nine-coordinate ground state leading to slow water-exchange rates [
15]. This trend in water-exchange rates is the opposite of what was reported for complexes
40 and
66–
71 likely because both polarity and bulkiness of ligand side chains contribute to water-exchange rates and the difficulty of separating one from the other.
In general, water-exchange rates of Eu
III-containing DOTA-tetraamide-based complexes tend to be slower when the polarity of ligand side chains is greater (primary amides < alkyl substituents < carboxylates < phosphonates) [
64]. An example of this trend can be observed in the differences in water-exchange rates among complexes
66,
69,
77, and
78 [primary amide-containing complex
66 > alkyl substituent-containing complex
77 > carboxylate-ester-containing complex
69 > phosphonate-ester-containing complex
78] (
Table 4) [
27,
65,
66]. The slowing of water-exchange rates as a function of polarity can be attributed to the stabilization of the nine-coordinate ground state through hydrogen-bond interactions. Although polar ligand side chains are expected to result in slow water-exchange rates, it was observed that complexes bearing negatively charged side chains display faster water-exchange rates than their neutral analogs [
64]. For example, a 2-fold difference in water-exchange rate was observed between negatively charged carboxylate-side-chain-containing analog
76 and neutral ethyl-ester-side-chain-containing complex
79 [
15]. In another example, a 52-fold difference in water-exchange rate was observed between neutral phosphate-ester-side-chain-containing complex
78 and negatively charged phosphonate-side-chain-containing analog
80 [
27,
66]. The slower water-exchange rates of neutral ester-containing complexes relative to their negatively charged analogs is likely due to the ability of ethyl groups to block incoming water molecules, subsequently lowering the number of water molecules available for exchange [
15]. The observation of complexes with negatively charged side chains leading to fast water-exchange rates is consistent with section 2.2 that relates negatively charged complexes to fast water-exchange rates.
Based on the idea of polar side chains leading to slow water-exchange rates, a recent study explored the ability of polar polyethylene glycol (PEG) to slow water-exchange rates as a function of length of PEG [
67]. A series of polyaminopolycarboxylate-type complexes,
81–
84, were used in this study. A 4-fold difference in water-exchange rate was observed between complex
81 (without PEG) and
84 (with a PEG length of 110 monomer units) (
Table 5) [
67]. The difference in water-exchange rates as a function of length of PEG is likely due to the formation of a hydrogen-bond network that stabilizes the nine-coordinate ground state, and longer PEG lengths would enable more hydrogen bonding. It is important to note that the influence of steric blocking caused by PEG was minimal based on the similar water-coordination numbers (
q) obtained for complexes
81–
84 (
Table 5) [
67]. The slowing of water-exchange rates as a function of PEG length in the polyaminopolycarboxylate-type system is similar to observations in studies with HOPO-based PEG conjugates
85–
87 [
68,
69]. Moreover, a 4-fold difference in water-exchange rate was observed between parent HOPO complex
4 without PEG and HOPO complex
87 with the PEG moiety with 123 monomer units (
Table 5) [
68,
69]. The observed difference in water-exchange rate is likely due to hydrogen-bonding and steric interactions brought about by PEG moieties.
Based on the studies described in this section, LnIII-containing complexes with bulky, hydrophobic side chains are desirable in designing T1-shortening agents, because they are expected to lead to fast water-exchange rates. On the other hand, LnIII-containing complexes with neutral hydrophilic side chains are suitable for developing PARACEST agents because hydrophilic side chains tend to result in slow water-exchange rates.
