α(δ')-Michael Addition of Alkyl Amines to Dimethyl (E)-hex-2-en-4-ynedioate: Synthesis of α,β-Dehydroamino Acid Derivatives

Abstract
:
1. Introduction
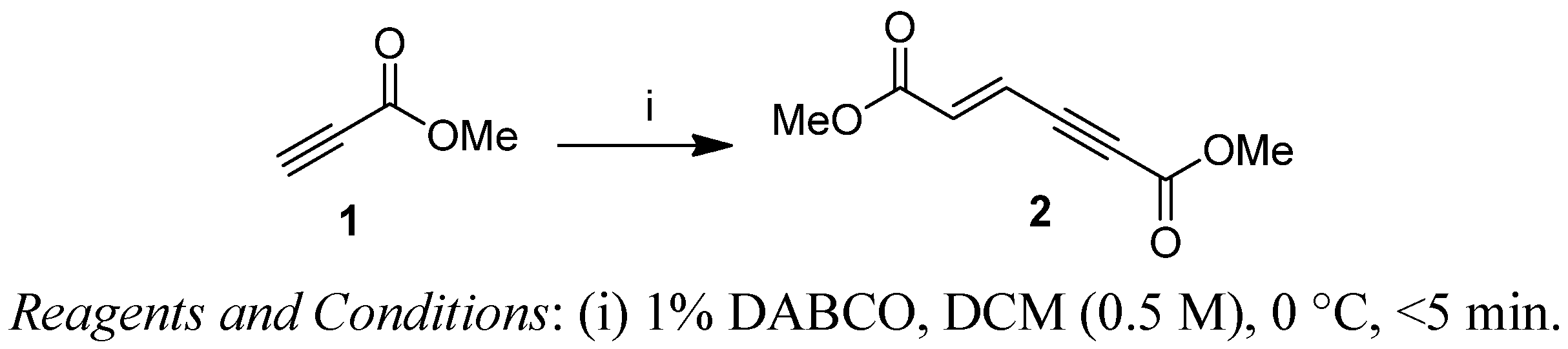
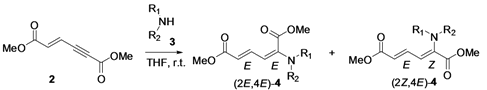
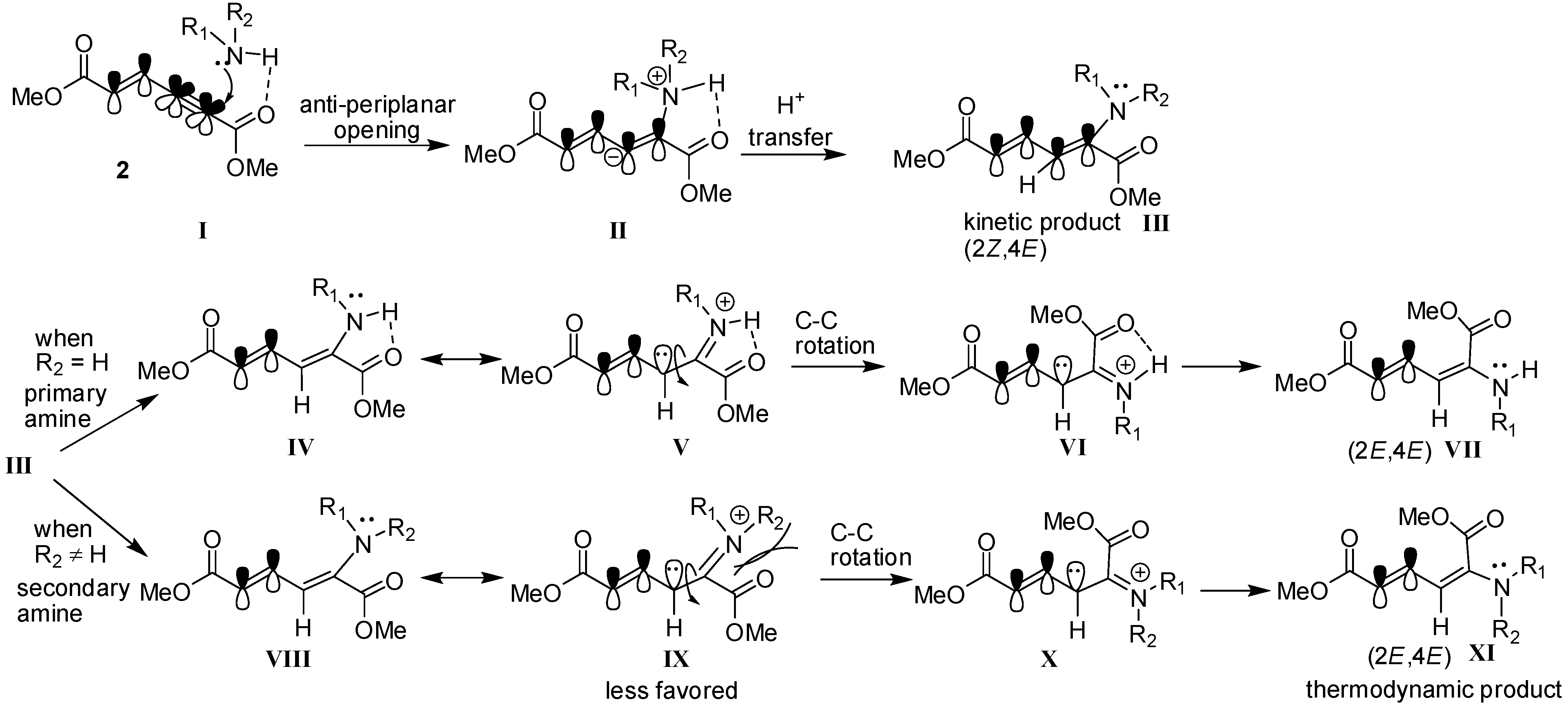
2. Results and Discussion

{kind=link}
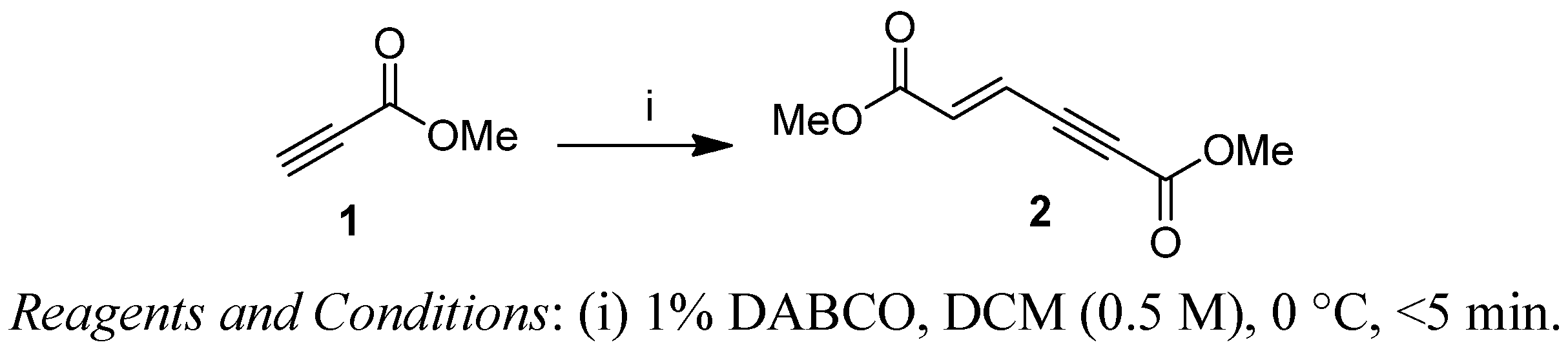{kind=link}
{kind=link}
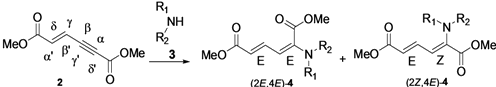 | |||||
|---|---|---|---|---|---|
| Entry | Amine | Solvent | Reaction conditions | Yields of 4 (%) b | |
| (2E,4E) | (2Z,4E) | ||||
| 1 | n-Octylamine | THF | rt | 82 | 00 |
| 2 | n-Octylamine | THF | 60 °C | 81 | 00 |
| 3 | n-Octylamine | Benzene | rt | 52 | 00 |
| 4 | n-Octylamine | Toluene | rt | 49 | 00 |
| 5 | n-Octylamine | DCM | rt | 46 | 00 |
| 6 | n-Octylamine | MeCN | rt | 65 | 00 |
| 7 | Piperidine | THF | rt | 64 | 16 |
| 8 | Piperidine | THF | 60 °C | 54 | 14 |
| 9 | Piperidine | Benzene | rt | 68 | 11 |
| 10 | Piperidine | Toluene | rt | 60 (68) | 08 (09) |
| 11 | Piperidine | DCM | rt | 58 (63) | 12 (13) |
| 12 | Piperidine | MeCN | rt | 48 (51) | 08 (09) |
 | |||
|---|---|---|---|
| Entry | R1 | Product (2E,4E) | Yield (%) b |
| 1 | n-Hexyl-3a | 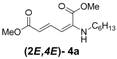 | 67 |
| 2 | n-Octyl-3b | 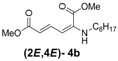 | 82 |
| 3 | n-Decyl-3c | 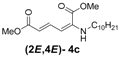 | 73 |
| 4 | n-Dodecyl-3d | 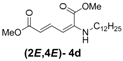 | 73 |
| 5 c | Benzyl- 3e |  | 73 (75) c |
| 6 | iso-Propyl-3f |  | 60 (64) |
| 7 | tert-Butyl-3g |  | 25 (58) |
| 8 | Allyl- 3h | 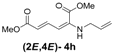 | 68 |
| 9 | Ethanol- 3i | 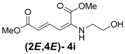 | 78 (80) |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R1R2 | Major Product (2E,4E) | Yield (%) b | Minor Product (2Z,4E) | Yield (%) b | Ratio of (2E,4E)/(2Z,4E) |
| 1 | Diethyl 3j | 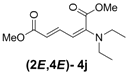 | 53 (58) | 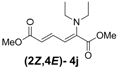 | 14 (15) | 3.8:1 |
