Syntheses of Four Enantiomers of 2,3-Diendo- and 3-Endo-aminobicyclo[2.2.2]oct-5-ene-2-exo-carboxylic Acid and Their Saturated Analogues


Abstract
:1. Introduction
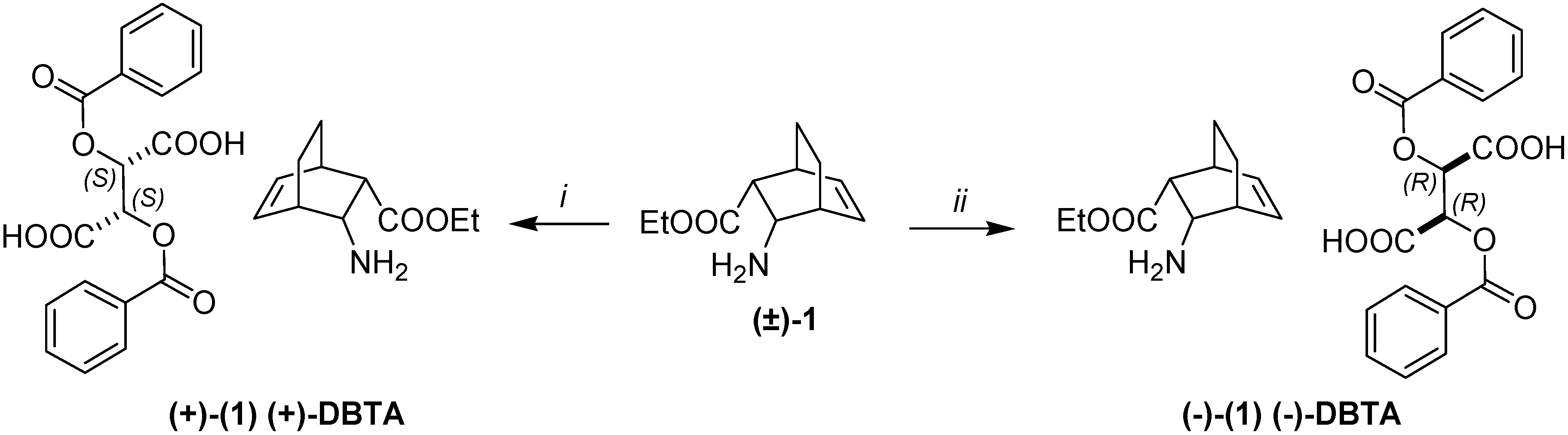
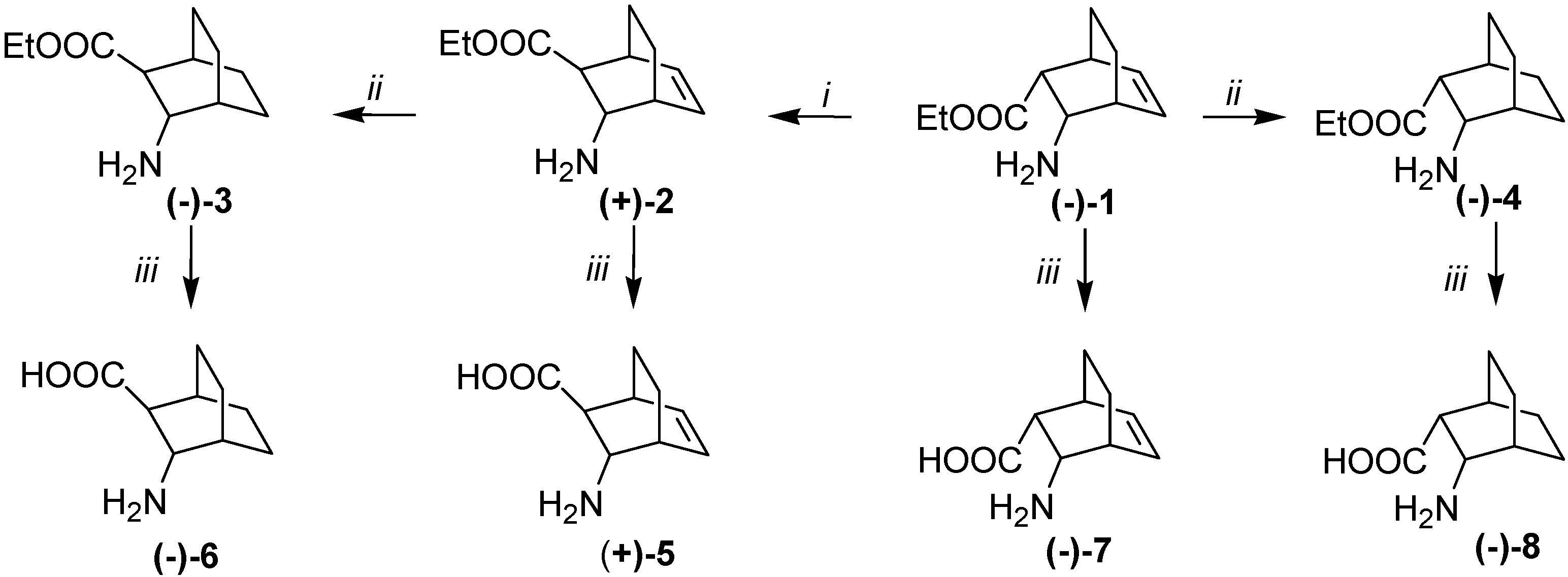
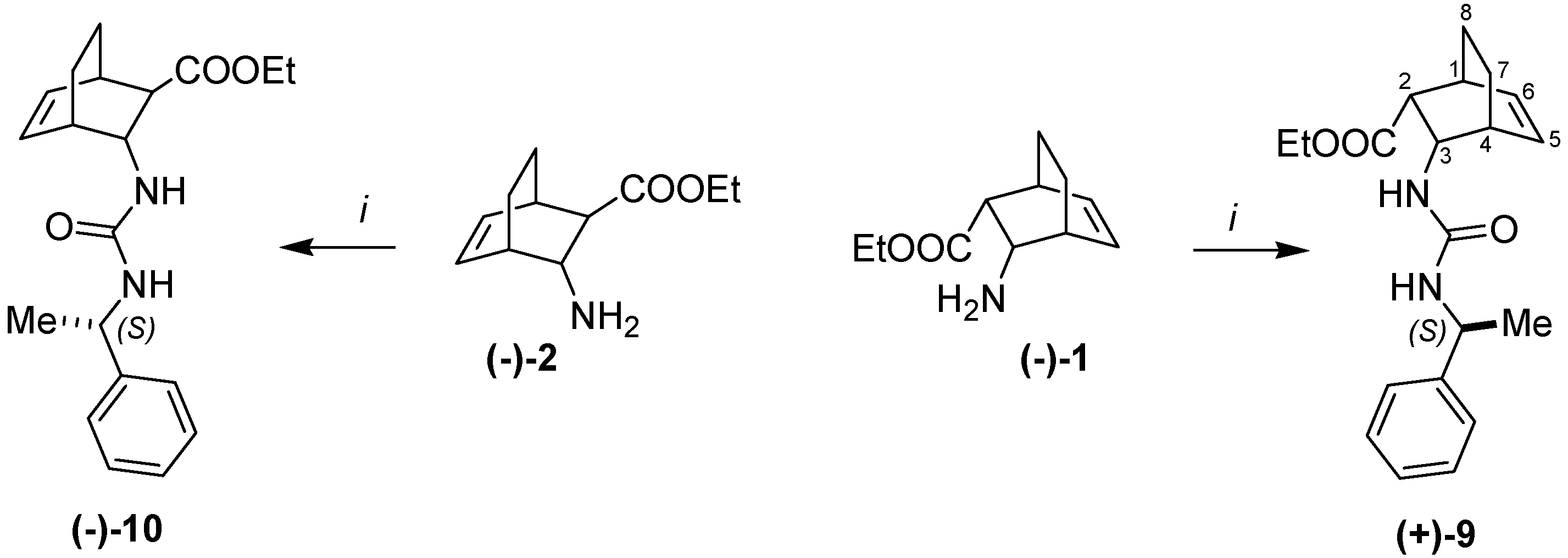
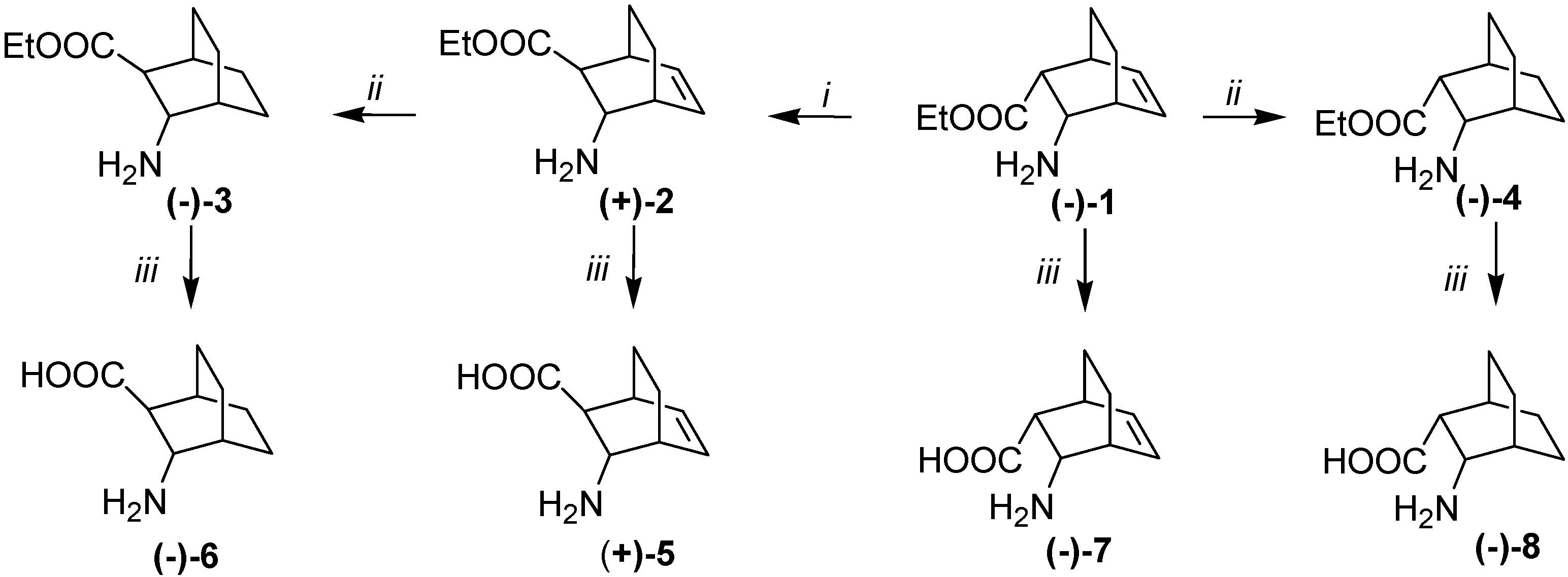
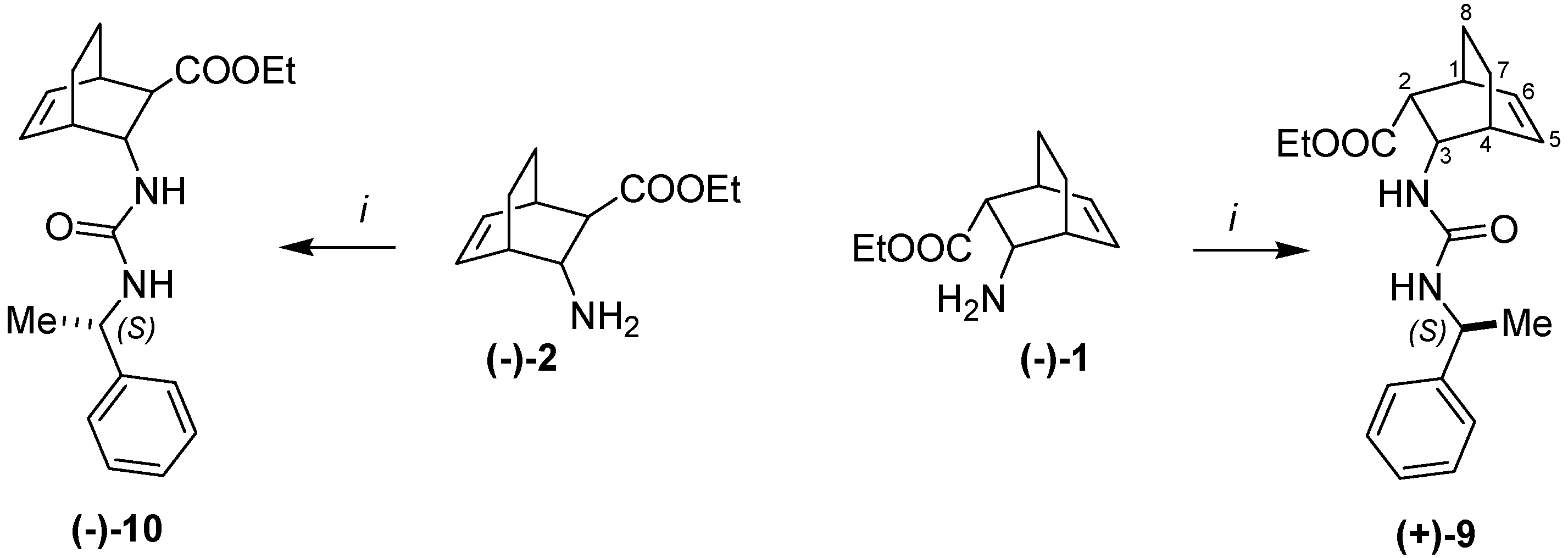
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Resolving agent | Yield (%) | de b (%) | S factor c |
|---|---|---|---|---|
| 1 | L-tartaric acid | 23 | 48 | 0.22 |
| 2 | S-mandelic acid | 30 | 45 | 0.27 |
| 3 | L-DBTA a | 47 | 70 | 0.66 |
| 4 | L-DBTA | 45 | 84 | 0.76 |


3. Experimental
3.1. General
3.2. Determination of de Values of O,O'-dibenzoyltartaric Acid (DBTA) Salts of 1
3.3. A Typical Resolution Procedure
 = –59.9 (c, 0.5, EtOH), de = 96%.; (–)-1.HCl salt mp 192–194 °C, = –14.8 (c, 0.6, EtOH), ee = 96%. The 1H- and 13C-NMR spectroscopic and elemental analysis data on the enantiomeric derivative were in accordance with those for racemic (±)-1·HCl [26]. = +63.5 (c, 0.5, EtOH), de = 98%.; (+)-1.HCl salt, mp 192–194 °C, = +15.3 (c, 0.6, EtOH), ee = 98%. The 1H- and 13C-NMR spectroscopic and elemental analysis data on the enantiomeric derivative were in accordance with those for racemic (±)-1·HCl [26].
= –59.9 (c, 0.5, EtOH), de = 96%.; (–)-1.HCl salt mp 192–194 °C, = –14.8 (c, 0.6, EtOH), ee = 96%. The 1H- and 13C-NMR spectroscopic and elemental analysis data on the enantiomeric derivative were in accordance with those for racemic (±)-1·HCl [26]. = +63.5 (c, 0.5, EtOH), de = 98%.; (+)-1.HCl salt, mp 192–194 °C, = +15.3 (c, 0.6, EtOH), ee = 98%. The 1H- and 13C-NMR spectroscopic and elemental analysis data on the enantiomeric derivative were in accordance with those for racemic (±)-1·HCl [26].3.4. Isomerization of Amino Esters (–)-1 and (+)-1
