Synthesis of Cycloveratrylene Macrocycles and Benzyl Oligomers Catalysed by Bentonite under Microwave/Infrared and Solvent-Free Conditions


Abstract
:1. Introduction
2. Results and Discussion

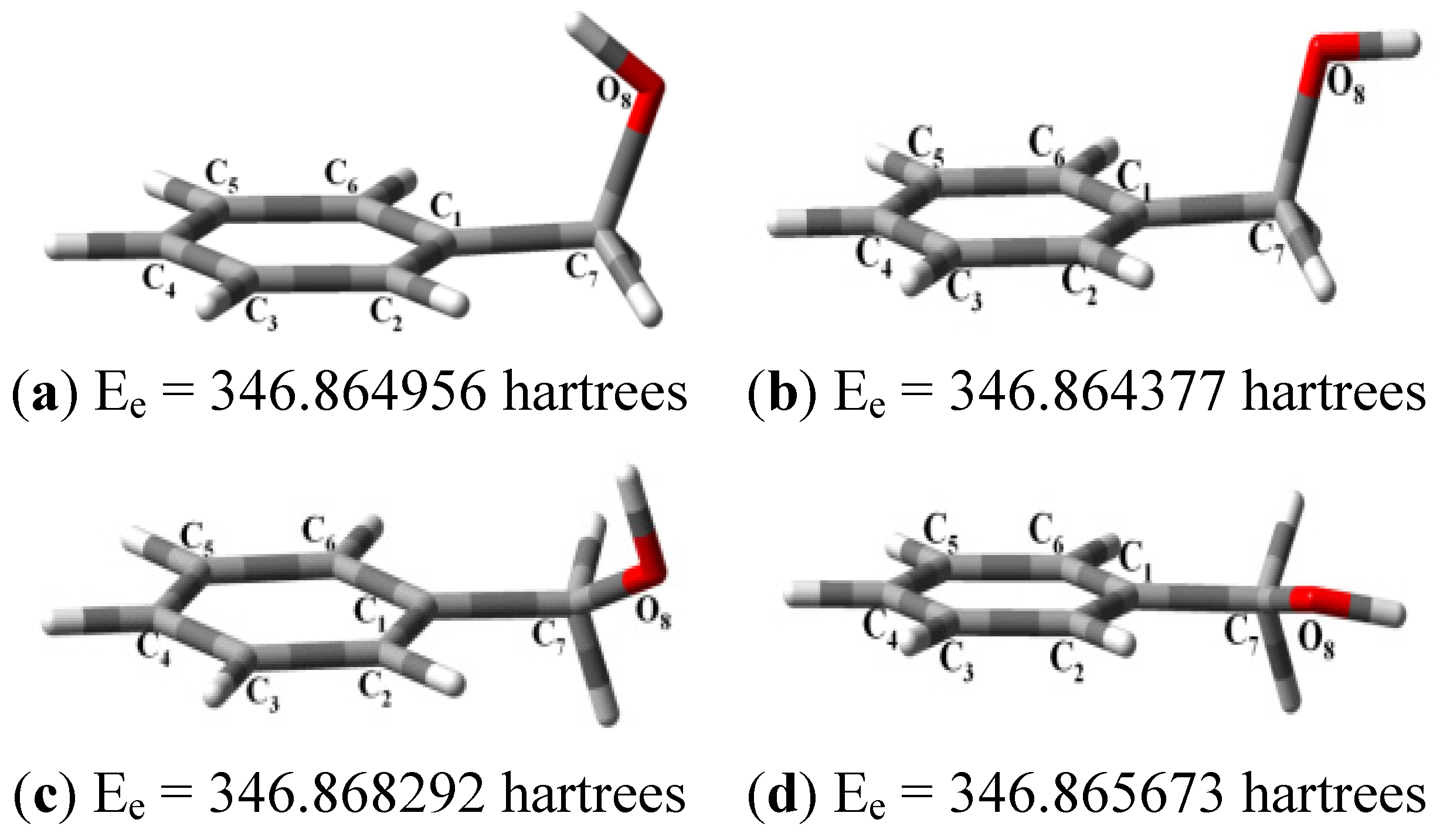{kind=link}
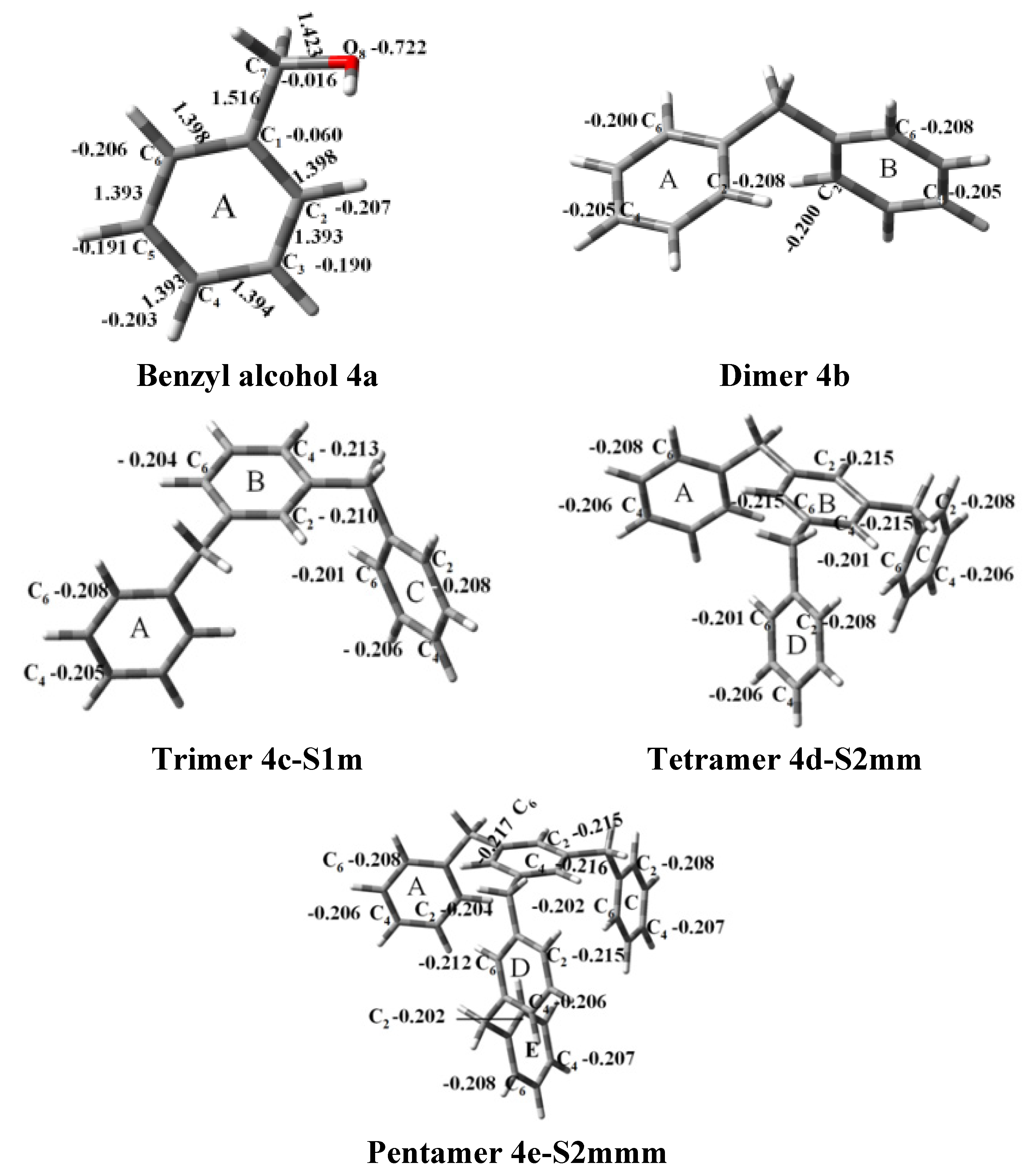{kind=link}
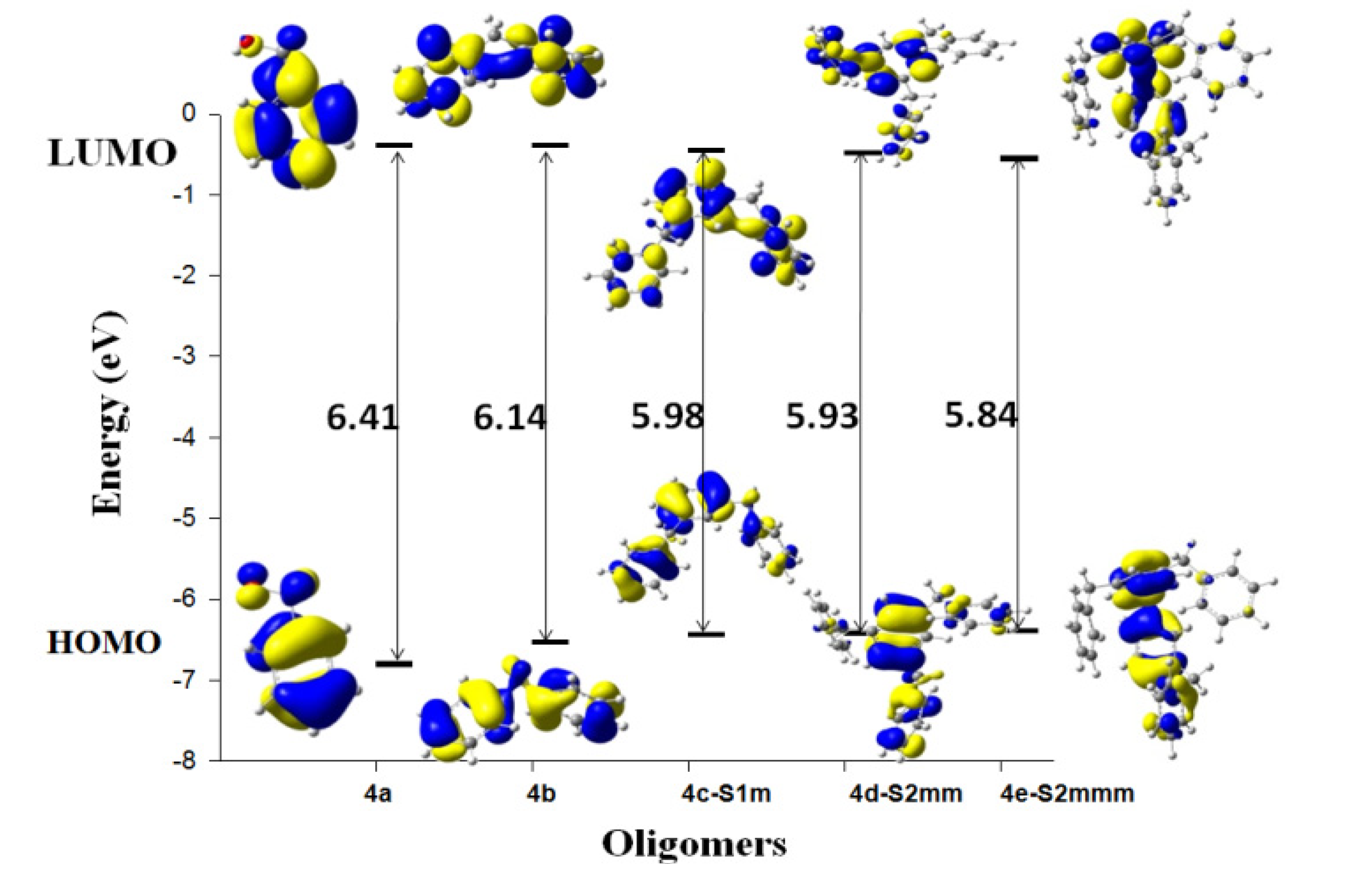{kind=link}
{kind=link}
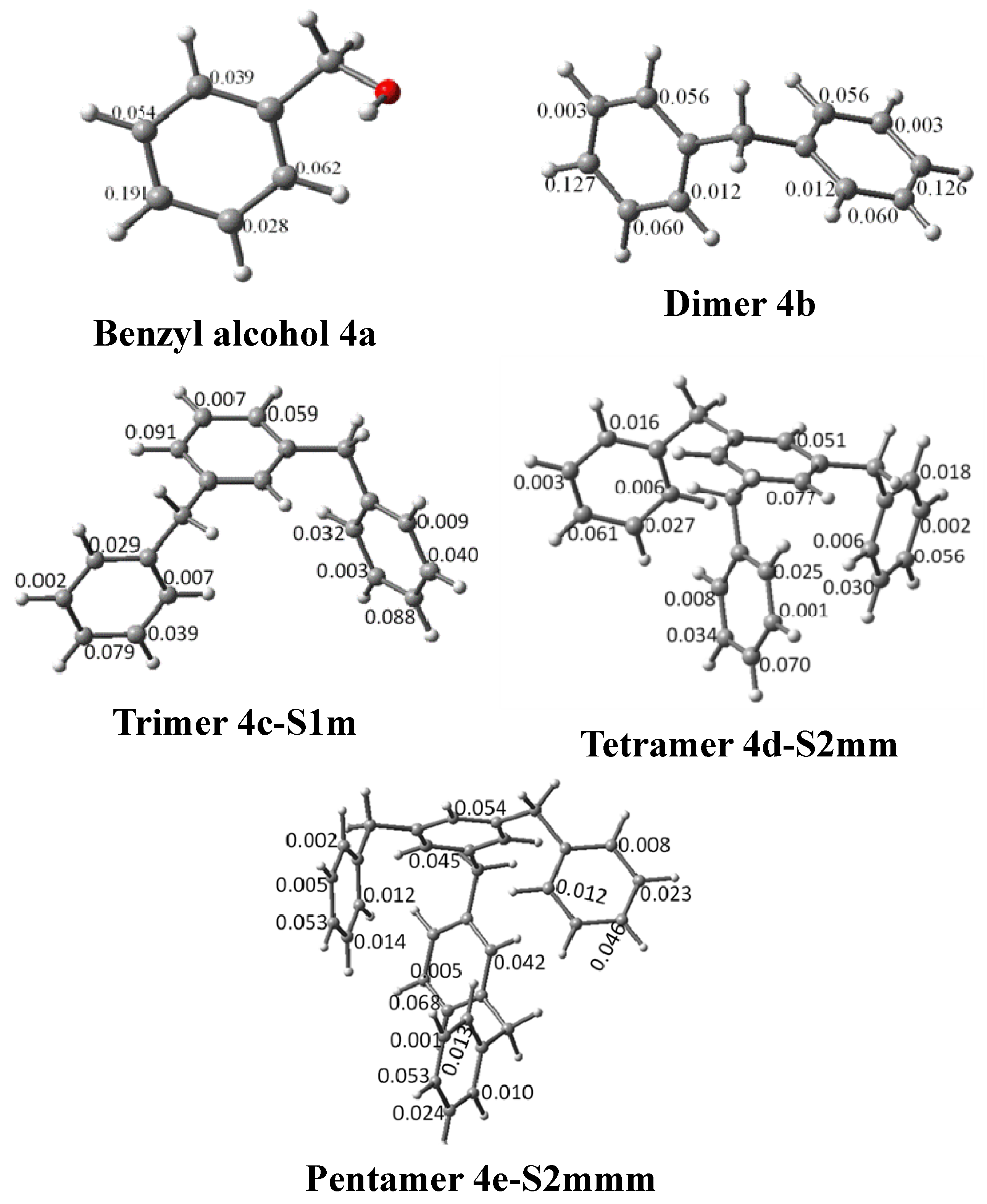{kind=link}
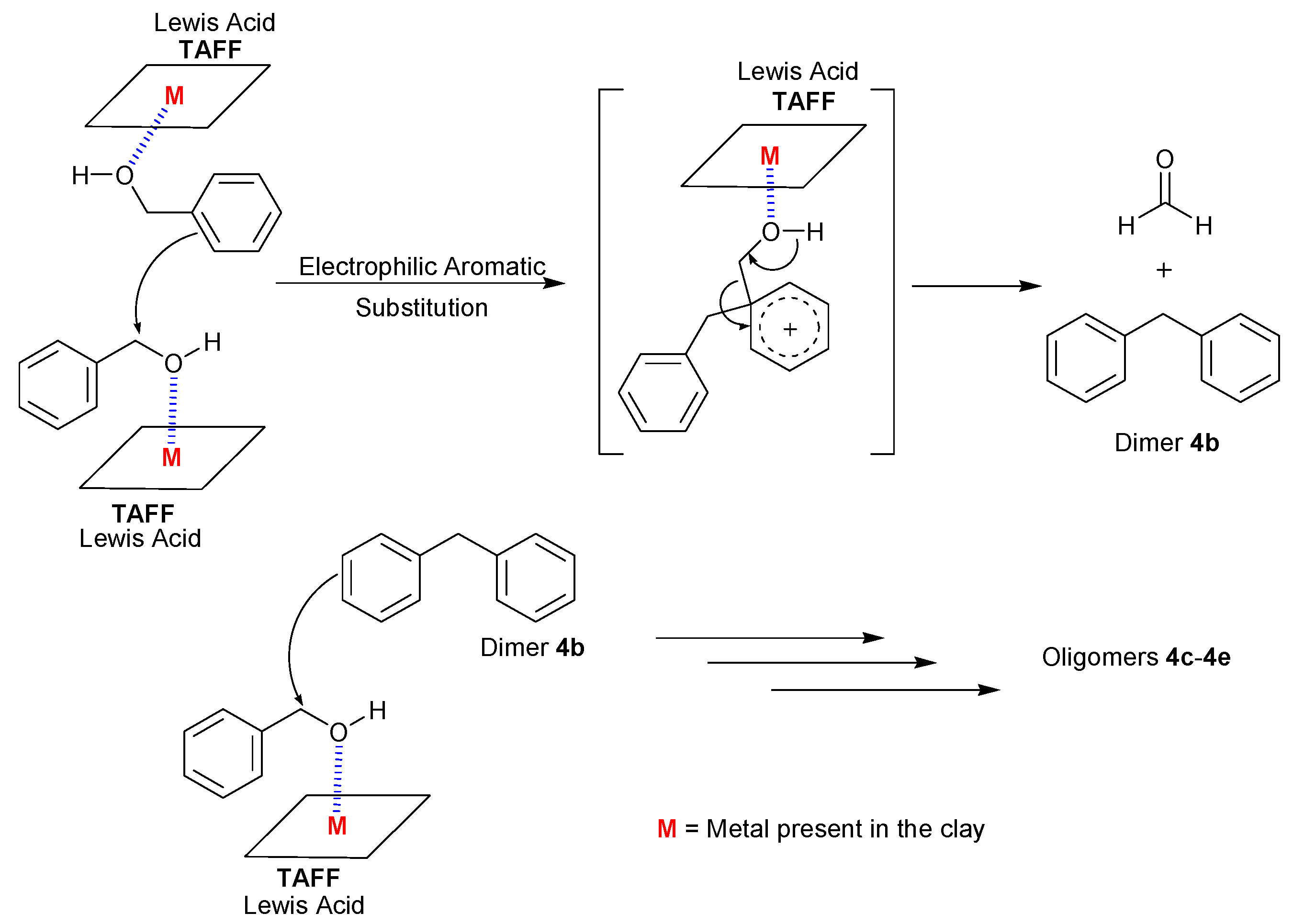{kind=link}
| Entry | Alcohol | Products | Yield% b | ||||
|---|---|---|---|---|---|---|---|
| MW c | IR d | ||||||
| 1 |  |  | 85 | 80 | |||
 | 1 | 3 | |||||
 | 2 | 5 | |||||
 | 4 | 5 | |||||
 | 8 | 7 | |||||
| 2 |  |  | 90 | 88 | |||
 | 10 | 12 | |||||
| 3 |  |  | 80 | 75 | |||
 | 5 | 5 | |||||
 | 15 | 20 | |||||
| Entry | Alcohol | Products | Yield% b | ||
|---|---|---|---|---|---|
| MW c | IR d | ||||
| 1 |  |  | 4b n = 0 | 0 | 0 |
| 4c n = 1 | 60 | 58 | |||
| 4d n = 2 | 40 | 22 | |||
| 4e n = 3 | 0 | 20 | |||
| 2 |  |  | 5b n = 0 | 0 | 0 |
| 5c n = 1 | 55 | 0 | |||
| 5d n = 2 | 28 | 0 | |||
| 5e n = 3 | 12 | 0 | |||
| 5f n = 4 | 5 | 0 | |||
 | 5g n = 0 | 0 | 65 | ||
| 5h n = 1 | 0 | 25 | |||
| 5i n = 2 | 0 | 10 | |||
| 3 |  |  | 6b n = 0 | 0 | 0 |
| 6c n = 1 | 35 | 8 | |||
| 6d n = 2 | 65 | 92 | |||
| Alcohol | Products | High resolution | Elemental composition | Yield% a | ||
|---|---|---|---|---|---|---|
| MW | IR | |||||
 |  | 4c n = 1 | 258.1385 b/258.1409 c (−8.9) d | C20H18 | 60 | 58 |
| 4d n = 2 | 348.1884 b/348.1878 c (1.6) d | C27H24 | 40 | 22 | ||
| 4e n = 3 | 438.2355 b/438.2348 c (1.8) d | C34H30 | 0 | 20 | ||
 |  | 5c n = 1 | 300.1885 b/300.1878 c (1.8) d | C23H24 | 55 | 0 |
| 5d n = 2 | 404.2498 b/404.2504 c (1.8) d | C31H32 | 28 | 0 | ||
| 5e n = 3 | 508.3136 b/508.3130 c (1.6) d | C39H40 | 12 | 0 | ||
| 5f n = 4 | 612.3762 b/612.3756 c (1.6) d | C47H48 | 5 | 0 | ||
 | 5g n = 0 | 226.1346 b/226.1358 (−5.4) d | C16H18O | 0 | 65 | |
| 5h n = 1 | 330.1990 b/330.1984 c (1.9) d | C24H26O | 0 | 25 | ||
| 5i n = 2 | 434.2614 b/434.2610 c (1.8) d | C32H34O | 0 | 10 | ||
 |  | 6c n = 1 | 348.1755 b/348.1757 c (3.8) d | C23H24O3 | 35 | 8 |
| 6d n = 2 | 468.2345 b/468.2343 c (4.6) d | C31H32O4 | 65 | 92 | ||
 |  |  |  |
| 4b | 4c-S1o | 4c-S1m | 4c-S1p |
 |  |  |  |
| 4d-S1oo | 4d-S1om | 4d-S1op | 4d-S1mm |
 |  |  |  |
| 4d-S1mp | 4d-S1pp | 4d-S2om | 4d-S2op |
 |  |  |  |
| 4d-S2mm | 4e-S1ooo | 4e-S1oom | 4e-S1oop |
 |  |  |  |
| 4e-S1omo | 4e-S1omm | 4e-S1omp | 4e-S1opo |
 |  |  |  |
| 4e-S1opm | 4e-S1opp | 4e-S1mom | 4e-S1mop |
 |  |  |  |
| 4e-S1mmm | 4e-S1mmp | 4e-S1mpm | 4e-S1mpp |
 |  |  |  |
| 4e-S1pop | 4e-S1pmp | 4e-S1ppp | 4e-S2ooo |
 |  |  |  |
| 4e-S2oom | 4e-S2oop | 4e-S2omo | 4e-S2omm |
 |  |  |  |
| 4e-S2omp | 4e-S2moo | 4e-S2mom | 4e-S2mop |
 |  |  |  |
| 4e-S2mmo | 4e-S2mmm | 4e-S2mmp | 4e-S2poo |
 |  |  |  |
| 4e-S2pom | 4e-S2pop | 4e-S3oom | 4e-S3omp |
 |  |  |  |
| 4e-S3mmp | 4e-S4moo | 4e-S4mom | 4e-S4mop |
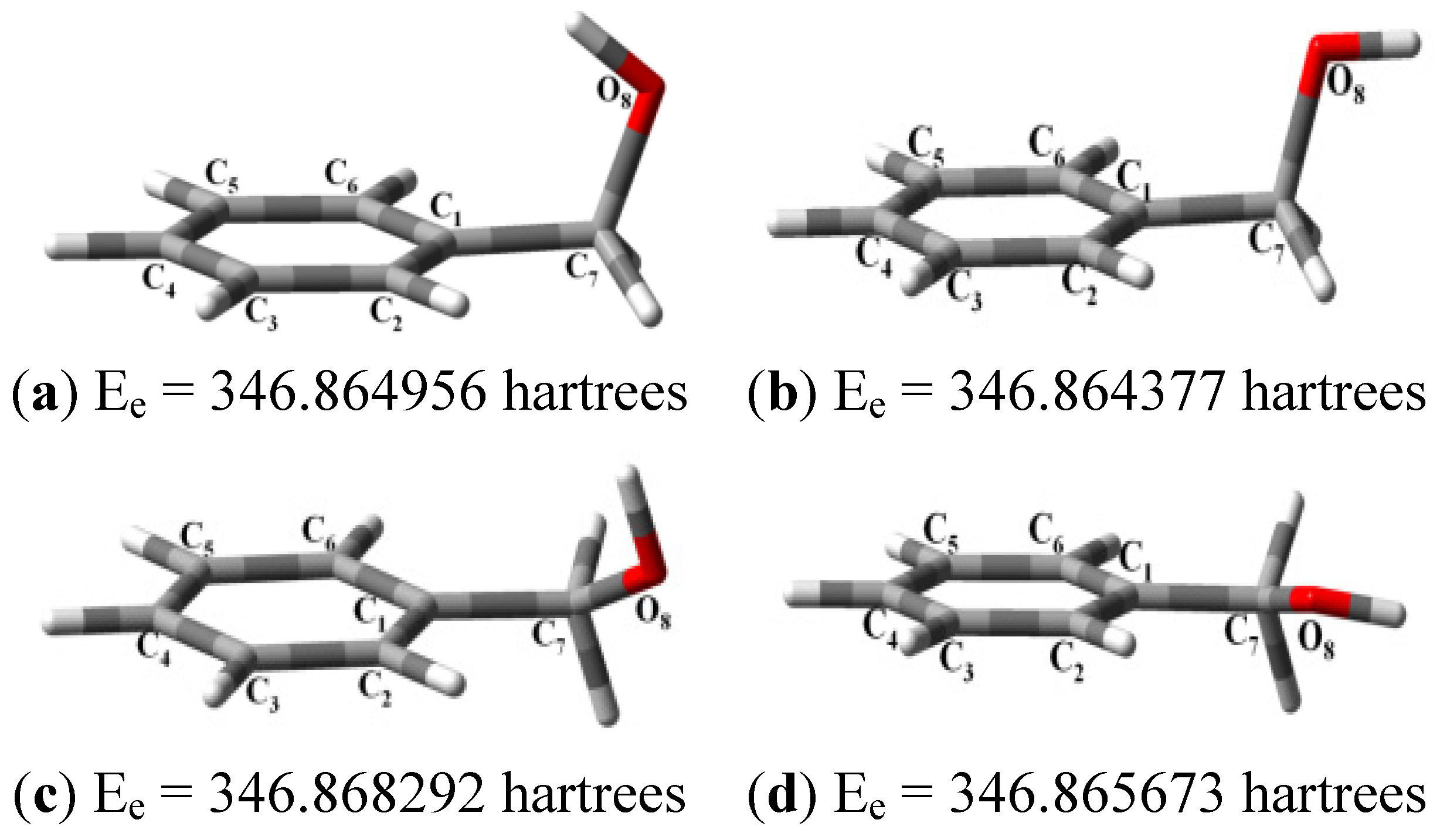
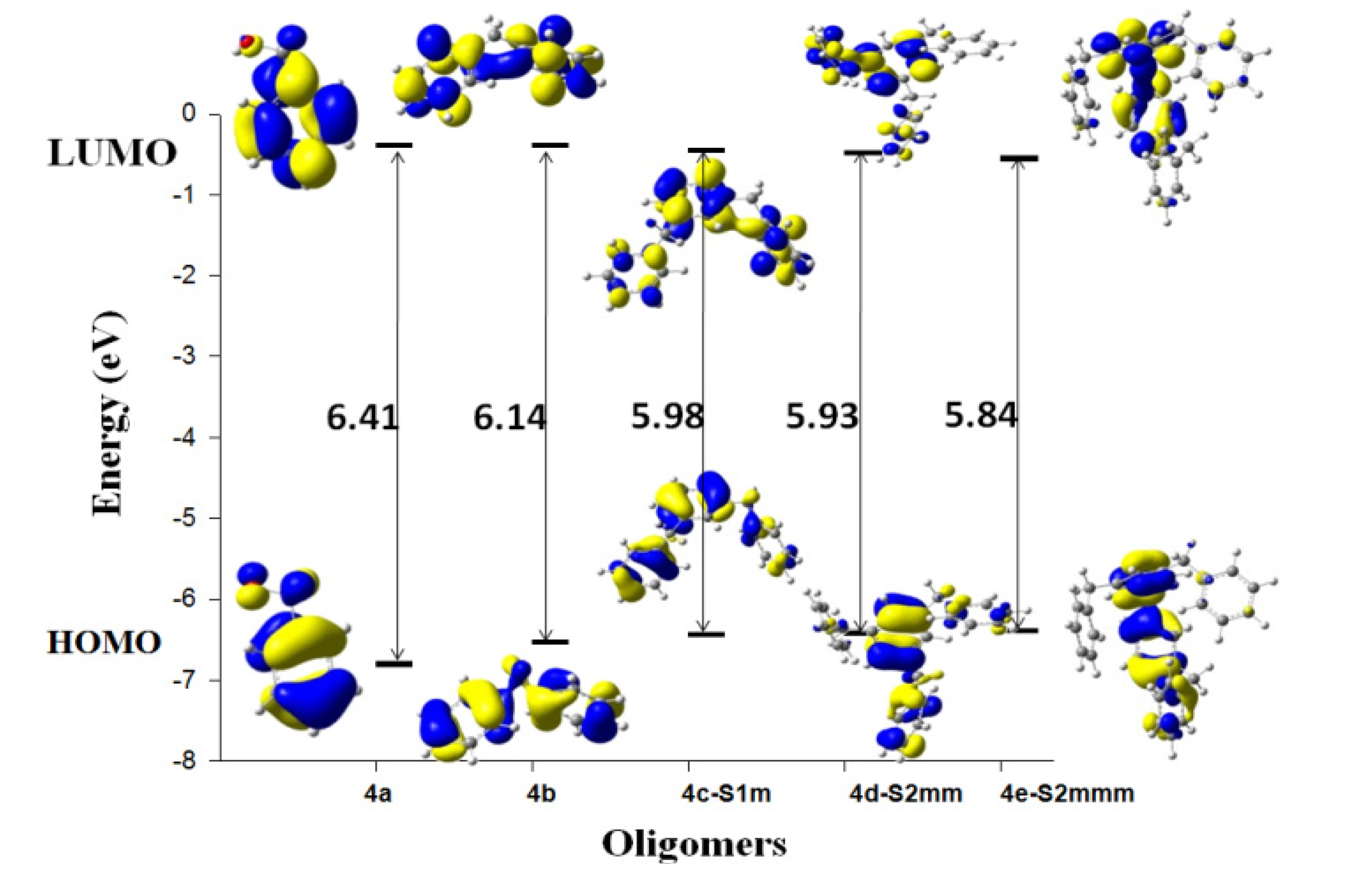
| Structure | Ee (Hartrees) | Erel (Kcal/mol) | HOMO (eV) | LUMO (eV) | GAP (eV) | IP (eV) | EA (eV) | Χ (eV) | η (eV) | µ (eV) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | - | - | −6.81 | −0.40 | 6.41 | 8.68 | −1.66 | 3.60 | 5.26 | −3.60 | |
