A UPLC-MS/MS Method for Qualification of Quercetin-3-O-β- D-glucopyranoside-(4®1)-α-L-rhamnoside in Rat Plasma and Application to Pharmacokinetic Studies

Abstract
:1. Introduction
2. Results and Discussion
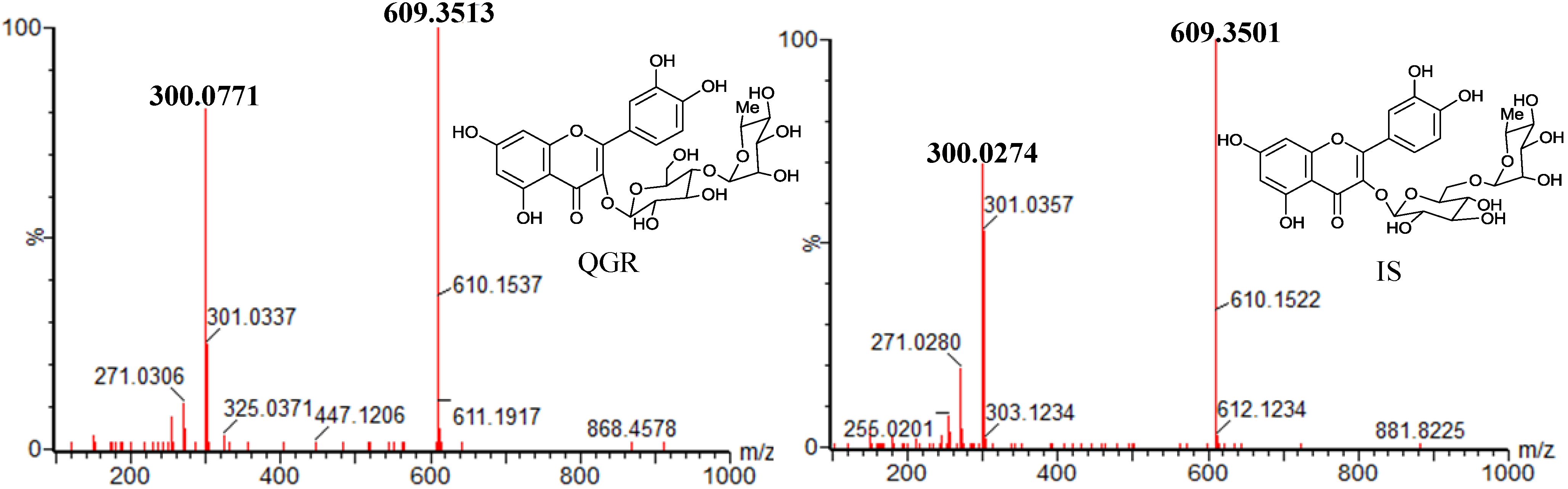
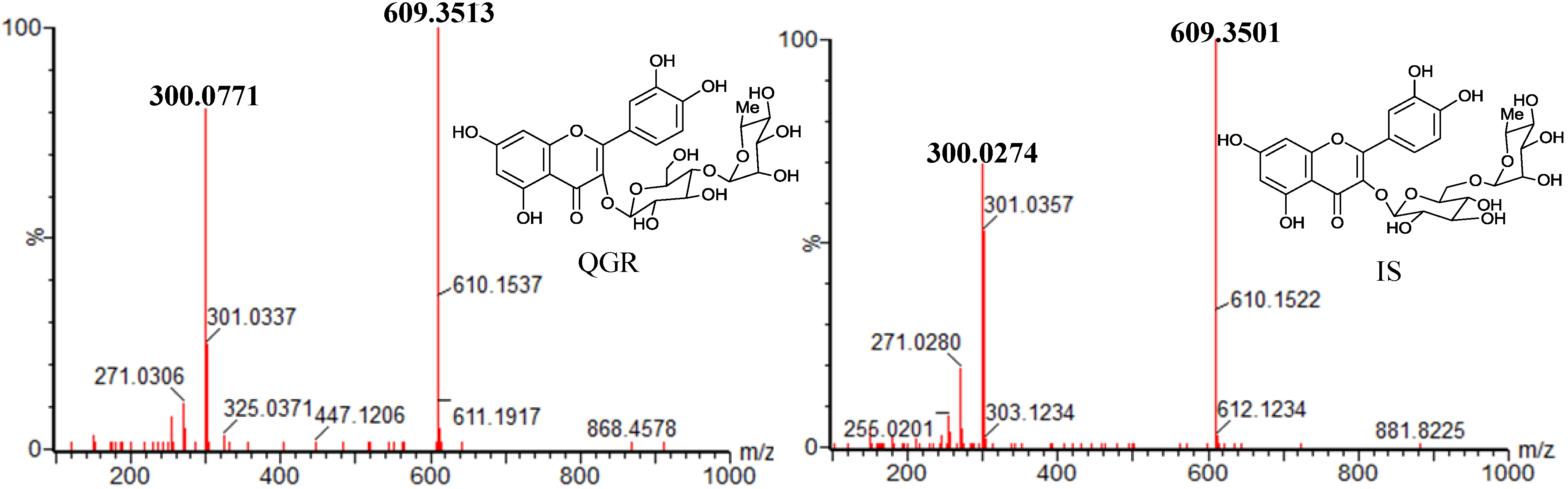
2.1. Optimization of the Chromatographic Conditions

{kind=link}
{kind=link}
{kind=link}
| Compds. | MW | MRM transitions | Cone voltage (V) | Collision energies (eV) | Rt (min) | Ion Mode |
|---|---|---|---|---|---|---|
| IS | 610 | 609.350 > 300.027 | 52 | 34 | 1.64 | ESI− |
| QGR | 610 | 609.351 > 300.077 | 50 | 34 | 3.23 | ESI− |
2.2. Method Validation
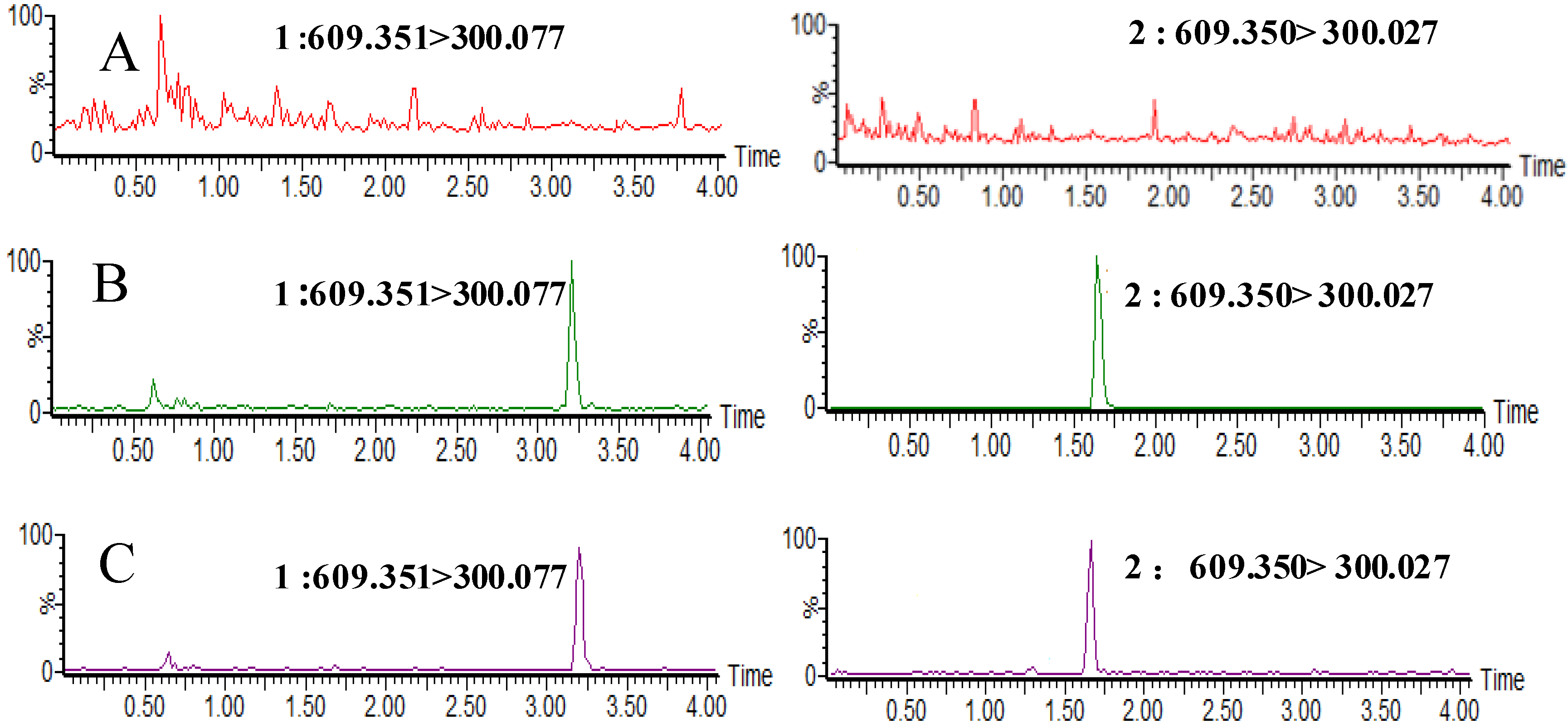
2.2.1. Selectivity and Specificity
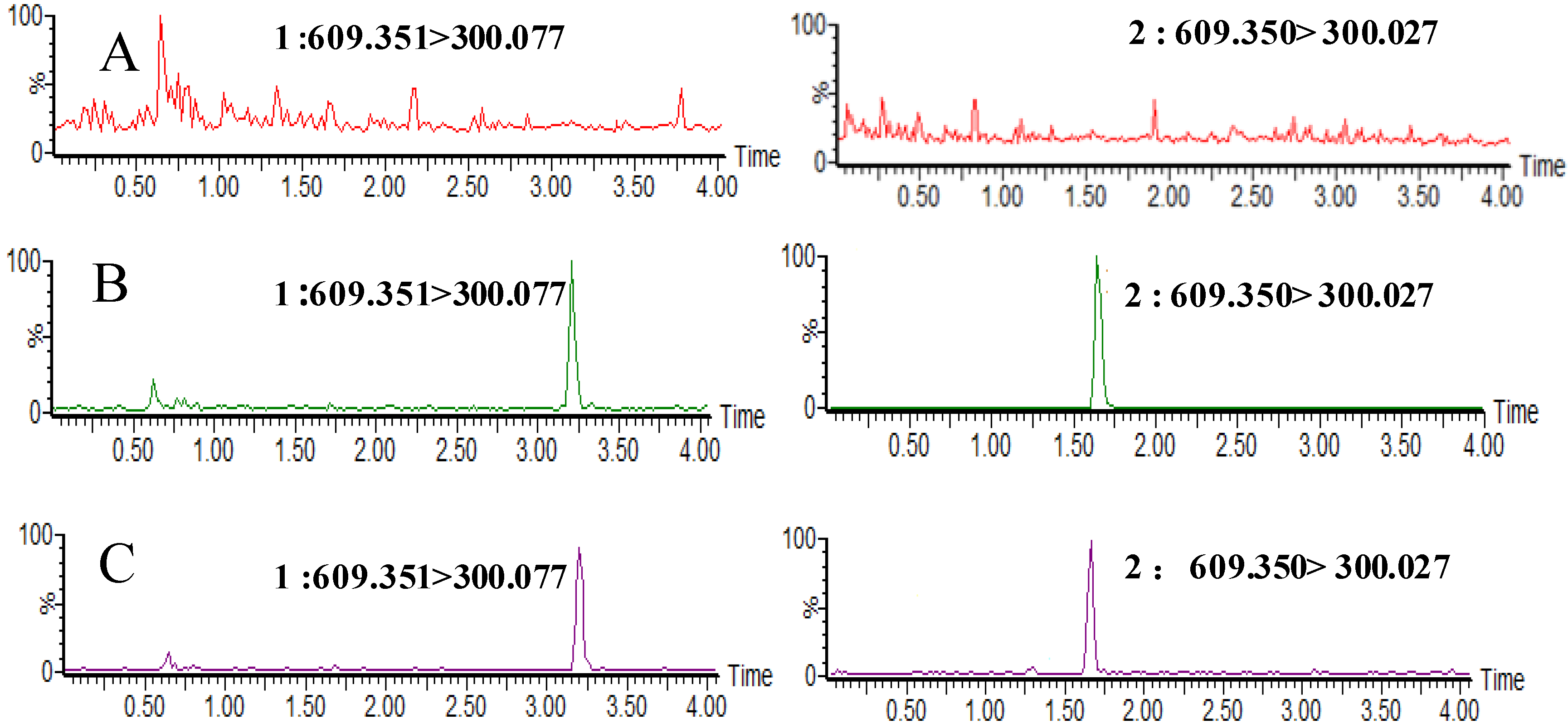
2.2.2. Matrix Effect
2.2.3. Extraction Recovery
| Spiked plasma concentration (ng/mL) | Measured concentration (ng/mL) | Extraction recovery (%) | RSD (%) |
|---|---|---|---|
| 14.8 | 11.3 ± 0.6 | 76.8 ± 4.6 | 5.8 |
| 740.0 | 570.5 ± 7.1 | 77.1 ± 3.8 | 6.7 |