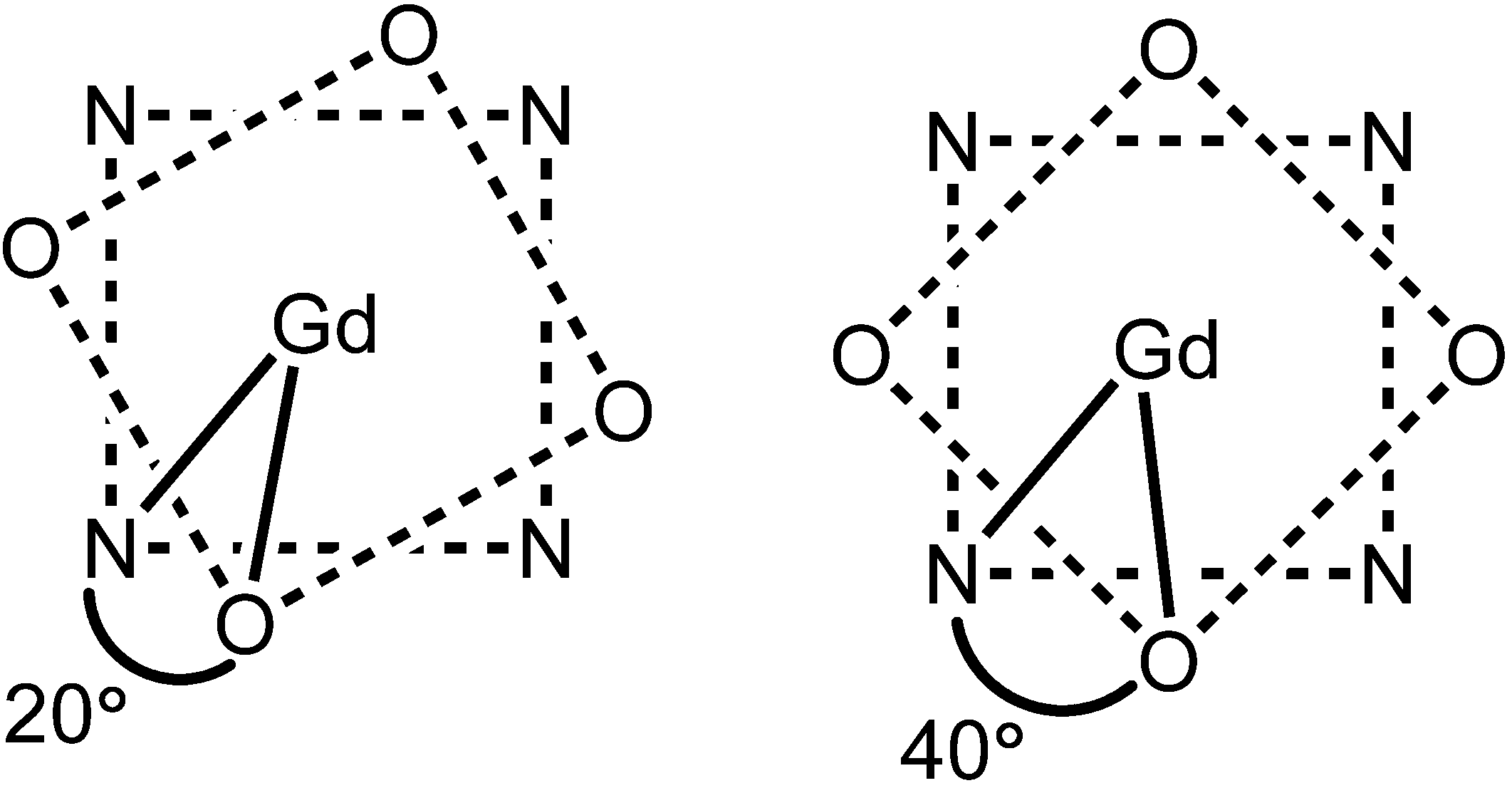
2.5. Modification of TSAP/SAP Ratio for DOTA-Type Complexes
The water-exchange rates of DOTA-type Ln
III-containing complexes are also governed by the ratio of the twisted-square-antiprism (TSAP) to square-antiprism (SAP) isomers. In this section, the influence of the TSAP/SAP ratio on water-exchange rate will be discussed using complexes
18–
20 and
88–
97 (
Figure 4,
Figure 8 and
Table 6). DOTA-type complexes exist as two diasteriomers in aqueous solution, namely TSAP and SAP that differ in the arrangement of their carboxylate arms and macrocyclic rings. Specifically, these two isomers differ in the twist angle between macrocyclic nitrogen plane and carboxylate oxygen plane: the TSAP isomer has a narrow O–Gd
III–N twist angle (20°), and the SAP isomer has a wide O–Gd
III–N twist angle (40°) (
Figure 9). These isomers have the ability to interconvert by ring inversion or arm rotation [
70].