| 4b | - | - | −6.54 | −0.40 | 6.14 | 8.18 | −1.17 | 3.51 | 4.67 | −3.51 | |
| 4c-S1o | −773.166658 | 1.19 | −6.55 | −0.52 | 6.04 | 8.08 | −0.84 | 3.58 | 4.43 | −3.58 | |
| 4c-S1m | −773.168557 | 0.00 | −6.44 | −0.46 | 5.98 | 7.83 | −0.87 | 3.48 | 4.35 | −3.48 | |
| 4c-S1p | −773.168276 | 0.18 | −6.36 | −0.45 | 5.92 | 7.76 | −0.87 | 3.44 | 4.31 | −3.44 | |
| 4d-S1oo | −1043.594698 | 2.68 | −6.50 | −0.54 | 5.96 | 7.78 | −0.68 | 3.55 | 4.23 | −3.55 | |
| 4d-S1om | −1043.597379 | 0.99 | −6.46 | −0.50 | 5.96 | 7.73 | −0.73 | 3.86 | 3.87 | −3.86 | |
| 4d-S1op | −1043.596796 | 1.36 | −6.37 | −0.52 | 5.86 | 7.68 | −0.66 | 3.51 | 4.17 | −3.51 | |
| 4d-S1mm | −1043.598465 | 0.31 | −6.43 | −0.53 | 5.90 | 7.67 | −0.69 | 3.49 | 4.18 | −3.49 | |
| 4d-S1mp | −1043.598569 | 0.25 | −6.37 | −0.50 | 5.87 | 7.62 | −0.73 | 3.44 | 4.18 | −3.44 | |
| 4d-S1pp | −1043.598254 | 0.45 | −6.30 | −0.46 | 5.84 | 7.49 | −0.69 | 3.40 | 4.09 | −3.40 | |
| 4d-S2om | −1043.593291 | 3.56 | −6.51 | −0.53 | 5.97 | 7.75 | −0.68 | 3.54 | 4.22 | −3.54 | |
| 4d-S2op | −1043.596965 | 1.25 | −6.40 | −0.53 | 5.87 | 7.70 | −0.71 | 3.50 | 4.21 | −3.50 | |
| 4d-S2mm | −1043.598963 | 0.00 | −6.42 | −0.49 | 5.94 | 7.69 | −0.74 | 3.47 | 4.22 | −3.47 | |
| 4e-S1ooo | −1314.022344 | 4.36 | −6.47 | −0.55 | 5.92 | 7.64 | −0.58 | 3.53 | 4.11 | −3.53 | |
| 4e-S1oom | −1314.024711 | 2.88 | −6.37 | −0.56 | 5.81 | 7.58 | −0.57 | 3.50 | 4.07 | −3.50 | |
| 4e-S1oop | −1314.024876 | 2.78 | −6.38 | −0.56 | 5.82 | 7.56 | −0.56 | 3.50 | 4.06 | −3.50 | |
| 4e-S1omo | −1314.024522 | 3.00 | −6.51 | −0.56 | 5.95 | 7.67 | −0.56 | 3.55 | 4.11 | −3.55 | |
| 4e-S1omm | −1314.027076 | 1.40 | −6.44 | −0.56 | 5.88 | 7.60 | −0.62 | 3.49 | 4.11 | −3.49 | |
| 4e-S1omp | −1314.027660 | 1.03 | −6.36 | −-0.53 | 5.83 | 7.53 | −0.57 | 3.48 | 4.05 | −3.48 | |
| 4e-S1opo | −1314.024289 | 3.14 | −6.42 | −0.55 | 5.87 | 7.64 | −0.53 | 3.56 | 4.08 | −3.56 | |
| 4e-S1opm | −1314.027017 | 1.43 | −6.35 | −0.55 | 5.80 | 7.56 | −0.55 | 3.50 | 4.05 | −3.50 | |
| 4e-S1opp | −1314.026242 | 1.92 | −6.32 | −0.51 | 5.81 | 7.52 | −0.59 | 3.46 | 4.05 | −3.46 | |
| 4e-S1mom | −1314.027453 | 1.16 | −6.35 | −0.54 | 5.81 | 7.55 | −0.58 | 3.48 | 4.07 | −3.48 | |
| 4e-S1mop | −1314.027335 | 1.23 | −6.31 | −0.49 | 5.81 | 7.51 | −0.61 | 3.45 | 4.06 | −3.45 | |
| 4e-S1mmm | −1314.028818 | 0.30 | −6.43 | −0.55 | 5.88 | 7.58 | −0.61 | 3.48 | 4.10 | −3.48 | |
| 4e-S1mmp | −1314.029167 | 0.08 | −6.33 | −0.51 | 5.82 | 7.49 | −0.61 | 3.44 | 4.05 | −3.44 | |
| 4e-S1mpm | −1314.028720 | 0.36 | −6.31 | −0.50 | 5.81 | 7.47 | −0.60 | 3.43 | 4.03 | −3.43 | |
| 4e-S1mpp | −1314.028319 | 0.62 | −6.27 | −0.49 | 5.78 | 7.39 | −0.58 | 3.41 | 3.99 | −3.41 | |
| 4e-S1pop | −1314.025814 | 2.19 | −6.38 | −0.52 | 5.86 | 7.48 | −0.56 | 3.46 | 4.02 | −3.46 | |
| 4e-S1pmp | −1314.028888 | 0.26 | −6.29 | −0.47 | 5.82 | 7.43 | −0.65 | 3.39 | 4.04 | −3.39 | |
| 4e-S1ppp | −1314.028174 | 0.71 | −6.23 | −0.47 | 5.76 | 7.34 | −0.59 | 3.37 | 3.96 | −3.37 | |
| 4e-S2ooo | −1314.021068 | 5.17 | −6.47 | −0.56 | 5.91 | 7.63 | −0.58 | 3.52 | 4.11 | −3.52 | |