| 14,800.0 | 11,055.6 ± 21.4 | 74.7 ± 4.5 | 4.5 |
2.2.4. The Lower Limit of Detection (LLOD), Quantification (LLOQ) and the Linearity
2.2.5. Precision and Accuracy
| Spiked concentration (ng/mL) | Intra-day | Inter-day | |||||
|---|---|---|---|---|---|---|---|
| Measured concentration (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | Measured concentration (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | ||
| 14.8 | 15.1 ± 0.7 | 8.1 | 7.3 | 15.2 ± 0.5 | 10.4 | 4.5 | |
| 740.0 | 736.7 ± 3.4 | 4.6 | −3.4 | 744.6 ± 4.7 | 6.7 | 4.6 | |
| 14,800.0 | 14,857 ± 41.2 | 6.9 | 2.8 | 14,841 ± 47.8 | 7.7 | 3.9 | |
2.2.6. Stability
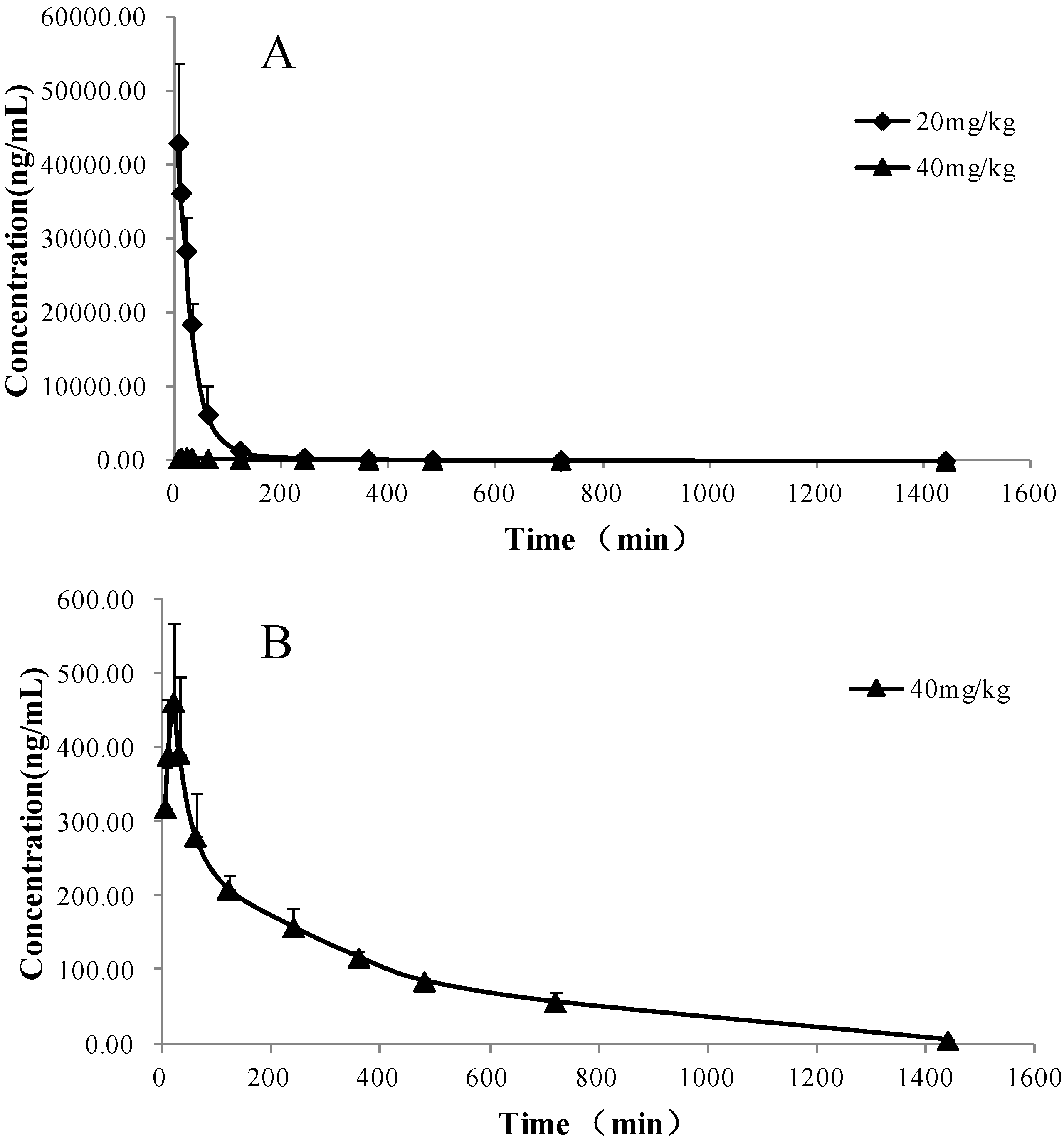
2.3. Pharmacokinetic Study
| Storage conditions | Concentration (ng/mL) | RSD (%) | RE (%) | |
|---|---|---|---|---|
| Spiked | Measured (mean ± SD) | |||
| At room temperature for 4 h | 14.8 | 14.7 ± 0.4 | 4.1 | -2.5 |
| 740.0 | 748.3 ± 3.1 | 4.6 | 2.8 | |
| 14,800.0 | 14,825 ± 20.5 | 6.1 | 4.5 | |