| 2 | Dipropyl 3k | 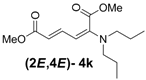 | 59 (62) | 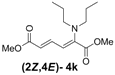 | 16 (17) | 3.7:1 |
| 3 | Pyrrolidine 3l |  | 56 | 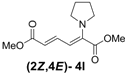 | 18 | 3.1:1 |
| 4 | Piperidine 3m | 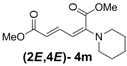 | 64 |  | 16 | 4:1 |
| 5 | Morpholine 3n | 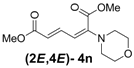 | 58 | 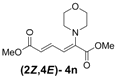 | 18 | 3.2:1 |
3. Experimental
3.1. General
3.2. General Experimental Procedure for Reactions with Primary Alkyl Amines
3.3. General Experimental Procedure for Reactions with Secondary Alkyl Amines
4. Conclusions
Supplementary Materials
Acknowledgments
References
- March, J. Advanced Organic Chemistry, 4th ed; Wiley: New York, NY, USA, 1992. [Google Scholar]
- Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis; Pergamon: Oxford, UK, 1992. [Google Scholar]
- Jung, M.E. Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Semmelhack, M.F., Eds.; Pergamon: Oxford, UK, 1991; Volume 4, pp. 1–67. [Google Scholar]
- Imanzadeh, G.; Ahmadi, F.; Zamanloo, M.; Mansoori, Y. Tetrabutylammonium Bromide Media Aza-Michael Addition of 1,2,3,6-Tetrahydrophthalimide to Symmetrical Fumaric Esters and Acrylic Esters under Solvent-Free Conditions. Molecules 2010, 15, 7353–7362. [Google Scholar] [CrossRef]
- Jiang, Z.-Y.; Yang, H.-M.; Ju, Y.-D.; Li, L.; Luo, M.-X.; Lai, G.-Q.; Jiang, J.-X.; Xu, L.-W. Organocatalytic Michael Addition of 1,3-Dicarbonyl Indane Compounds to Nitrostyrenes. Molecules 2010, 15, 2551–2563. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, Y.-Q.; Guo, S.-R. Silica Sulfuric Acid Promotes Aza-Michael Addition Reactions under Solvent-Free Condition as a Heterogeneous and Reusable Catalyst. Molecules 2009, 14, 4779–4789. [Google Scholar] [CrossRef]
- Escalante, J.; Carrillo-Morales, M.; Linzaga, I. Michael Additions of Amines to Methyl Acrylates Promoted by Microwave Irradiation. Molecules 2008, 13, 340–347. [Google Scholar] [CrossRef]