| 4e-S2oom | −1314.023710 | 3.51 | −6.43 | −0.53 | 5.91 | 7.58 | −0.60 | 3.49 | 4.09 | −3.49 | |
| 4e-S2oop | −1314.023301 | 3.76 | −6.35 | −0.54 | 5.81 | 7.53 | −0.55 | 3.49 | 4.04 | −3.49 | |
| 4e-S2omo | −1314.024372 | 3.09 | −6.35 | −0.55 | 5.80 | 7.61 | −0.57 | 3.52 | 4.09 | −3.52 | |
| 4e-S2omm | −1314.027225 | 1.30 | −6.34 | −0.52 | 5.83 | 7.54 | −0.60 | 3.47 | 4.07 | −3.47 | |
| 4e-S2omp | −1314.027168 | 1.34 | −6.33 | −0.53 | 5.80 | 7.51 | −0.57 | 3.47 | 4.04 | −3.47 | |
| 4e-S2moo | −1314.024830 | 2.81 | −6.32 | −0.55 | 5.77 | 7.55 | −0.56 | 3.50 | 4.05 | −3.50 | |
| 4e-S2mom | −1314.027493 | 1.13 | −6.35 | −0.52 | 5.82 | 7.53 | −0.62 | 3.45 | 4.07 | −3.45 | |
| 4e-S2mop | −1314.026932 | 1.49 | -6.33 | −0.54 | 5.79 | 7.48 | −0.57 | 4.46 | 4.03 | −3.46 | |
| 4e-S2mmo | −1314.027206 | 1.31 | -6.35 | −0.53 | 5.82 | 7.52 | −0.60 | 3.46 | 4.06 | −3.46 | |
| 4e-S2mmm | −1314.029301 | 0.00 | −6.40 | −0.56 | 5.84 | 7.59 | −0.59 | 3.50 | 4.09 | −3.50 | |
| 4e-S2mmp | −1314.028956 | 0.22 | −6.31 | −0.51 | 5.81 | 7.50 | −0.62 | 3.44 | 4.06 | −3.44 | |
| 4e-S2poo | −1314.024639 | 2.92 | −6.35 | −0.53 | 5.81 | 7.57 | −0.59 | 3.49 | 4.08 | −3.49 | |
| 4e-Spom | −1314.027489 | 1.14 | −6.34 | −0.51 | 5.83 | 7.52 | −0.62 | 3.45 | 4.07 | −3.45 | |
| 4e-S2pop | −1314.026941 | 1.48 | −6.33 | −0.54 | 5.79 | 7.50 | −0.56 | 3.47 | 4.03 | −3.47 | |
| 4e-S3oom | −1314.020185 | 5.72 | −6.47 | −0.58 | 5.89 | 7.70 | −0.56 | 3.57 | 4.13 | −3.57 | |
| 4e-S3omp | −1314.024768 | 2.84 | −6.35 | −0.60 | 5.75 | 7.59 | −0.53 | 3.53 | 4.06 | −3.53 | |
| 4e-S3mmp | −1314.023392 | 3.71 | −6.37 | −0.58 | 5.78 | 7.57 | −0.55 | 3.51 | 4.06 | −3.51 | |
| 4e-S4moo | −1314.020794 | 5.34 | −6.44 | −0.55 | 5.89 | 7.62 | −0.59 | 3.51 | 4.11 | −3.51 | |
| 4e-S4mom | −1314.023562 | 3.60 | −6.44 | −0.52 | 5.92 | 7.61 | −0.60 | 3.50 | 4.10 | −3.50 | |
| 4e-S4mop | −1314.023314 | 3.76 | −6.34 | −0.54 | 5.79 | 7.52 | −0.55 | 3.48 | 4.04 | −3.48 | |

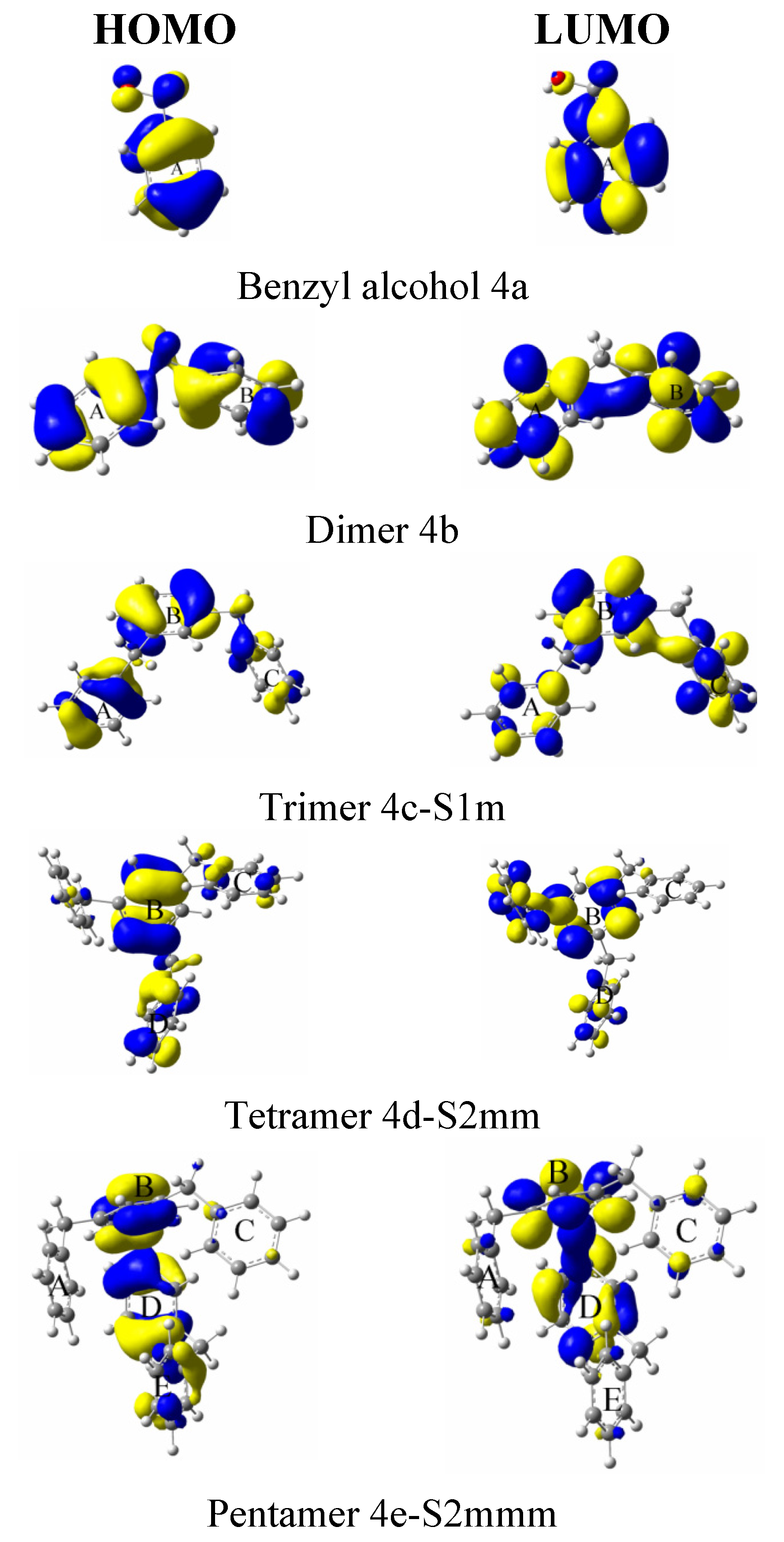
| System | Atom a | f- |
|---|---|---|
| 4a | Cp | 0.191 |
| 4b | Cp | 0.126 |
| Cp | 0.127 | |
| 4c-S1p | Cp | 0.079 |
| Cp | 0.078 | |
| 4c-S1m | Cp | 0.079 |
| Cp | 0.091 | |
| Cp | 0.088 | |
| 4c-S1o | Cp | 0.080 |
| Cp | 0.082 | |
| Cp | 0.083 | |
| Cp | 0.083 | |
| 4d-S1pp | Cp | 0.059 |
| Cp | 0.058 | |
| 4d-S2om | Cp | 0.051 |
| Cp | 0.056 | |
| Cp | 0.065 | |
| 4d-S1mp | Cp | 0.062 |
| Cp | 0.070 | |
| Cp | 0.058 | |
| 4d-S1op | Cp | 0.061 |
| Cp | 0.070 | |
| 4e-S1ppp | Cp | 0.044 |
| Cp | 0.044 | |
| 4e-S2mop | Cp | 0.050 |
| Cp | 0.056 | |
| 4e-S2pop | Cp | 0.045 |
| Cp | 0.057 | |
| 4e-S2omp | Cp | 0.046 |
| Cp | 0.051 | |
| 4e-S2mpp | Cp | 0.045 |
| Cp | 0.054 | |
| Cp | 0.050 | |
| 4e-S1opp | Cp | 0.48 |
| Cp | 0.045 | |
| 4e-S1pmp | Cp | 0.038 |
| Cp | 0.040 | |
| 4e-S1pop | Cp | 0.054 |
| Cp | 0.049 |
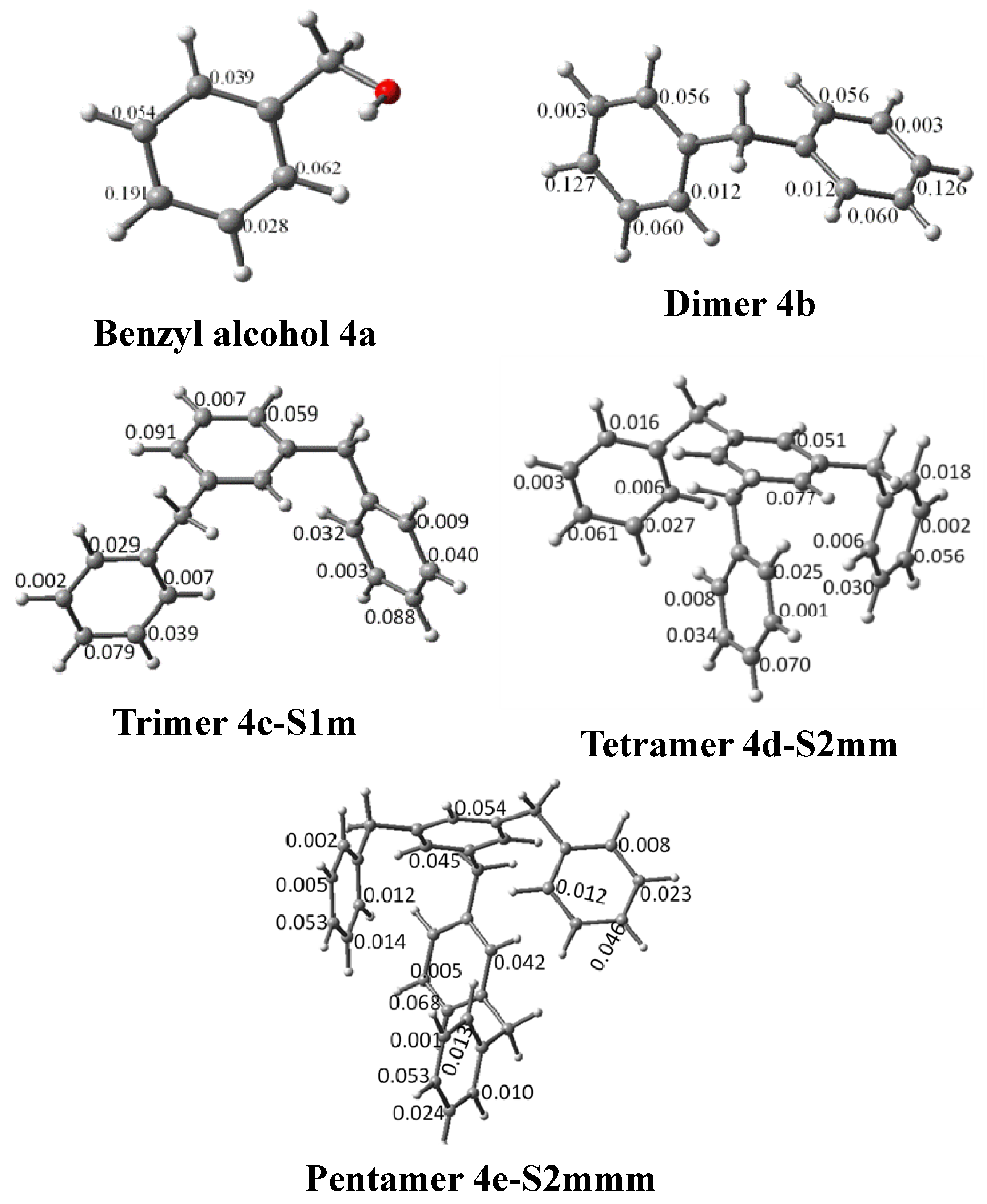
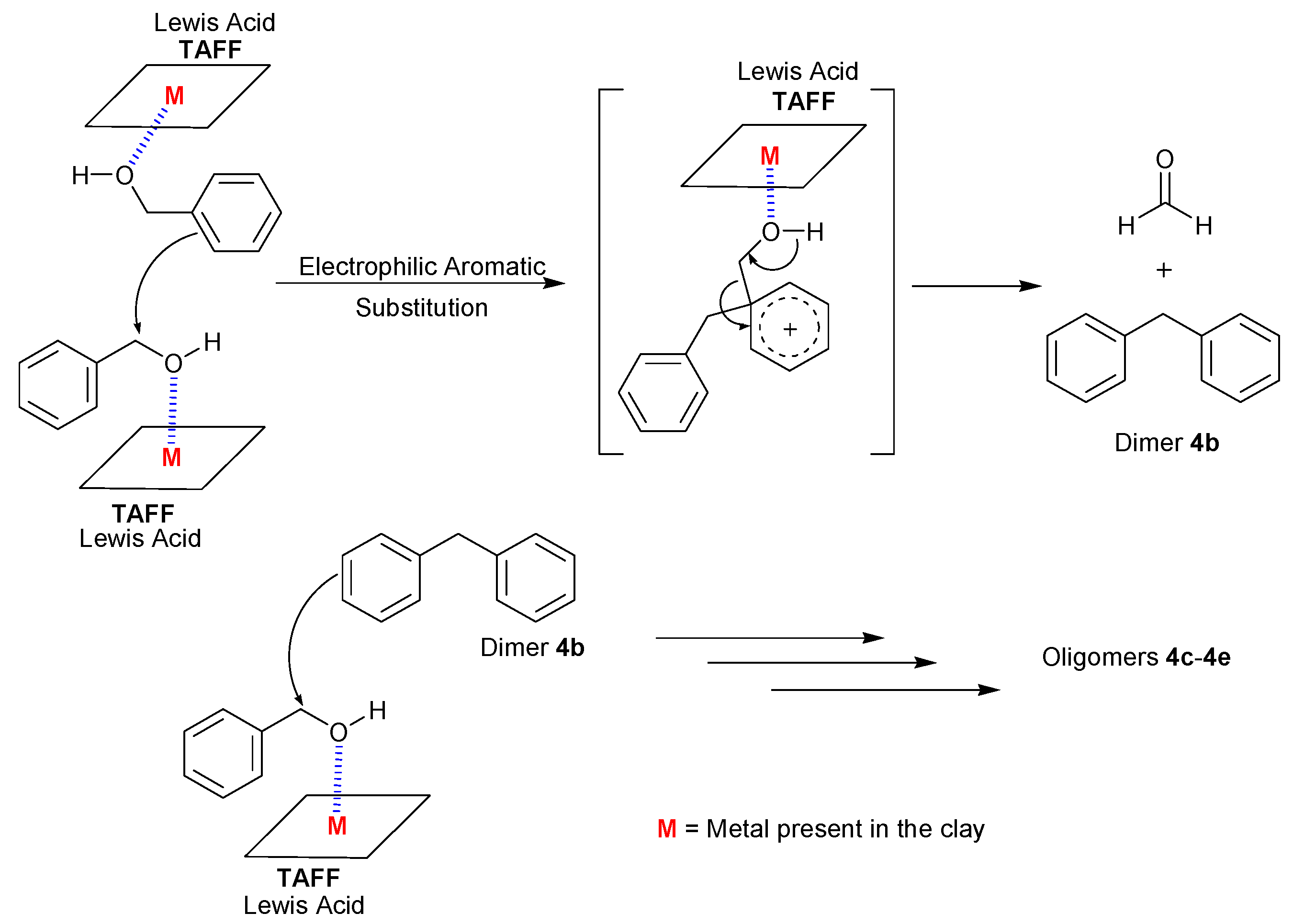
3. Experimental
3.1. Materials
3.2. Typical Procedure for the Catalytic Reaction under Microwave Conditions
3.3. Typical Procedure for the Catalytic Reaction under Infrared Conditions
3.4. Characterization Data
3.4.1. Synthesis of Cyclotripiperonylene (CTP, 1b)