| After three freeze/thaw cycles in plasma | 14.8 | 15.2 ± 0.6 | 4.7 | 4.1 |
| 740.0 | 734.7 ± 6.3 | 7.5 | −3.7 | |
| 14,800.0 | 14,822 ± 24.2 | 3.6 | 4.7 | |
| In the auto-sampler for 24 h | 14.8 | 15.3 ± 0.8 | 2.7 | 2.6 |
| 740.0 | 751.4 ± 8.7 | 2.7 | 3.9 | |
| 14,800.0 | 14,835 ± 28.9 | 3.2 | 6.8 | |
| Long-term stability (at −80 °C for 30 days) | 14.8 | 15.4 ± 1.1 | 8.5 | 4.1 |
| 740.0 | 761.7 ± 20.4 | 5.6 | 8.6 | |
| 14,800.0 | 14,871 ± 71.2 | 7.8 | 10.4 | |
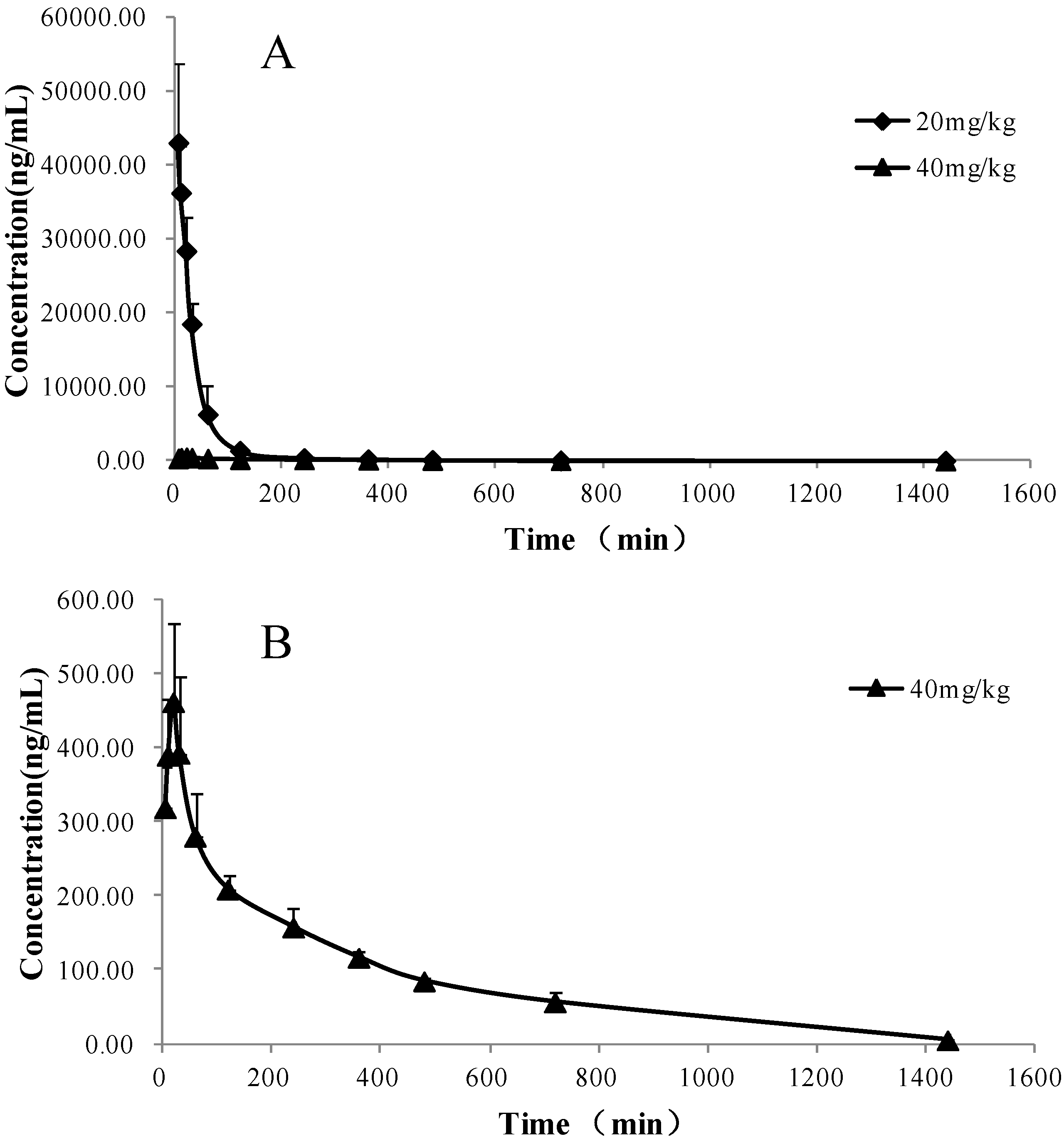
| PK parameters | Unit | Intravenous | Oral |
|---|---|---|---|
| t1/2 | min | 118.89 ± 5.65 | 236.87 ± 28.59 |
| AUC(0→t) | μg/mL × min | 1,775.96 ± 36.92 | 120.81 ± 11.38 |
| AUC(0→∞) | μg/mL × min | 1,790.24 ± 37.53 | 122.14 ± 16.15 |
| Tmax | min | - | 20 |
| Cmax | ng/mL | - | 495.69 ± 58.36 |
| F | % | - | 3.41 ± 1.21 |
3. Experimental
3.1. Chemicals and Reagents
3.2. UPLC-MS/MS Instrument and Conditions
3.3. Preparation of Calibration Standards and Quality Control Samples
3.4.Sample Preparation
3.5. Method Validation