= +47.9 (c, 0.5, EtOH), ee = 98%. 1H-NMR: δ = 0.98–1.09 (m, 1H, H-8), 1.18–1.26 (t, J = 7.1 Hz, 3H, CH3), 1.18–1.38 (m, 3H, H-7, H-7, H-8,), 2.282.32 (m, 1H, H-2), 2.82–2.91 (m, 2H, H-1, H-4), 3.64 (t, J = 3.2 Hz, 1H, H-3), 4.8–4.23 (m, 2H, OCH2), 6.18 (t, J = 7.1Hz, 1H, H-5) 6.53 (t, J = 7.5 Hz 1H, H-6) 7.95 (bs, 3H, NH3+) ppm. 13C-NMR: δ = 14.9, 19.9, 23.0, 33.3, 33.4, 49.3, 51.7, 61.6, 130.9, 136.6, 172.4 ppm. Anal. Calcd for C11H18ClNO2 (231,74): C, 57.02; H, 7.83; Cl 15,30; N, 6,04. Found: C, 56.81; H, 8.04; N, 6.05. = –43 (c, 0.4, EtOH), ee = 98%. The 1H- and 13C-NMR spectroscopic data on the enantiomeric derivative were consistent with the data on the antipode (+)-2. Anal. Calcd for C11H18ClNO2 (231.74): C, 57.02; H, 7.83; Cl 15.30; N, 6.04. Found: C, 56.94; H, 8.11; N, 6.12.3.5. General Procedure for Hydrogenation of Amino Esters (–)-1 (+)-1, (–)-2 and (+)-2 over Pd/C Catalyst in a Fixed-Bed Reactor
= –17.5 (c, 1.0, MeOH), ee = 99%. 1H-NMR: δ = 1.21 (t, J = 7.2, 3H, CH3) 1.25–1.86 (m, 9H, H-5, H-5, H-6, H-6, H-7, H-7, H-8, H-8, H-2), 1.90–1.95 (m, 1H, H-4), 2.54–2.58 (m, 1H, H-4), 3.54–3,64 (m, 1H, H-3), 4.06–4.21 (m, 2H, OCH2), 8.16 (bs, 3H, NH3+) ppm. 13C-NMR: δ = 14.7, 18.6, 21.0, 24.5, 25.3, 27.8, 28.8, 47.9, 50.5, 61.58, 166.4 ppm. Anal. Calcd for C11H20ClNO2 (233.74): C, 56.52; H, 8.62; Cl 15.17; N, 5.99. Found: C, 56.21; H, 8.39; N, 5.93. = +15,4 (c, 0.9, MeOH), ee = 99%. The 1H- and 13C-NMR spectroscopic data on the enantiomeric derivative were consistent with the data on the antipode (–)-3. Anal. Calcd for C11H20ClNO2 (233.74): C, 56.52; H, 8.62; Cl 15.17; N, 5.99. Found: C, 56.29; H, 8.88; N, 6.14. = –31.5 (c, 0.6, EtOH), ee = 99%. 1H-NMR: δ = 1.22 (t, J = 7.1, 3H, CH3) 1.29–1.98 (m, 10H, H-5, H-5, H-6, H-6, H-7, H-7, H-8, H-8, H-1, H-4), 3.00 (dd, J = 5.1, J = 1.1 H-2), 3.47 (d, J = 4.6 1H, H-3), 4.03–4.23 (m, 2H, OCH2), 8.00 (bs, 3H, NH3+) ppm. 13C-NMR: δ = 14.9, 18.7, 21.2, 24.5, 25.2, 28.0, 28.7, 43.4, 48.8, 61.4, 172.8 Anal. Calcd for C11H20ClNO2 (233.74): C, 56.52; H, 8.62; Cl 15.17; N, 5.99. Found: C, 56.76; H, 8.49; N, 6.17. The ee values for esters (–)-4 and (+)-4 were determined after derivatization with Ac2O in the presence of DMAP and pyridine (1:9) by using Perkin-Elmer GC instrumentation equipped with CP-Chirasil L-Val columns (30 m) with the temperature held at 140 °C for 5 min and then raised to 160 °C at 10 °C min−1, flow rate: 1 mL min−1, retention times: (–)-4: 9.62 min; (+)-4: 9.97 min. = +30.1 (c, 0.4, EtOH) ee = 99%. The 1H- and 13C-NMR spectroscopic data on the enantiomeric derivative were consistent with the data on the antipode (–)-4. Anal. Calcd for C11H20ClNO2 (233.74): C, 56.52; H, 8.62; Cl 15.17; N, 5.99. Found: C, 56.81; H, 8.75; N, 6.21.3.6. General Procedure for Hydrolyses of Amino Esters 1–4