3.4.2. Synthesis of Cyclotriveratrylene (2b) [4]
3.4.3. Synthesis of 1,2,3,6,7,8,11,12,13-Nonamethoxy-10,15-dihydro-5H-trbibenzo[a,d,g]cyclononene (3b) [60]
3.4.4. Synthesis of Oligotoluenes 4c–e
3.5. Theoretical Calculations


 is the external potential. Using a finite difference method the working equations for the calculation of chemical potential and chemical hardness can be given by:
is the external potential. Using a finite difference method the working equations for the calculation of chemical potential and chemical hardness can be given by:



 (for nucleophilic attack)
(for nucleophilic attack)
 (for electrophilic attack), and
(for electrophilic attack), and
 (for radical attack),
(for radical attack),
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Taylor, F.R.S.; Sá, J.; Hardacre, C. Friedel–Crafts alkylation of aromatics with benzyl alcohol over gold-modified silica. ChemCatChem 2011, 3, 119–121. [Google Scholar] [CrossRef]
- Salmón, M.; Miranda, R.; Nicolás-Vázquez, I.; Vargas-Rodriguez, M.Y.; Cruz-Borbolla, J.; Medrano, M.I.; Morales-Serna, J.A. Effects of bentonite on p-methoxybenzyl acetate: A theoretical model for oligomerization via an electrophilic-substitution mechanism. Molecules 2011, 16, 1761–1775. [Google Scholar] [CrossRef]
- Cruz-Almanza, R.; Shiba-Matzumoto, I.; Fuentes, A.; Martínez, M.; Cabrera, A.; Cárdenas, J.; Salmon, M. Oligomerization of benzylic alcohols and its mechanism. J. Mol. Catal. A 1997, 126, 161–168. [Google Scholar]
- Peterca, M.; Percec, V.; Imam, M.R.; Leowanawat, P.; Morimitsu, K.; Heiney, P.A. Molecular structure of helical supramolecular dendrimers. J. Am. Chem. Soc. 2008, 130, 14840–14852. [Google Scholar]
- Miranda, R.; Rios, H.; Delgado, F.; Castro, M.; Cogordan, A.; Salmón, M. Characterization of a bentonitic clay and its application as catalyst in the preparation of benzyltoluenes and oligotoluenes. Appl. Catal. A 2003, 244, 217–233. [Google Scholar] [CrossRef]
- Miranda, R.; Delgado, F.; Valasco, L.; Pérez, J.; Salmón, M. Mass spectrometric detection and identification of ortho, para-benzyltoluenes and oligotoluenes. Rapid Commun. Mass. Spectrom. 2000, 14, 188–193. [Google Scholar] [CrossRef]
- Hardie, M. Recent advances in the chemistry of cyclotriveratrylene. J. Chem. Soc. Rev. 2010, 39, 516–527. [Google Scholar] [CrossRef]
- Brotin, T.; Dutasta, J.P. Cryptophanes and their complexes—present and future. Chem. Rev. 2009, 109, 88–130. [Google Scholar] [CrossRef]
- Coquiére, D.; Le Gac, S.; Darbost, U.; Sénéque, O.; Jabin, I.; Reinaud, O. Biomimetic and self-assembled calix[6]arene-based receptors for neutral molecules. Org. Biomol. Chem. 2009, 7, 2485–2500. [Google Scholar] [CrossRef]
- Foster, J.A.; Steed, J.W. Exploiting cavities in supramolecular gels. Angew. Chem. Int. Ed. 2010, 49, 6718–6724. [Google Scholar] [CrossRef]
- Ménand, M.; Jabin, I. Acid–base controllable recognition properties of a highly versatile calix[6]crypturea. Chem. Eur. J. 2010, 16, 2159–2169. [Google Scholar] [CrossRef]
- Taratula, O.; Hill, P.A.; Khan, N.S.; Carroll, P.J.; Dmochowski, I.J. Crystallographic observation of 'induced fit' in a cryptophane host-guest model system. Nat. Commun. 2010, 1, 1–7. [Google Scholar]