3.5.1. Selectivity
3.5.2. Linearity
3.5.3. Limit of Detection and Lower Limit of Quantification
3.5.4. Precision and Accuracy
3.5.5. Extraction Recovery and Matrix Effect
3.5.6. Stability
3.6. Pharmacokinetic Study in Rats
4. Conclusions
Acknowledgments
References
- Van Beek, T.A.; Montoro, P. Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J. Chromatogr. A 2009, 1216, 2002–2032. [Google Scholar] [CrossRef]
- Lin, L.Z.; Chen, P.; Ozcan, M.; Harnly, J.M. Chromatographic profiles and identification of new phenolic components of Ginkgo biloba leaves and selected products. J. Agric. Food Chem. 2008, 56, 6671–6679. [Google Scholar] [CrossRef]
- Vesna, I.; Mira, P.; Nada, N.; Mirjana, R.; Vesna, V. The effect of Ginkgo Biloba (EGb 761) on epileptic activity in rabbits. Molecules 2008, 13, 2509–2520. [Google Scholar] [CrossRef]
- Hasler, A.; Sticher, O.; Meier, B. High-performance liquid chromatographic determination of five widespread flavonoid aglycones. J. Chromatogr. A 1990, 508, 236–240. [Google Scholar] [CrossRef]
- Hasler, A.; Sticher, O. Identification and determination of the flavonoids from Ginkgo biloba by high-performance liquid chromatography. J. Chromatogr. A 1992, 605, 41–48. [Google Scholar]
- Van Beek, T.A. Chemical analysis of Ginkgo biloba leaves and extracts. J. Chromatogr. A 2002, 967, 21–55. [Google Scholar] [CrossRef]
- Yao, X.; Shang, E.X.; Zhou, G.S.; Tang, Y.P.; Guo, S.; Su, S.L.; Jin, C.; Qian, D.W.; Qin, Y.; Duan, J.A. Comparative characterization of total flavonol glycosides and terpene lactones at different ages, from different cultivation sources and genders of Ginkgo biloba leaves. Int. J. Mol. Sci. 2012, 12, 10305–10315. [Google Scholar]
- Wang, C.G.; Dai, Y.; Li, D.L.; Ma, K.Y. Ginkgo biloba leaf extract action in scavenging free radicals and reducing mutagenicity and toxicity of cigarette smoke in vivo. J. Environ. Sci. Health A Tox. Hazard. Subst. Environ. Eng. 2010, 45, 498–505. [Google Scholar] [CrossRef]