- Chen, H.; Zhong, X.; Wei, J. Stereoselective Syntheses of Fluorescent Non-Natural Aromatic Amino Acids Based on Asymmetric Michael Additions. Molecules 2007, 12, 1170–1182. [Google Scholar] [CrossRef]
- Davies, S.G.; Lee, J.A.; Roberts, P.M.; Thomson, J.E.; Yin, J. Double Asymmetric Induction as a Mechanistic Probe: The Doubly Diastereoselective Conjugate Addition of Enantiopure Lithium Amides to Enantiopureα,β-Unsaturated Esters and Enantiopureα,β-Unsaturated Hydroxamates. Tetrahedron 2011, 67, 6382–6403. [Google Scholar] [CrossRef]
- Davies, S.G.; Smith, A.D.; Price, P.D. The Conjugate Addition of Enantiomerically Pure Lithium Amides as Homochiral Ammonia Equivalents: Scope, Limitations and Synthetic Applications. Tetrahedron Asymmetry 2005, 16, 2833–2891. [Google Scholar] [CrossRef]
- Lewandowska, E. Substitution at the α-Carbons of α,β-Unsaturated Carbonyl Compounds: Anti-Michael Addition. Tetrahedron 2007, 63, 2107–2122. [Google Scholar] [CrossRef]
- Bi, X.; Zhang, J.; Liu, Q.; Tan, J.; Li, B. IntramolecularAza-Anti-Michael Addition of an Amide Anion to Enones: A Regiospecific Approach to Tetramic Acid Derivatives. Adv. Synth. Catal. 2007, 349, 2301–2306. [Google Scholar] [CrossRef]
- Li, Y.; Xu, X.; Tan, J.; Liao, P.; Zhang, J.; Liu, Q. Polarity-Reversible Conjugate Addition Tuned by Remote Electronic Effects. Org. Lett. 2010, 12, 244–247. [Google Scholar] [CrossRef]
- Ballini, R.; Bazán, N.A.; Bosica, G.; Palmieri, A. Uncatalyzed, Anti-Michael Addition of Amines to β-Nitroacrylates: Practical, Eco-Friendly Synthesis of β-Nitro-α-Amino Esters. Tetrahedron Lett. 2008, 49, 3865–3867. [Google Scholar] [CrossRef]
- Ballini, R.; Bosica, G.; Palmieri, A.; Bakhtiari, K. Solvent-Free, Anti-Michael Addition of Active Methylene Derivatives to β-Nitroacrylates: Eco-Friendly, Chemoselective Synthesis of PolyfunctionalizedNitroalkanes. Synlett 2009, 2009, 268–270. [Google Scholar]
- Wilson, J.E.; Sun, J.; Fu, G.C. StereoselectivePhosphine-Catalyzed Synthesis of Highly Functionalized Diquinanes. Angew.Chem. Int. Ed. Engl. 2010, 49, 161–163. [Google Scholar] [CrossRef]