- Sanseverino, J.; Chambron, J.C.; Aubert, E.; Espinosa, E. Sulfur-incorporating cyclotriveratrylene analogues: The synthesis of cyclotrithioguaiacylene. J. Org. Chem. 2011, 76, 1914–1917. [Google Scholar] [CrossRef]
- Osner, Z.R.; Nyamjav, D.; Holz, R.C.; Becker, D.P. Direct patterning of a cyclotriveratrylene derivative for directed self-assembly of C60. Nanotechnology 2011, 22, 275–611. [Google Scholar]
- Cornut, D.; Marrot, J.; Jabin. , I. Acid–base modulation of a versatile heteroditopic calix[6]arene based receptor. Org. Biomol. Chem. 2011, 9, 6373–6384. [Google Scholar] [CrossRef]
- Lindsey, A.S. The structure of cyclotriveratrylene (10,15-dihydro-2,3,7,8,12,13-hexamethoxy-5H-tribenzo[a,d,g]cyclononene) and related compounds. J. Chem. Soc. 1965, 1685–1692. [Google Scholar] [CrossRef]
- Manville, J.F.; Troughton, G.E. Synthesis, structure, and conformation of 10,15-dihydro-1,6,11-trihydroxy-2,7,12-trimethoxy-4,9,14-trimethyl-5H-tribenzo[a,d,g] cyclononene and its tripropyl analog. J. Org. Chem. 1973, 38, 4278–4281. [Google Scholar] [CrossRef]
- Sato, T.; Uno, K. Medium-sized cyclophanes. Part XV. 10,15-Dihydro-5H-tribenzo-[a,d,g]cyclononene and analogues. J. Chem. Soc. Perkin Trans. 1973, 1, 895–900. [Google Scholar] [CrossRef]
- Canceill, J.; Gabard, J.; Collet, A. (C3)-Tris-(O-allyl)-cyclotriguaiacylene, a key intermediate in cyclotriveratrylene chemistry. Short and efficient synthesis of cyclotriguaiacylene. J. Chem. Soc. Chem. Commun. 1983. [Google Scholar] [CrossRef]
- Scott, J.L.; MacFarlane, D.R.; Raston, C.L.; Teoh, C.M. Clean efficient syntheses of cyclotriveratrylene (CTV) and tris-(O-allyl)CTV in an ionic liquid. Green Chem. 2000, 2, 123–126. [Google Scholar] [CrossRef]
- Vargas-Rodríguez, Y.M.; Vargas, M.; Miranda, R.; Francisco, B.; Noguez, O.; Morales-Serna, J.A.; Salmon, M. Synthesis of benzyl chlorides and cycloveratrylene macrocycles using benzylic alcohols under homogeneous catalysis by HCl/dioxane. Org. Commun. 2012, 5, 58–63. [Google Scholar]
- Berger, N.; Jay, P. A new impregnant for HV power capacitors. IEEE Trans. Electr. Insul. 1986, 21, 59–63. [Google Scholar] [CrossRef]
- Kunio, S.; Takao, T.; Toshikatsu, S. Alkyldiphenylmethane Solvents for Pesticides. Jpn. Kokai Tokkyo Koho JP 09025202. 28 January 1997. [Google Scholar]
- Pisanenko, D.A.; Avilov, B.O.; Likhnitskii, K.V. Corrosion-protecting properties of products of pyridine quaternization with chloromethylated benzyltoluenes. Russ. J. Appl. Chem. 2010, 83, 1663–1665. [Google Scholar] [CrossRef]
- Varma, R.S. Clay and clay-supported reagents in organic synthesis. Tetrahedron 2002, 58, 1235–1255. [Google Scholar] [CrossRef]
- Dasgupta, S.; Török, B. Application of clay catalysts in organic synthesis. A review. Org. Prep. Proced. Int. 2008, 40, 1–65. [Google Scholar] [CrossRef]
- Sartori, G.; Maggi, R. Update 1 of: Protection (and deprotection) of functional groups in organic synthesis by heterogeneous catalysis. Chem. Rev. 2010, 110. [Google Scholar] [CrossRef]
- Nagendrappa, G. Organic synthesis using clay and clay-supported catalysts. Appl. Clay Sci. 2011, 53, 106–138. [Google Scholar] [CrossRef]