= +51 (c, 0.3, H2O). 1H-NMR: δ = 0.95–1.55 (m, 4H, H-7, H-7, H-8, H-8), 2.13–2.21 (m, 1H, H-2), 2.75–2.85 (m, 1H, H-1) 2.85–2.95 (m, 1H, H-4), 3.63 (t, J = 3.0 Hz, 1H, H-3), 3.76 (bs, 3H, NH3+) 6.17 (t, J = 7.1 Hz, 1H, H-5) 6.52 (t, J = 7.4 Hz 1H, H-6) ppm. 13C-NMR: δ = 20.0, 23.2, 33.3, 33,6, 49.9, 52.0, 130.9, 136.8, 173.9 ppm. Anal. Calcd for C9H13NO2 (167.09): C, 64.65; H, 7.84; N, 8.38. Found: C, 64.71; H, 7.98; N, 8.45. = –64 (c, 0.5, H2O). The 1H- and 13C-NMR spectroscopic data on the enantiomeric derivative are consistent with the data of the antipode (+)-5. Anal. Calcd for C9H13NO2 (167.09): C, 64.65; H, 7.84; N, 8.38. Found: C, 64.83; H, 7.99; N, 8.61. = –35.4 (c, 0.5, H2O). 1H-NMR: δ = 1.39–1.75 (m, 8H, H-5, H-5, H-6, H-6, H-7, H-7, H-8, H-8), 1.87–1.91 (m, 1H, H-4), 2.07–2.11 (m, 1H, H-1) 2.34 (d, J = 6.1, 1H, H-2), 3.90 (d, J = 6.1 Hz, 1H, H-3). 13C-NMR: δ = 18.4, 20.9, 24.2, 25.5, 28.1, 29.2, 51.0, 52.9, 180.9 ppm. Anal. Calcd for C9H15NO2 (169.11): C, 63.88; H, 8.93; N, 8.28. Found: C, 64.07; H, 9.11; N, 8.08. = +35.1 (c, 0.4, H2O). The 1H- and 13C-NMR spectroscopic data on the enantiomeric derivative were consistent with the data on the antipode (–)-6. Anal. Calcd for C9H15NO2 (169.11): C, 63.88; H, 8.93; N, 8.28. Found: C, 64.12; H, 9.05; N, 8.41. = –3 (c, 0.4, H2O). The 1H- and 13C-NMR spectroscopic and elemental analysis data on the enantiomeric derivative were in accordance with those for racemic (±)-7 [29]. = +3.2 (c, 0.4, H2O) The 1H- and 13C-NMR spectroscopic and elemental analysis data on the enantiomeric derivative were in accordance with those for racemic (±)-7 [29]. = –49 (c, 0.3, H2O). The 1H- and 13C-NMR spectroscopic and elemental analysis data on the enantiomeric derivative were in accordance with those for racemic (±)-8 [29]. = +46.5 (c, 0.3, H2O). The 1H- and 13C-NMR spectroscopic and elemental analysis data on the enantiomeric derivative were in accordance with those for racemic (±)-8 [29].3.7. Syntheses of Ureas (+)-9 and (–)-10