Figure 8.
Representative complexes
88–
97 that relate the influence of TSAP/SAP ratios with water-exchange rates of DOTA-type complexes [
41,
71,
72,
73,
74,
75,
76,
77,
78].
Figure 8.
Representative complexes
88–
97 that relate the influence of TSAP/SAP ratios with water-exchange rates of DOTA-type complexes [
41,
71,
72,
73,
74,
75,
76,
77,
78].
Table 6.
Water-exchange rates and TSAP/SAP ratios of Gd
III-containing complexes (
18–
20 and
88–
97) discussed in
Section 2.5.
Table 6.
Water-exchange rates and TSAP/SAP ratios of GdIII-containing complexes (18–20 and 88–97) discussed in Section 2.5.
| Complex | kex (×106 s−1) | TSAP/SAP ratio | kex (×106 s−1) for TSAP | kex (×106 s−1) for SAP | References |
|---|
| 18 | 80 | 1.3 | nr | nr | [41] |
| 19 | 20 | 0.56 | nr | nr | [41] |
| 20 | 4.4 | 0.39 | nr | nr | [41] |
| 88 | 140 a | 0.78 b | 330 | 0.43 | [71] |
| 89 | 31 a | 0.77 b | 70 | 0.74 | [71] |
| 90 | 71 | 1.5 | nr | nr | [72] |
| 91 | 61.7 | 1.5 b | nr | nr | [73] |
| 92 | 58 | ¥ | nr | nr | [74] |
| 93a | 15.4 | 2 | nr | nr | [75] |
| 93b | 9.0 | 0.75 | nr | nr | [75] |
| 93c | 3.45 | 0.20 | nr | nr | [75] |
| 94 | 5 | 0.08 | nr | nr | [76] |
| 95 | 67 | ¥ | nr | nr | [77] |
| 96 | 8.3 | 0 | nr | nr | [77] |
| 97 | 4.5 (37 °C) | 0.4 | 110 | 1.6 | [78,79] |
Figure 9.
Top-down view of (left) TSAP and (right) SAP isomers with the O–GdIII–N twist angles indicated. In these views, the oxygen plane is above the GdIII ion and the nitrogen plane is below the GdIII ion.
Figure 9.
Top-down view of (left) TSAP and (right) SAP isomers with the O–GdIII–N twist angles indicated. In these views, the oxygen plane is above the GdIII ion and the nitrogen plane is below the GdIII ion.
In general, the measured water-exchange rate of DOTA-type complexes is the weighted average of the water-exchange rates of the TSAP and SAP isomers. Between the two isomers, the TSAP isomer tends to display a water-exchange rate that is approximately 50–200 times faster than the SAP isomer in DOTA-tetraamide type complexes [
70,
80]. The faster water-exchange rate in the TSAP isomer relative to the SAP isomer is likely due to the higher amount of steric encumbrance at the site of water coordination, which facilitates the dissociation of coordinated water similar to fast water-exchange rates (discussed in
Section 2.3) due to the steric hindrance at the site of water-coordination. Because the TSAP and SAP isomers display different water-exchange rates, the water-exchange rates of DOTA-type complexes can be tuned by changing the relative abundance of the two isomers in solution using coordination-chemistry-based strategies. Complexes with sterically bulky substituents tend to favor the TSAP isomer, while complexes without bulky substituents favor the SAP isomer. However, it is important to note that relative abundance of the two isomers also depends on other factors including ionic radius, solvent, ionic strength of the solution, and salt composition [
81,
82,
83].