- Fernandes, C.I.; Nunes, C.D.; Vaz, P.D. Clays in organic synthesis–preparation and catalytic applications. Curr. Org. Synth. 2012, 9, 670–694. [Google Scholar] [CrossRef]
- Bishwa Bidita Varadwajab, G.; Parida, K.M. Montmorillonite supported metal nanoparticles: An update on syntheses and applications. RSC Adv. 2013, 3, 13583–13593. [Google Scholar] [CrossRef]
- Miranda, R.; Arroyo, G.A.; Penieres, G.; Delgado, F.; Cabrera, A.; Alvarez, C.; Salmon, M. Preparative heterocyclic chemistry using Tonsil a bentonitic clay; 1981 to 2003. Trends Heterocycl. Chem. 2003, 9, 195–235. [Google Scholar]
- Osnaya, R.; Arroyo, G.A.; Parada, L.; Delgado, F.; Trujillo, J.; Salmón, M.; Miranda, R. Biginelli vs. Hantzsch esters study under infrared radiation and solventless conditions (MX-870EP). ARKIVOC 2003, xi, 112–117. [Google Scholar]
- Salmón, M.; Angeles, E.; Miranda, R. Bromine/bentonite earth system, promoter of phenylmethanes from toluene. Chem. Commun. 1990, 17, 1188–1190. [Google Scholar]
- Salmón, M.; Zavala, N.; Cabrera, A.; Cardenas, J.; Gaviño, R.; Miranda, R.; Martinez, M. Aromatic substitution reactions of benzyl derivatives with a bentonite clay. J. Mol. Catal. A: Chem. 1995, 104, L127–L129. [Google Scholar] [CrossRef]
- Guerrero, R.R.; Cárdenas, J.; Bautista, L.; Vargas, M.; Labastida, E.V.; Salmón, M. Catalytic synthesis of 1,3,5-triphenylbenzenes, β-methylchalcones and 2,4,6-triphenyl pyrylium salts, promoted by a super acid triflouromethane sulfonic clay from acetophenones. J. Mex. Chem. Soc. 2006, 50, 114–118. [Google Scholar]
- Cabrera, A.; Peon, J.; Velasco, L.; Miranda, R.; Salmón, A.; Salmon, M. Clay-mediated cyclooligomerization of olefin oxides: A one-pot route to crown ethers. J. Mol. Catal. A: Chem. 1995, 104, L5–L7. [Google Scholar] [CrossRef]
- Salmon, M.; Zavala, N.; Martinez, M.; Miranda, R.; Cruz, R.; Cardenas, J.; Gaviño, R.; Cabrera, A. Cyclic and linear oligomerization reaction of 3,4,5-trimethoxybenzyl alcohol with a bentonite-clay. Tetrahedron Lett. 1994, 35, 5797–5800. [Google Scholar] [CrossRef]
- Miranda, R.; Osnaya, R.; Garduno, R.; Delgado, F.; Alvarez, C.; Salmón, M. A general alternative to obtain S.S-acetals using Taff, a bentonitic clay. Synth. Commun. 2001, 31, 1587–1597. [Google Scholar] [CrossRef]
- Morales-Serna, J.A.; López-Duran, L.E.; Castro, M.; Sansores, L.E.; Zolotukhin, M.; Salmón, M. Oligomerization of 3,5-dimethyl benzyl alcohol promoted by clay: Experimental and theoretical study. Molecules 2010, 15, 8156–8168. [Google Scholar] [CrossRef]
- Granados-Oliveros, G.; Gómez-Vidales, V.; Nieto-Camacho, A.; Morales-Serna, J.A.; Cárdenas, J.; Salmón, M. Photoproduction of H2O2 and hydroxyl radicals catalysed by natural and super acid-modified montmorillonite and its oxidative role in the peroxidation of lipids. RCS Adv. 2013, 3, 937–944. [Google Scholar]
- Pineiro, M.; Pinho e Melo, T.M.V.D. Microwave-assisted 1,3-Dipolar cycloaddition: An eco-friendly approach to five-membered heterocycles. Eur. J. Org. Chem. 2009, 31, 5287–5307. [Google Scholar]
- Caddick, S.; Fitzmaurice, R. Microwave enhanced synthesis. Tetrahedron 2009, 65, 3325–3355. [Google Scholar] [CrossRef]