= +8 (c, 0.3, EtOH). 1H-NMR: δ = 0.99–1.07 (m 1H, H-7) 1.11–1.20 (m, 1H, H-7) 1.16 (t, J = 7.2, 3H, CH3) 1.23 (d, J = 7.1 (3H, CH3) 1.43–1.53 (m, 2H, H-8), 2.44 (m, 1H, H-1) 2.66 (m, 1H, H-4) 2.96 (d, J = 9.8, 1H, H-2) 3.90–4.06 (m, 2H, OCH2) 4.3 (td, J = 10.1, J = 1.5, 1H, H-3) 4.68 (m, 1H, CH) 5.34 (d, J = 10.4 H, NH) 6.12 (t, J = 7.2, 1H, H-5, 6.47 (t, J = 7.4, 1H, H-6) 6.54 (d, J = 8.1 1H, NH) 7.16–7.32 (m, 5H, Ar) 13C-NMR: δ = 14.8, 22.7, 24.4, 25.3, 32.6, 36.9, 49.3, 51.3, 51.4, 60.4, 126.5, 127.2, 129.0, 130.5, 137.1, 146.6, 157.0, 173.0. Anal. Calcd for C20H26N2O3 (342.43): C 70.15, H 7.65; N 8.18. Found C 70.22, H 7.92, N 8.12. = –101 (c, 0.2, EtOH) 1H-NMR: δ = 0.92–1.06 (m 1H, H-7) 1.19–1.23 (m, 1H, H-7) 1.16 (t, J = 7.1, 3H, CH3) 1.28 (d, J = 6.9 (3H, CH3) 1.36–1.52 (m, 2H, H-8), 1.95 (m, 1H, H-2) 2.58 (m, 1H, H-1) 2.70 (m, 1H, H-4) 4,08–4.23 (m, 3H, OCH2, H-3) 4.64 (m, 1H, CH) 5.54 (d, J = 7.9 H, NH) 6.12–6,21 (m, 2H, H-5, NH), 6.44 (t, J = 7.3, 1H, H-6) 7.14–7.35 (m, 5H, Ar) 13C-NMR: δ = 14.9, 20.3, 23.3, 24.3, 33.6, 36.0, 49.3, 51.5, 53.1, 60.8, 126.6, 127.3, 129.1, 132.8, 135.7, 146.5, 157.3, 173.8, Anal. Calcd for C20H26N2O3 (342.43): C 70.15, H 7.65, N 8.18. Found C 70.31, H 7.79, N 8.27.3.8. Racemic Compounds
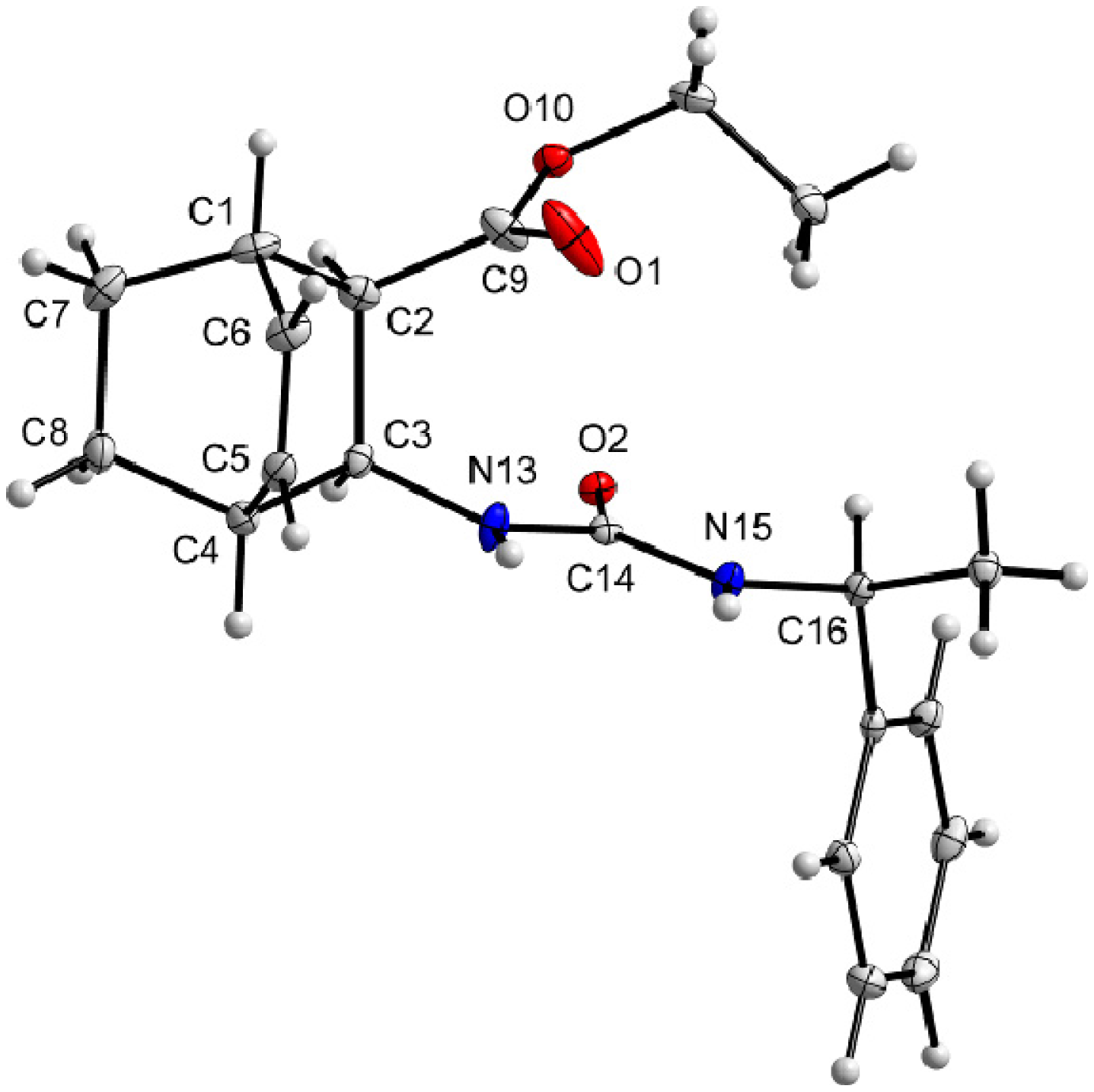
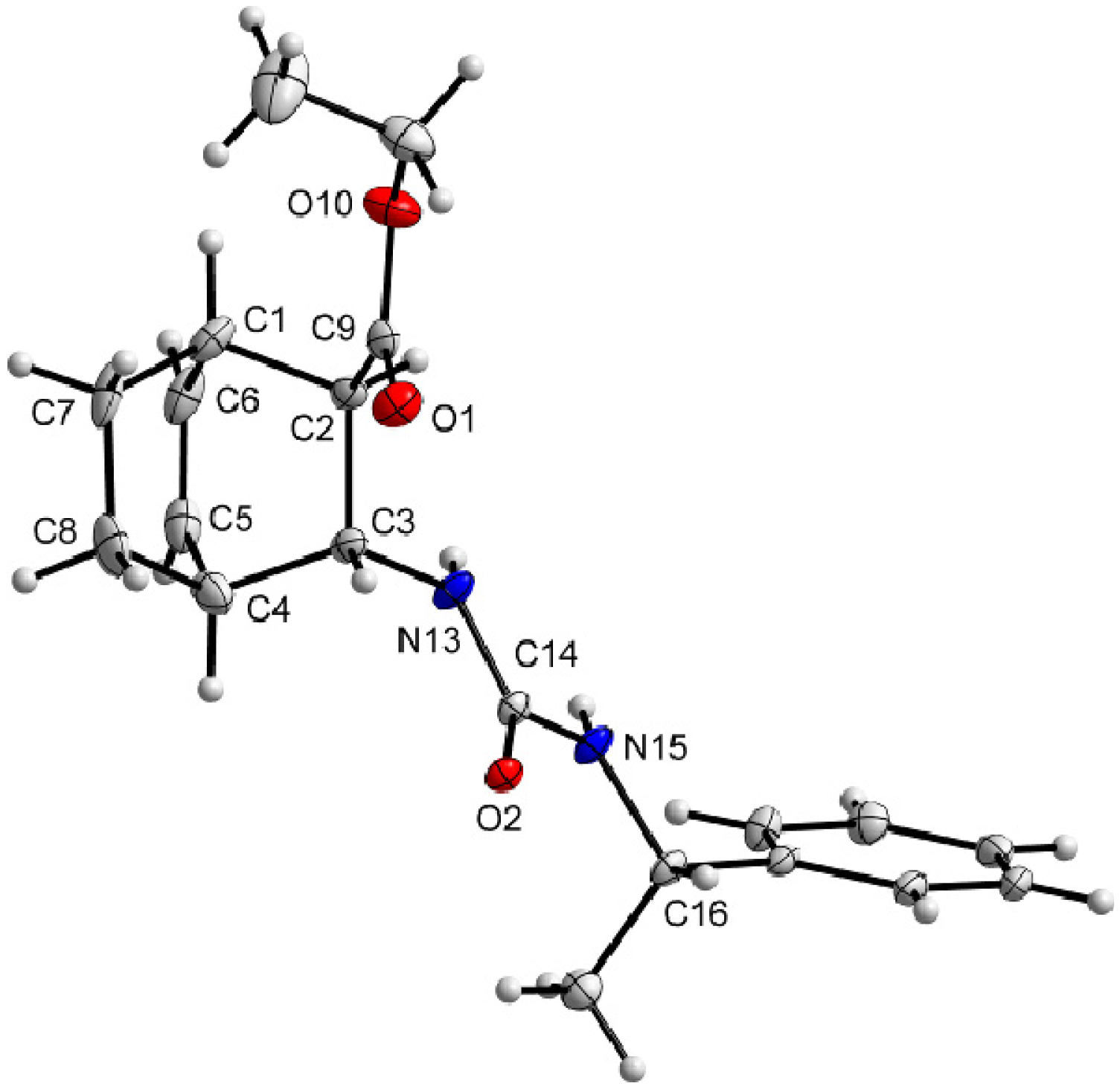
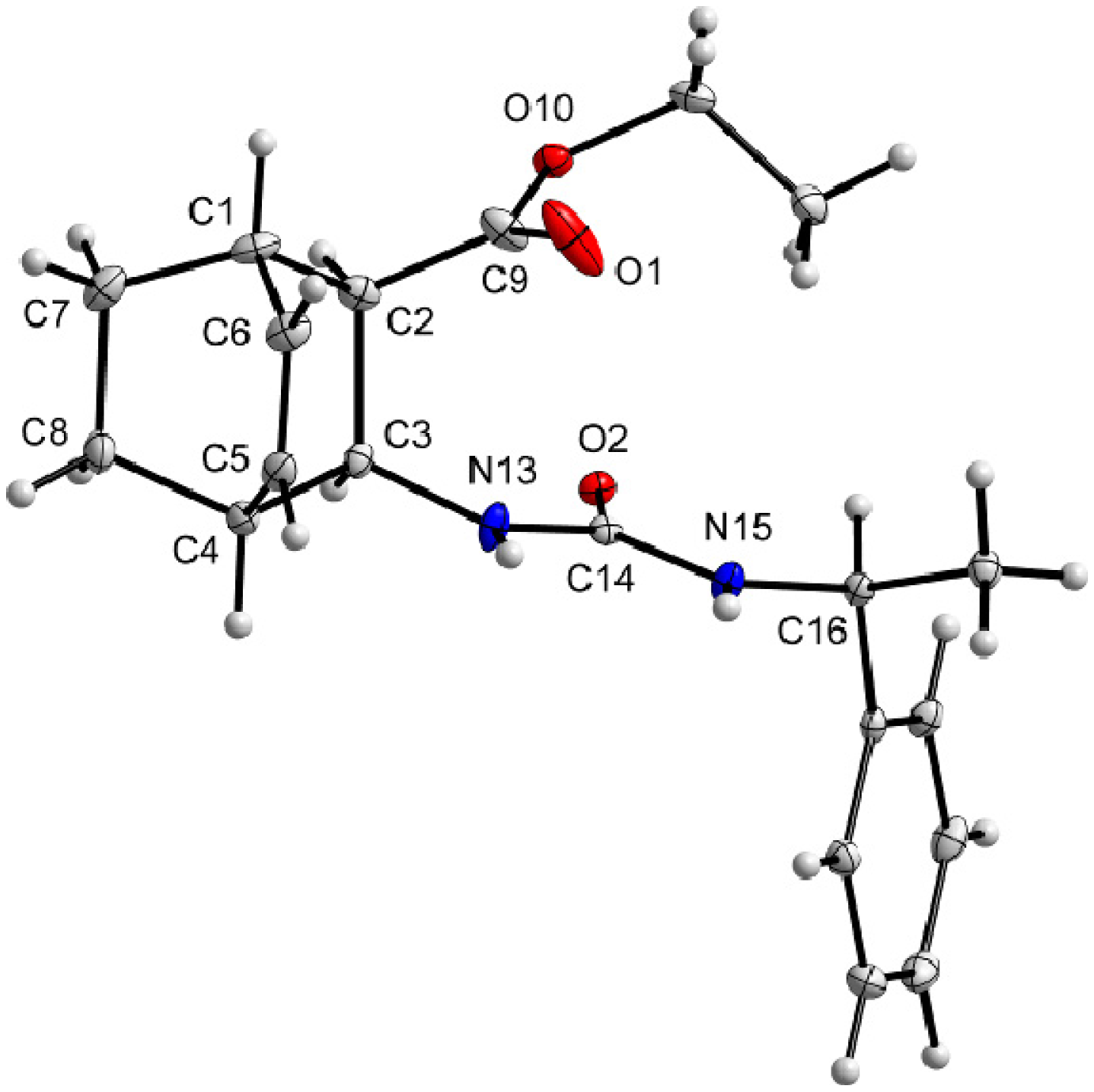
3.9. X-Ray Crystallographic Studies
| Compound | (+)-9 | ( –)-10 |
|---|---|---|
| Formula | C20H26N2O3 | C20H26N2O3 |
| Mr | 342.43 | 342.43 |
| Crystal system | orthorhombic | orthorhombic |
| Space group (no.) | P212121(19) | P212121 (19) |
| a (Å) | 12.0973(2) | 8.9562(3) |
| b (Å) | 17.4189(2) | 18.4189(6) |
| c (Å) | 17.5471(2) | 22.9915(3) |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 90 | 90 |
| V (Å) | 3697.55(9) | 3792.75(18) |
| Z | 8 | 8 |
| Dc (g cm−3) | 1.230 | 1.199 |
| T (K) | 123 | 123 |
| μ(CuKα) | 0.665 | 0.648 |
| Observed reflections | 6269 | 6726 |
| Rint | 0.0279 | 0.0213 |
| Parameters | 467 | 477 |
| Flack’s parameter | 0.05(17) | 0.0(2) |