- Hyun, S.K.; Jung, H.A.; Chung, H.Y.; Choi, J.S. In vitro peroxynitrite scavenging activity of 6-hydroxykynurenic acid and other flavonoids from Gingko biloba yellow leaves. Arch. Pharm. Res. 2006, 29, 1074–1079. [Google Scholar] [CrossRef]
- Ou, H.C.; Lee, W.J.; Lee, I.T.; Chiu, T.H.; Tsai, K.L.; Lin, C.Y.; Sheu, W.H. Ginkgo biloba extract attenuates oxLDL-induced oxidative functional damages in endothelial cells. J. Appl. Physiol. 2009, 106, 1674–1685. [Google Scholar] [CrossRef] [Green Version]
- Trompezinski, S.; Bonneville, M.; Pernet, I.; Denis, A.; Schmitt, D.; Viac, J. Gingko biloba extract reduces VEGF and CXCL-8/IL-8 levels in keratinocytes with cumulative effect with epigallocatechin-3-gallate. Arch. Dermatol. Res. 2010, 302, 183–189. [Google Scholar] [CrossRef]
- Shi, C.; Zhao, L.; Zhu, B.; Li, Q.; Yew, D.T.; Yao, Z.; Xu, J. Dosage effects of EGb761 on hydrogen peroxide-induced cell death in SH-SY5Y cells. Chem. Biol. Interact. 2009, 180, 389–397. [Google Scholar] [CrossRef]