To study the influence of steric bulk on amide nitrogen atoms on the relative abundance of the TSAP and SAP isomers, complexes
88 and
89 were investigated. The TSAP/SAP ratios of 0.78 and 0.77 were measured for Eu
III-containing complexes
88 and
89, respectively. These ratios are 2-times greater than Eu
III-containing complex
1 that has a TSAP/SAP ratio of 0.36 [
71]. The water-exchange rates obtained for the Gd
III-containing complexes
88 and
89 indicated that the TSAP isomers displayed 782- and 95-times faster water-exchange rates, respectively, than the SAP isomers [
71].
In addition to introducing steric bulk on amide nitrogen atoms, other bulky DOTA-type complexes increase the population of the TSAP isomer relative to the SAP isomer, leading to faster water-exchange rates. This idea can be exemplified using phosphonate-containing DOTA-type systems [
41,
72,
73]. In one example, a TSAP/SAP ratio of approximately 1.5 was observed for phosphonate-containing complex
90 and resulted in a 17-fold faster water-exchange rate relative to that of complex
1 [
72]. In another example, a TSAP/SAP ratio of 1.5 for phosphonate-containing complex
91, which is 4-times greater than that of complex
1, resulted in a water-exchange rate that is 15-times faster than complex
1 [
73]. Interestingly, the series of phosphonate-containing complexes
18–
20 were used to demonstrate that the TSAP/SAP ratios are larger with complexes of more negative charge. The TSAP/SAP ratios were 1.3 and 0.56 in negatively charged complexes
18 and
19, respectively, and 0.39 in neutral complex
20 suggesting that the TSAP isomer dominates in complexes with more negative charge. The difference in the populations of the TSAP isomers was reflected in the water-exchange rates of complexes
18–
20: 18- and 4.5-fold differences in water-exchange rates were observed in complexes
18 and
19, respectively, relative to complex
20 [
41]. The differences in water-exchange rates of complexes
18–
20 are likely due to the difference in the TSAP isomer population, the difference in complex charge as described in
Section 2.2, or both factors. In addition to bulky phosphonate-containing complexes, complex
92 with a bulky bis-methylene picolinate platform was explored. Complex
92 displayed a water-exchange rate that is 14-times faster than that of complex
1 [
74]. The faster water-exchange rate is likely because complex
92 exists exclusively as the distorted TSAP isomer [
74].
In addition to incorporating steric bulk on the ends of the arms farthest from the macrocycle, DOTA-type complexes with substituents at the α-positions of the pendent arms increase the abundance of the TSAP isomer. As an example, Parker and co-workers observed that a Gd
III-containing DOTA analog with propionate groups in the α-positions of the pendent arms contains the three isomers
93a,
93b, and
93c. In this series, the water-exchange rate increases with increasing TSAP/SAP ratios [
75]. Interconversion between TSAP and SAP isomers in complexes
93a and
93b was reported to occur only through ring inversion because the bulky propionate group in the α-position of the pendent arm hinders arm rotation affording TSAP/SAP ratios that are 10- and 3.8-times higher, respectively, than in complex
93c. The low TSAP/SAP ratio in complex
93c is likely due to interconversion between TSAP and SAP isomers through arm rotation as well as ring inversion. The differences in the TSAP/SAP ratios are likely responsible for the 4.5- and 2.6-fold faster water-exchange rates of complexes
93a and
93b, respectively, relative to complex
93c [
75]. In addition to blocking arm rotation, complexes containing bulky substituents on the macrocycle were synthesized with the objective of blocking ring inversion in DOTA-type complexes. A study of complex
94 with a
p-nitrobenzyl group on the macrocycle demonstrated that TSAP to SAP interconversion through ring inversion can be blocked. However, arm rotation persisted affording a TSAP/SAP ratio of 0.08 resulting in a water-exchange rate that is only 1.2-times faster than that of complex
1 [
76]. Based on the observations that arm rotation and ring inversion can be blocked by arm and ring substitutions, respectively, complexes with both arm and ring substitutions were reported to control isomer type. Furthermore, controlling the chirality at five carbons led to preference for a single isomer. For example, complexes
95 (
S-
SSSS) and
96 (
S-
RRRR) with substituents on the macrocycle as well as the pendant arms were reported to exist exclusively as TSAP and SAP isomers, respectively [
77]. The water-exchange rate of
95 (
S-
SSSS) in the TSAP form was 8-times faster than the water-exchange rate of
96 (
S-
RRRR) in the SAP form [
77]. These examples substantiate that complexes existing exclusively in the TSAP form have faster water-exchange rates relative to complexes existing in the SAP form.