| R1a | 0.0406 (0.0386) b | 0.0521 (0.0480)b |
| wR2c | 0.0991 (0.0969) | 0.1266 (0.1230) |
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Konishi, M.; Nishio, M.; Saitoh, K.; Miyaki, T.; Oki, T.; Kawaguchi, H. Cispentacin: A new antifungal antibiotic I. Production, isolation, physico-chemical properties and structure. J. Antibiot. 1989, 42, 1749–1755. [Google Scholar] [CrossRef]
- Oki, T.; Hirano, M.; Tomatsu, K.; Numata, K.; Kamei, H. Cispentacin, a new antifungal antibiotic II. In vitro and in vivo antifungal activities. J. Antibiot. 1989, 42, 1756–1762. [Google Scholar] [CrossRef]
- Kuhl, A.; Hahn, M.G.; Dumić, M.; Mittendorf, J. Alicyclic β-amino acids in medicinal chemistry. Amino Acids 2005, 29, 89–100. [Google Scholar] [CrossRef]
- Pou, A.; Moyano, A. Stereoselective organocatalytic approach to α,β-disubstituted β-amino acids: A sort enantioselective synthesis of Cispentacin. Eur. J. Org. Chem. 2013, 3103–3111. [Google Scholar] [CrossRef]
- Martinek, T.A.; Fülöp, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef]
- Horne, W.S. Peptide and peptoid foldamers in medicinal chemistry. Expert Opin. Drug Disc. 2011, 6, 1247–1262. [Google Scholar] [CrossRef]
- Sathe, M.; Thavaselvam, D.; Srivastava, A.K.; Kaushik, M.P. Synthesis and antimalarial evaluation of cyclic β-amino acid-containing dipeptides. Molecules 2008, 13, 432–443. [Google Scholar] [CrossRef]
- Keresztes, A.; Szücs, M.; Borics, A.; Kövér, K.E.; Forró, E.; Fülöp, F.; Tömböly, C.; Péter, A.; Páhi, A.; Fábián, G.; et al. New endomorphin analogues containing alicyclic β-amino acids: Influence on bioactive conformation and pharmacological profile. J. Med. Chem. 2008, 51, 4270–4279. [Google Scholar] [CrossRef]
- Horne, W.S.; Gellman, S.H. Foldamers with heterogeneous backbones. Acc. Chem. Res. 2008, 41, 1399–1408. [Google Scholar] [CrossRef]
- Seebach, D.; Gardiner, J. β-Peptidic peptidomimetics. Acc. Chem. Res. 2008, 41, 1366–1375. [Google Scholar] [CrossRef]
- Martín-Vilà, M.; Muray, E.; Aguado, G.P.; Alvarez-Larena, A.; Branchadell, V.; Minguillón, C.; Giralt, E.; Ortuňo, R.M. Enantioselective synthetic approaches to cyclopropane and cyclobutane β-amino acids: Synthesis and structural study of a conformationally constrained β-dipeptide. Tetrahedron-Asymmetr. 2000, 11, 3569–3584. [Google Scholar] [CrossRef]