- Kappe, C.O. Microwave dielectric heating in synthetic organic chemistry. Chem. Soc. Rev. 2008, 37, 1127–1139. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Varma, R.S. Microwave-assisted organic synthesis and transformations using benign reaction media. Acc. Chem. Res. 2008, 41, 629–639. [Google Scholar] [CrossRef]
- Zhang, C.; Liao, L.; Gong, S. Recent developments in microwave-assisted polymerization with a focus on ring-opening polymerization. Green Chem. 2007, 9, 303–314. [Google Scholar] [CrossRef]
- Flores-Conde, M.I.; Reyes, L.; Herrera, R.; Rios, H.; Vazquez, M.A.; Miranda, R.; Tamariz, J.; Delgado, F. Highly regio- and stereoselective Diels-Alder cycloadditions via two-step and multicomponent reactions promoted by infrared irradiation under solvent-free conditions. Int. J. Mol. Sci. 2012, 13, 2590–2617. [Google Scholar] [CrossRef]
- Córdova, M.O.N.; Ramírez, C.I.F.; Bejarano, B.V.; Razo, G.A.A.; Flores, F.J.P.; Tellez, V.C.; Ruvalcaba, R.M. Comparative study using different infrared zones of the solventless activation of organic reactions. Int. J. Mol. Sci. 2011, 12, 8575–8580. [Google Scholar] [CrossRef]
- Noguez, M.O.; Marcelino, V.; Rodríguez, H.; Martín, O.; Martínez, J.O.; Arroyo, G.A.; Pérez, F.J.; Suárez, M.; Miranda, R. Infrared assisted production of 3,4-Dihydro-2(1H)-pyridones in solvent-free conditions. Int. J. Mol. Sci. 2011, 12, 2641–2649. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, J.; Li, J.H. Infrared heat aided solid state synthesis of pyrroles from 1,4-diketones and ammonium acetate. J. Heterocycl. Chem. 2012, 49, 204–207. [Google Scholar] [CrossRef]
- Qin, W.P.; Zhang, D.S.; Zhao, D.; Wang, L.L.; Zheng, K.Z. Near-infrared photocatalysis based on YF3: Yb3+,Tm3+/TiO2 core/shell nanoparticles. Chem. Commun. 2010, 46, 2304–2306. [Google Scholar]
- Li, C.; Wang, F.; Zhu, J.; Yu, J.C. NaYF4:Yb,Tm/CdS composite as a novel near-infrared-driven photocatalyst. Appl. Catal. B: Environ. 2010, 100, 433–439. [Google Scholar] [CrossRef]
- Ji, H.; Li, L.; Xu, X.; Ham, S.; Hammad, L.A.; Birney, D.M. Multiphoton infrared initiated thermal reactions of esters: Pseudopericyclic eight-centered cis-elimination. J. Am. Chem. Soc. 2009, 131, 528–537. [Google Scholar]
- Trætteberg, M.; Østensen, H.; Seip, R. The molecular structure and conformation of gaseous benzyl alcohol by electron diffraction. Acta Chem. Scand. 1980, 34a, 449–454. [Google Scholar] [CrossRef]
- Gross, K.C.; Seybold, P.G.; Peralta-Inga, Z.; Murray, J.S.; Politzer, P. Comparison of quantum chemical parameters and Hammett constants in correlating pKa values of substituted anilines. J. Org. Chem. 2001, 66, 6919–6925. [Google Scholar] [CrossRef]
- Thompson, D.J.; Fanning, M.O.; Hodnett, B.K. Modelling the active sites in vanadyl pyrophosphate. J. Mol. Catal. A: Chem. 2003, 198, 125–137. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F.A. Natural population analysis@fa@f). J. Chem. Phys. 1985, 83, 735–747. [Google Scholar] [CrossRef]
- Weinhold, F.A. Natural bond orbital methods. In Encyclopedia of Computational Chemistry, 1st ed.; Schleyer, P.v.R., Allinger, N.L., Clark, T., Gasteiger, J., Kollman, P.A., Schaefer, H.F., III., Schreiner., P.R., Eds.; John Wiley & Sons: Chichester, UK, 1998; pp. 1792–1811. [Google Scholar]