In addition to experimental studies carried out to tune TSAP/SAP ratios in DOTA-type complexes, a computational study was carried out on complex
97 to understand the rationale behind the TSAP isomer leading to fast water-exchange rates [
78]. This computational study suggested that the TSAP isomer displays hydrogen-bond interactions between pendant arms and second-sphere water, enabling the coordinated water to exchange readily with bulk water, leading to fast water-exchange rates. On the other hand, the SAP isomer is expected to form hydrogen-bond interactions between second-sphere and inner-sphere water, stabilizing the ground state in dissociative exchange processes and leading to slow water-exchange rates. Based on this study, increasing the ability of pendant arms to form hydrogen bonds without lengthening side chains is likely to result in large proportions of the TSAP isomer with DOTA-type complexes. Although the predictions of this study at first glance appear to be in contrast with the studies discussed in section 2.4, the difference is likely because the side chains of complexes
81–
87 are long compared to side chain of complex
97. The length of the side chain influences how far from the Ln
III ion hydrogen bonding occurs, and this distance is an important difference between the complexes described in sections 2.4 and 2.5.
Complexes with large TSAP/SAP ratios are expected to lead to fast water-exchange rates necessary for conventional T1-shortening agents because the TSAP isomers display fast water-exchange rates. Large TSAP/SAP ratios are likely to form with complexes that contain bulky substituents on the α-position of pendant arms, on the macrocycle, and on amide nitrogen atoms as well as from complexes with side chains that facilitate hydrogen-bonding interactions near the inner-sphere water. On the other hand, for the design of PARACEST agents, low TSAP/SAP ratios are desirable because the SAP isomers display water-exchange rates that are 1–2 orders of magnitude slower than the TSAP isomers. Complexes with low TSAP/SAP ratios are likely with low steric strain on pendant arms and the macrocycle as well as on amide nitrogen atoms. Also, side chains that hinder hydrogen-bond interactions close to the inner-sphere water might favor the SAP isomer leading to slow water-exchange rates useful for PARACEST agents.