- Zhu, X.-F.; Henry, C.E.; Kwon, O. Stable Tetravalent PhosphoniumEnolate Zwitterions. J. Am. Chem. Soc. 2007, 129, 6722–6723. [Google Scholar]
- Lecerclé, D.; Sawicki, M.; Taran, F. Phosphine-Catalyzed α-P-Addition on Activated Alkynes: A new route to P−C−P backbones. Org. Lett. 2006, 8, 4283–4285. [Google Scholar] [CrossRef]
- Shim, J.-G.; Park, J.C.; Cho, C.S.; Shim, S.C.; Yamamoto, Y. Catalytic and Highly Regiospecific Carbon–Carbon Bond Formation at α-Position of Michael Acceptor by Palladium Complex. Chem. Commun. 2002, 2002, 852–853. [Google Scholar]
- Klumpp, G.W.; Mierop, A.J.C.; Vrielink, J.J.; Brugman, A.; Schakel, M. Anti-Michael Carbolithiation of Silicon and Phenyl-Substituted α,β-Unsaturated Secondary Amides. J. Am. Chem. Soc. 1985, 107, 6740–6742. [Google Scholar] [CrossRef]
- Aurell, M.J.; Bañuls, M.J.; Mestres, R.; Muñoz, E. On the Mechanism of the Addition of Organolithium Reagents to Cinnamic Acids. Tetrahedron 2001, 57, 1067–1074. [Google Scholar]
- Chatfield, D.C.; Augsten, A.; D’Cunha, C.; Lewandowska, E.; Wnuk, S.F. Theoretical and Experimental Study of the Regioselectivity of Michael Additions. Eur. J. Org. Chem. 2004, 2004, 313–322. [Google Scholar]
- Lewandowska, E.; Chatfield, D.C. Regioselectivity of Michael Additions to 3-(Pyridin-3-yl or Pyrimidin-2-yl)-Propenoates and their N-Oxides—Experimental and Theoretical Studies. Eur. J. Org. Chem. 2005, 2005, 3297–3303. [Google Scholar] [CrossRef]
- Yuan, H.; Zheng, Y.; Zhang, J. Mechanism Study of the Intramolecular Anti-Michael Addition of N-Alkylfurylacrylacetamides. J. Org. Chem. 2012, 77, 8744–8749. [Google Scholar]
- Trost, B.M.; Dake, G.R. Nucleophilic α-Addition to Alkynoates. A Synthesis of Dehydroamino Acids. J. Am. Chem. Soc. 1997, 119, 7595–7596. [Google Scholar] [CrossRef]
- Deng, J.-C.; Chuang, S.-C. Three-Component and Nonclassical Reaction of Phosphines with Enynes and Aldehydes: Formation of γ-Lactones Featuring α-Phosphorus Ylides. Org. Lett. 2011, 13, 2248–2251. [Google Scholar]