- Gauzy, C.; Pereira, E.; Faure, S.; Aitken, D.J. Synthesis of (+)-(1S,2R) and (–)-(1R,2S)-2-aminocyclobutane-1-carboxylic acids. Tetrahedron Lett. 2004, 45, 7095–7097. [Google Scholar] [CrossRef]
- Fernandez, C.; Pereira, E.; Faure, S.; Aitken, D.J. Expedient preparation of all isomers of 2-aminocyclobutanecarboxylic acid in enantiomerically pure form. J. Org. Chem. 2009, 74, 3217–3220. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. The first direct enzymatic hydrolysis of alicyclic β-amino esters: A route to enantiopure cis and trans β-amino acids. Chem. Eur. J. 2007, 13, 6397–6401. [Google Scholar] [CrossRef]
- Priego, J.; Flores, P.; Ortiz-Nava, C.; Escalante, J. Synthesis of enantiopure cis- and trans-2-aminocyclohexane-1-carboxylic acids from octahydroquinazolin-4-ones. Tetrahedron- Asymmetr. 2004, 15, 3545–3549. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Vapour-assisted enzymatic hydrolysis of β-lactams in a solvent-free system. Tetrahedron-Asymmetr. 2008, 19, 1005–1009. [Google Scholar] [CrossRef]
- Palkó, M.; Benedek, G.; Forró, E.; Wéber, E.; Hänninen, M.; Sillanpää, R. Synthesis of mono- and dihydroxy-substituted 2-aminocyclooctanecarboxylic acid enantiomers. Tetrahedron-Asymmetr. 2010, 21, 957–961. [Google Scholar] [CrossRef]
- Bolm, C.; Schiffers, I.; Atodiresei, I.; Hackenberger, P.R. An alkaloid-mediated desymmetrization of meso-anhydrides via a nucleophilic ring opening with benzyl alcohol and its application in the synthesis of highly enantiomerically enriched β-amino acids. Tetrahedron-Asymmetr. 2003, 14, 3455–3467. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Synthesis of enantiopure 1,4-ethyl- and 1,4-ethylene-bridged cispentacin by lipase-catalyzed enantioselective ring opening of β-lactams. Tetrahedron-Asymmetr. 2004, 15, 573–575. [Google Scholar] [CrossRef]
- Todorov, P.T.; Wesselinova, D.W.; Pavlov, N.D.; Martinez J.; Calmes, M.; Naydenova, E.D. Cytotoxic activity of new racemic and optically active N-phosphonoalkyl bicyclic β-amino acids against human malignant cell lines. Amino Acids 2012, 43, 1445–1450. [Google Scholar] [CrossRef]