- Pearson, R.G. Chemical Hardness: Applications from Molecules to Solids; Wiley-VCH: Weinheim, Germany, 1997; pp. 9–197. [Google Scholar]
- Miranda, R.; Escobar, J.; Delgado, F.; Salmón, M.; Cabrera, A. Catalytic promotion of piperonyl alcohol to trimethylendioxyorthocyclophane by bentonitic earth, or by hydrochloric acid. J. Mol. Catal. A: Chem. 1999, 150, 299–305. [Google Scholar] [CrossRef]
- Ding, Y.; Li, B.; Zhang, G. Improved preparation of racemic 1,2,3,6,7,8,11,12,13-nonamethoxy-10,15-dihydro-5H-tribenzo[a,d,g]cyclononene. ARKIVOC 2007, xiv, 322–326. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11 − 18. J. Chem. Phys. 1980, 72, 5639–5649. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Jamorski, C.; Casida M.E.; Salahub, D.R. Dynamic polarizabilities and excitation spectra from a molecular implementation of time-dependent density-functional response theory: N2 as a case study. J. Chem. Phys. 1996, 104, 5134–5148. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Schleyer, P.v.R. An ab initio study resulting in a greater understanding of the HSAB principle. J. Am. Chem. Soc. 1994, 116, 1067–1071. [Google Scholar] [CrossRef]
- Bulat, F.A.; Chamorro, E.; Fuentealba, P.; Toro-Labbé, A. Condensation of frontier molecular orbital Fukui functions. J. Phys. Chem. A 2004, 108, 342–349. [Google Scholar] [CrossRef]
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar]
- Sample Availability: Samples of the compounds 1b–f, 2b–c and 3b–d are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miranda, R.; Valencia-Vázquez, O.; Maya-Vega, C.A.; Nicolás-Vázquez, I.; Vargas-Rodriguez, Y.M.; Morales-Serna, J.A.; García-Ríos, E.; Salmón, M. Synthesis of Cycloveratrylene Macrocycles and Benzyl Oligomers Catalysed by Bentonite under Microwave/Infrared and Solvent-Free Conditions. Molecules 2013, 18, 12820-12844. https://doi.org/10.3390/molecules181012820
Miranda R, Valencia-Vázquez O, Maya-Vega CA, Nicolás-Vázquez I, Vargas-Rodriguez YM, Morales-Serna JA, García-Ríos E, Salmón M. Synthesis of Cycloveratrylene Macrocycles and Benzyl Oligomers Catalysed by Bentonite under Microwave/Infrared and Solvent-Free Conditions. Molecules. 2013; 18(10):12820-12844. https://doi.org/10.3390/molecules181012820
Chicago/Turabian StyleMiranda, René, Omar Valencia-Vázquez, Carlos Abel Maya-Vega, Inés Nicolás-Vázquez, Yolanda Marina Vargas-Rodriguez, José Antonio Morales-Serna, Eréndira García-Ríos, and Manuel Salmón. 2013. "Synthesis of Cycloveratrylene Macrocycles and Benzyl Oligomers Catalysed by Bentonite under Microwave/Infrared and Solvent-Free Conditions" Molecules 18, no. 10: 12820-12844. https://doi.org/10.3390/molecules181012820
APA StyleMiranda, R., Valencia-Vázquez, O., Maya-Vega, C. A., Nicolás-Vázquez, I., Vargas-Rodriguez, Y. M., Morales-Serna, J. A., García-Ríos, E., & Salmón, M. (2013). Synthesis of Cycloveratrylene Macrocycles and Benzyl Oligomers Catalysed by Bentonite under Microwave/Infrared and Solvent-Free Conditions. Molecules, 18(10), 12820-12844. https://doi.org/10.3390/molecules181012820