- Sample Availability: Samples of quercetin-3-O-β-D-glucopyranoside-(4→1)-α-L-rhamnoside and rutin are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yao, X.; Zhou, G.; Tang, Y.; Li, Z.; Su, S.; Qian, D.; Duan, J.-A. A UPLC-MS/MS Method for Qualification of Quercetin-3-O-β- D-glucopyranoside-(4®1)-α-L-rhamnoside in Rat Plasma and Application to Pharmacokinetic Studies. Molecules 2013, 18, 3050-3059. https://doi.org/10.3390/molecules18033050
Yao X, Zhou G, Tang Y, Li Z, Su S, Qian D, Duan J-A. A UPLC-MS/MS Method for Qualification of Quercetin-3-O-β- D-glucopyranoside-(4®1)-α-L-rhamnoside in Rat Plasma and Application to Pharmacokinetic Studies. Molecules. 2013; 18(3):3050-3059. https://doi.org/10.3390/molecules18033050
Chicago/Turabian StyleYao, Xin, Guisheng Zhou, Yuping Tang, Zhenhao Li, Shulan Su, Dawei Qian, and Jin-Ao Duan. 2013. "A UPLC-MS/MS Method for Qualification of Quercetin-3-O-β- D-glucopyranoside-(4®1)-α-L-rhamnoside in Rat Plasma and Application to Pharmacokinetic Studies" Molecules 18, no. 3: 3050-3059. https://doi.org/10.3390/molecules18033050