- André, C.; Calmes, M.; Escale, F.; Amblard, M.; Martinez, J.; Songis, O. New 1,3-amino alcohols derived from enantiopure bridgehead β-aminobicyclo[2.2.2]oct-5-ene-2-carboxylic acids. Amino Acids 2012, 43, 415–421. [Google Scholar] [CrossRef]
- Calmes, M.; Escale, F.; Didierjean, C.; Martinez, J. Synthesis of enantiopure trans-N-Boc-3-aminobicyclo[2.2.2]octane-2-carboxylic acids and their bicyclic 1,3-amino alcohol derivatives via the [4+2] cycloaddition of 1,3-cyclohexadiene to a chiral β-nitroacrylate. Chirality 2011, 23, 245–249. [Google Scholar] [CrossRef]
- Ruebsam, F.; Murphy, D.E.; Tran, C.V.; Li, L.S.; Zhao, J.; Dragovich, P.S.; McGuire, H.M.; Xiang, A.X.; Sun, Z.; Ayida, B.K.; et al. Discovery of tricyclic 5,6-dihydro-1H-pyridin-2-ones as novel, potent, and orally bioavailable inhibitors of HCV NS5B polymerase. Bioorg. Med. Chem. Lett. 2009, 19, 6404–6412. [Google Scholar] [CrossRef]
- Bunue, E.; Cativiela, C.; Diaz-de-Villages, M.D.; Galvez, J.A. A new conformationally restricted aspartic acid analogue with a bicyclo[2.2.2]octanes skeleton. Acta Cryst. C 1996, 52, 2292–2294. [Google Scholar] [CrossRef]
- Pope, W.J.; Peachey, S.J. The application of powerful optically active acids to the resolution of externally compensated basic substances. Resolution of tetrahydroquinaldine. J. Chem. Soc. Trans. 1899, 75, 1066–1093. [Google Scholar] [CrossRef]
- Sakurai, R.; Yuzawa, A.; Sakai, K. Practical resolution of 3-aminopyrrolidine via diastereomeric salt formation with (S)-2-methoxy-2-phenylacetic acid. Tetrahedron-Asymmetr. 2008, 19, 1622–1625. [Google Scholar] [CrossRef]
- Faigl, F.; Fogassy, E.; Nógrádi, M.; Pálovics, E.; Schindler, J. Strategies in optical resolution: A practical guide. Tetrahedron-Asymmetr. 2008, 19, 519–536. [Google Scholar] [CrossRef]
- Leeman, M.; Kaptein, B.; Kellogg, R.M. Nucleation inhibition in attrition-enhanced Pope-Peachey type of diastereomeric resolutions. Tetrahedron-Asymmetr. 2009, 20, 1363–1364. [Google Scholar] [CrossRef]