- Chuang, S.-C.; Deng, J.-C.; Chan, F.-W.; Chen, S.-Y.; Huang, W.-J.; Lai, L.-H.; Rajeshkumar, V. [3+2] Cycloaddition of Dialkyl (E)-Hex-2-en-4-ynedioates to [60]Fullerene by Phosphane-Promoted Tandem α(d')-Michael Additions. Eur. J. Org. Chem. 2012, 2012, 2606–2613. [Google Scholar]
- Kazmaier, U. Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A.B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; Volume 2, pp. 3–34. [Google Scholar]
- Mathur, P.; Ramakumar, S.; Chauhan, V.S. Peptide Design Using α,β-Dehydroamino Acids: From β-Turns to Helical Hairpins. Pept. Sci. 2004, 76, 150–161. [Google Scholar] [CrossRef]
- Ferreira, P.M.T.; Maia, H.L.S.; Monteiro, L.S.; Sacramento, J. High Yielding Synthesis of Dehydroamino Acid and Dehydropeptide Derivatives. J. Chem. Soc. Perkin Trans. 1 1999, 1999, 3697–3703. [Google Scholar]
- Ramachandran, P.V.; Rudd, M.T.; Reddy, M.V.R. Stereoselective Synthesis of Hex-2-(E)-en-4-yn-1,6-dioates and E,Z-Muconic Acid Diesters via Organo-Catalyzed Self-Coupling of Propiolates. Tetrahedron Lett. 2005, 46, 2547–2549. [Google Scholar] [CrossRef]
- Zhou, L.-H.; Yu, X.-Q.; Pu, L. Reactivity of a PropiolateDimer with Nucleophiles and an Efficient Synthesis of Dimethyl α-Aminoadipate. Tetrahedron Lett. 2010, 51, 425–427. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chavan, A.S.; Deng, J.-C.; Chuang, S.-C. α(δ')-Michael Addition of Alkyl Amines to Dimethyl (E)-hex-2-en-4-ynedioate: Synthesis of α,β-Dehydroamino Acid Derivatives. Molecules 2013, 18, 2611-2622. https://doi.org/10.3390/molecules18032611
Chavan AS, Deng J-C, Chuang S-C. α(δ')-Michael Addition of Alkyl Amines to Dimethyl (E)-hex-2-en-4-ynedioate: Synthesis of α,β-Dehydroamino Acid Derivatives. Molecules. 2013; 18(3):2611-2622. https://doi.org/10.3390/molecules18032611
Chicago/Turabian StyleChavan, Arjun S., Jie-Cheng Deng, and Shih-Ching Chuang. 2013. "α(δ')-Michael Addition of Alkyl Amines to Dimethyl (E)-hex-2-en-4-ynedioate: Synthesis of α,β-Dehydroamino Acid Derivatives" Molecules 18, no. 3: 2611-2622. https://doi.org/10.3390/molecules18032611