{kind=link}
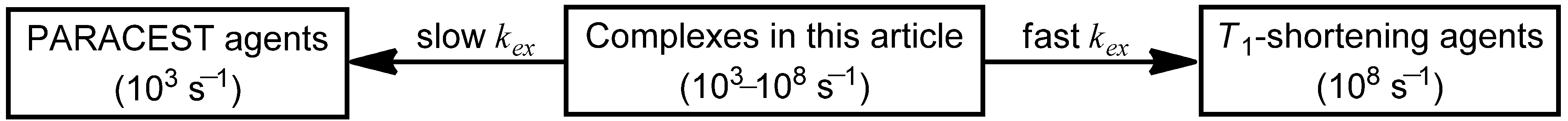{kind=link}
{kind=link}
{kind=link}
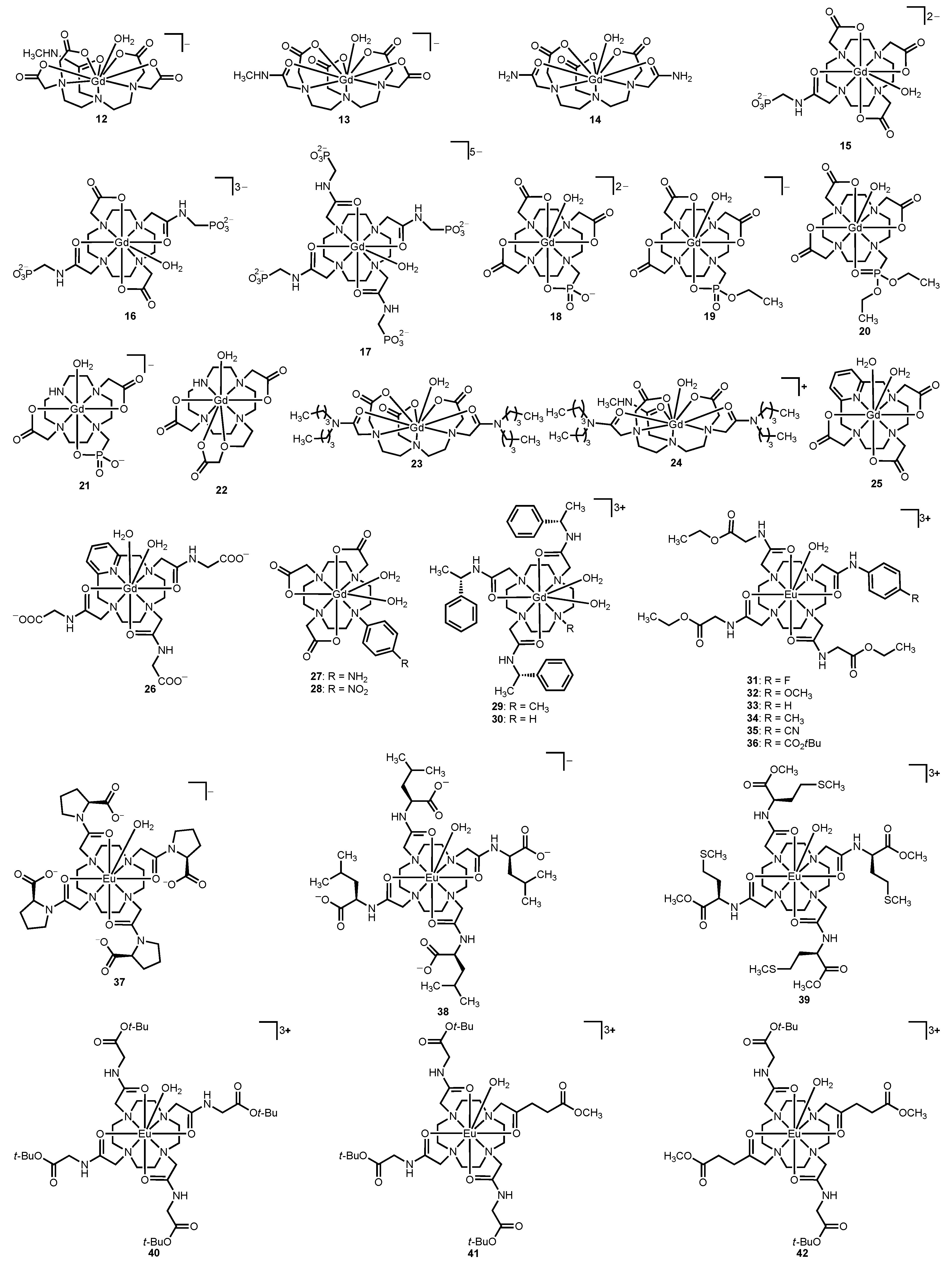