- Palkó, M.; Sohár, P.; Fülöp, F. Synthesis and transformations of di-endo-3-aminobicyclo-[2.2.2]oct-5-ene-2-carboxylic acid derivatives. Molecules 2011, 16, 7691–7705. [Google Scholar] [CrossRef]
- Fogassy, E.; Lopata, A.; Faigl, F.; Darvas, F.; Ács, M.; Tőke, L. A quantitative approach to optical resolution. Tetrahedron Lett. 1980, 21, 647–650. [Google Scholar] [CrossRef]
- Forró, E. New gas chromatographic method for the enantioseparation of β-amino acids by a rapid double derivatization technique. J. Chromatogr. A 2009, 1216, 1025–1029. [Google Scholar] [CrossRef]
- Moriconi, E.I.; Crawford, W.C. Reaction of chlorosulfonyl isocyanate with bridge bi- and tricyclic olefins. J. Org. Chem. 1968, 33, 370–378. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO, Agilent Technologies UK Ltd.: Yarnton, UK, 2011.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Pennington, W.T. DIAMOND-visual crystal structure information system. J. Appl. Cryst. 1999, 32, 1028–1029. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–10 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Palkó, M.; Hänninen, M.M.; Sillanpää, R.; Fülöp, F. Syntheses of Four Enantiomers of 2,3-Diendo- and 3-Endo-aminobicyclo[2.2.2]oct-5-ene-2-exo-carboxylic Acid and Their Saturated Analogues. Molecules 2013, 18, 15080-15093. https://doi.org/10.3390/molecules181215080
Palkó M, Hänninen MM, Sillanpää R, Fülöp F. Syntheses of Four Enantiomers of 2,3-Diendo- and 3-Endo-aminobicyclo[2.2.2]oct-5-ene-2-exo-carboxylic Acid and Their Saturated Analogues. Molecules. 2013; 18(12):15080-15093. https://doi.org/10.3390/molecules181215080
Chicago/Turabian StylePalkó, Márta, Mikko M. Hänninen, Reijo Sillanpää, and Ferenc Fülöp. 2013. "Syntheses of Four Enantiomers of 2,3-Diendo- and 3-Endo-aminobicyclo[2.2.2]oct-5-ene-2-exo-carboxylic Acid and Their Saturated Analogues" Molecules 18, no. 12: 15080-15093. https://doi.org/10.3390/molecules181215080
APA StylePalkó, M., Hänninen, M. M., Sillanpää, R., & Fülöp, F. (2013). Syntheses of Four Enantiomers of 2,3-Diendo- and 3-Endo-aminobicyclo[2.2.2]oct-5-ene-2-exo-carboxylic Acid and Their Saturated Analogues. Molecules, 18(12), 15080-15093. https://doi.org/10.3390/molecules181215080