{kind=link}
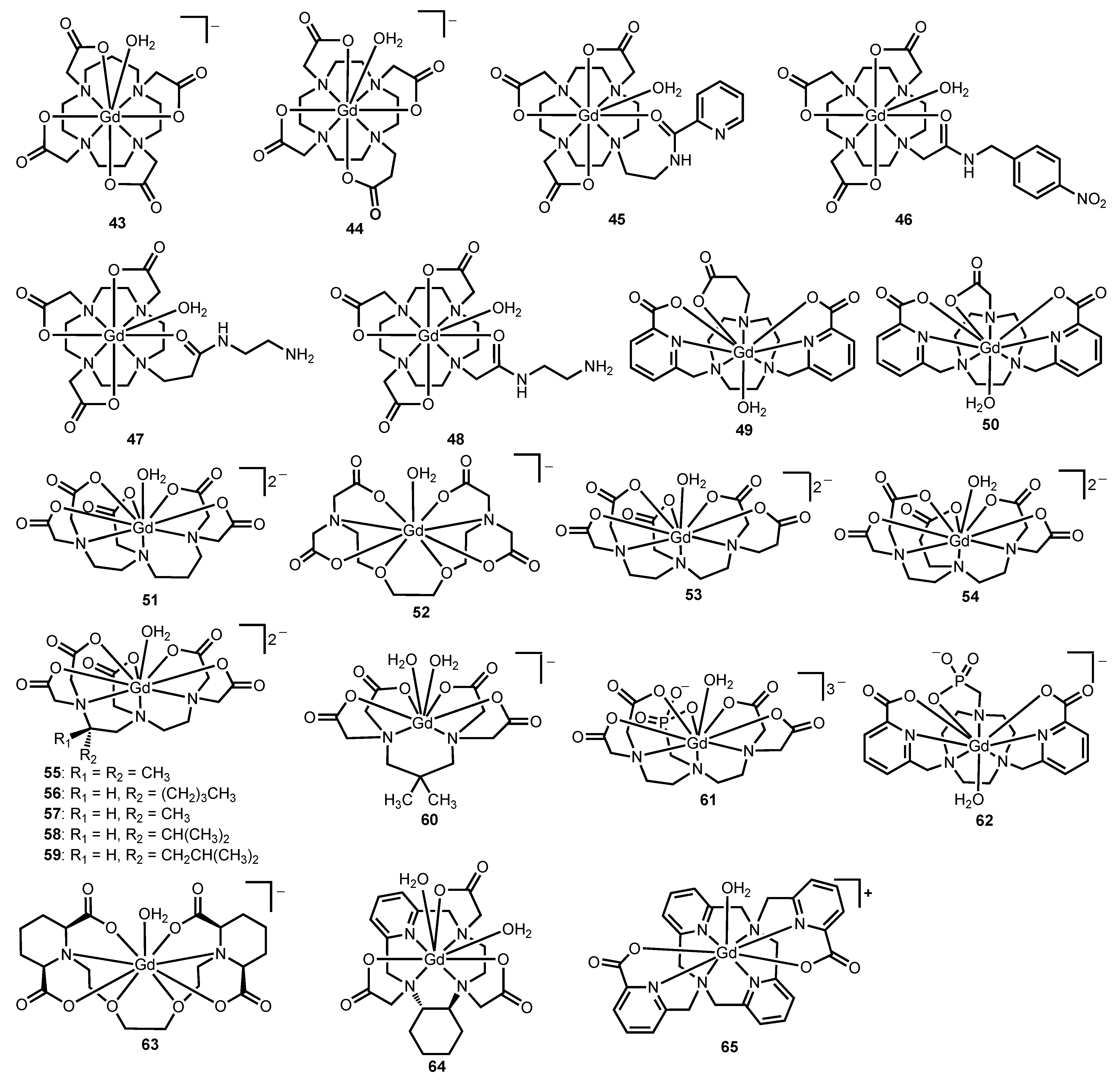{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}