Ginsenoside Rb1 Attenuates Oxygen-Glucose Deprivation-Induced Apoptosis in SH-SY5Y Cells via Protection of Mitochondria and Inhibition of AIF and Cytochrome c Release

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
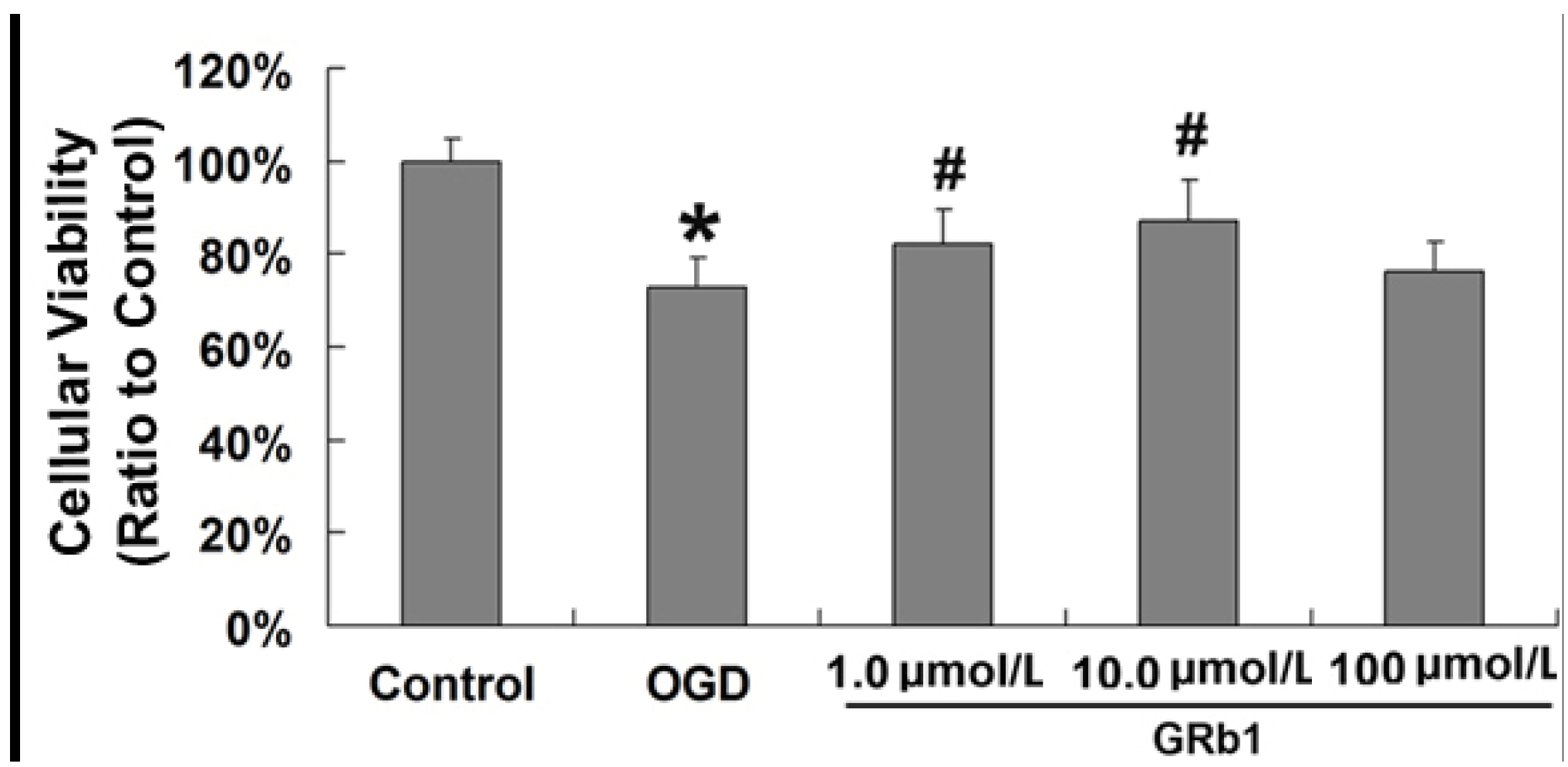
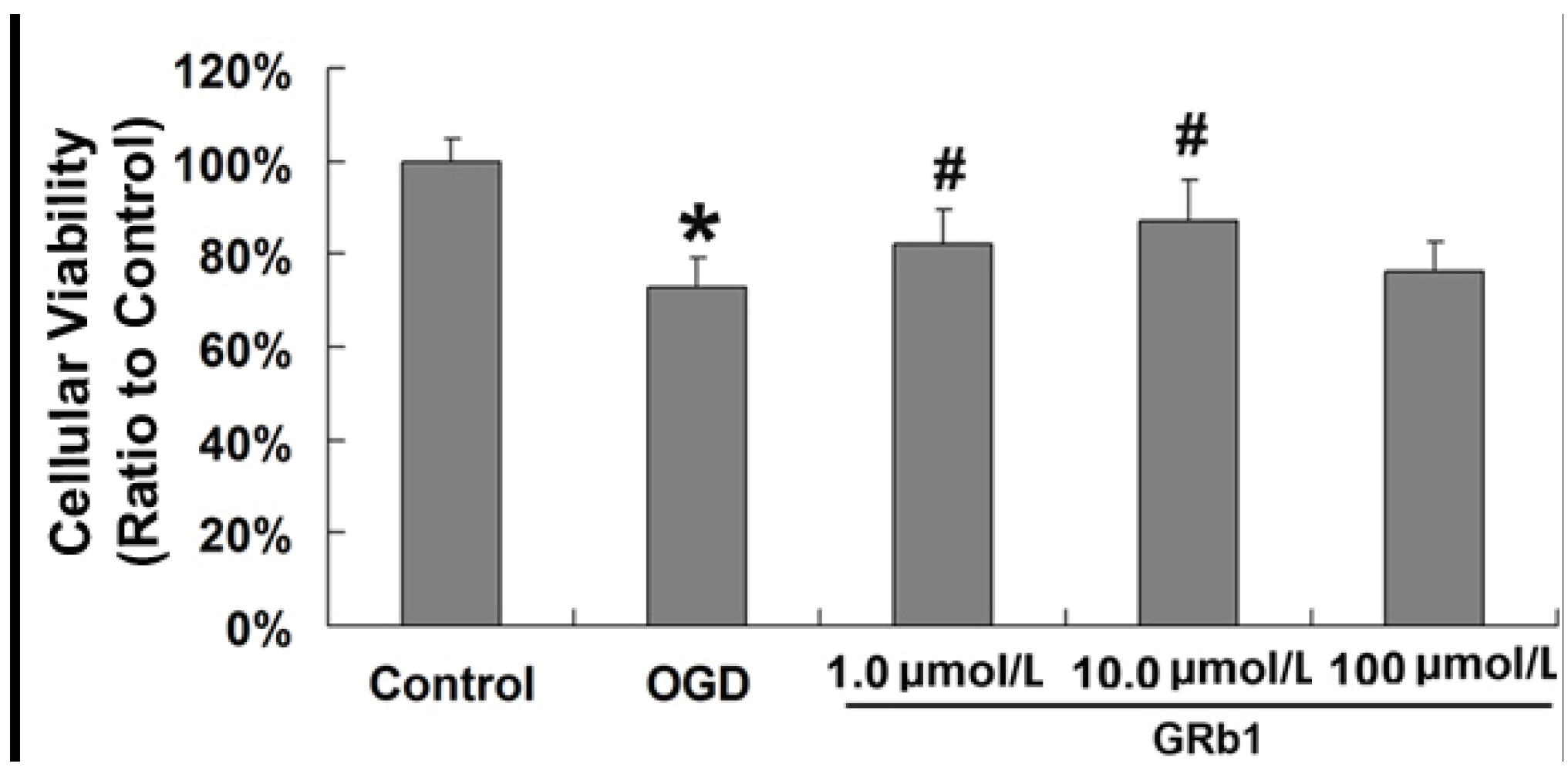
2.1. Ginsenoside Rb1 Decreased Cell Death Caused by OGD
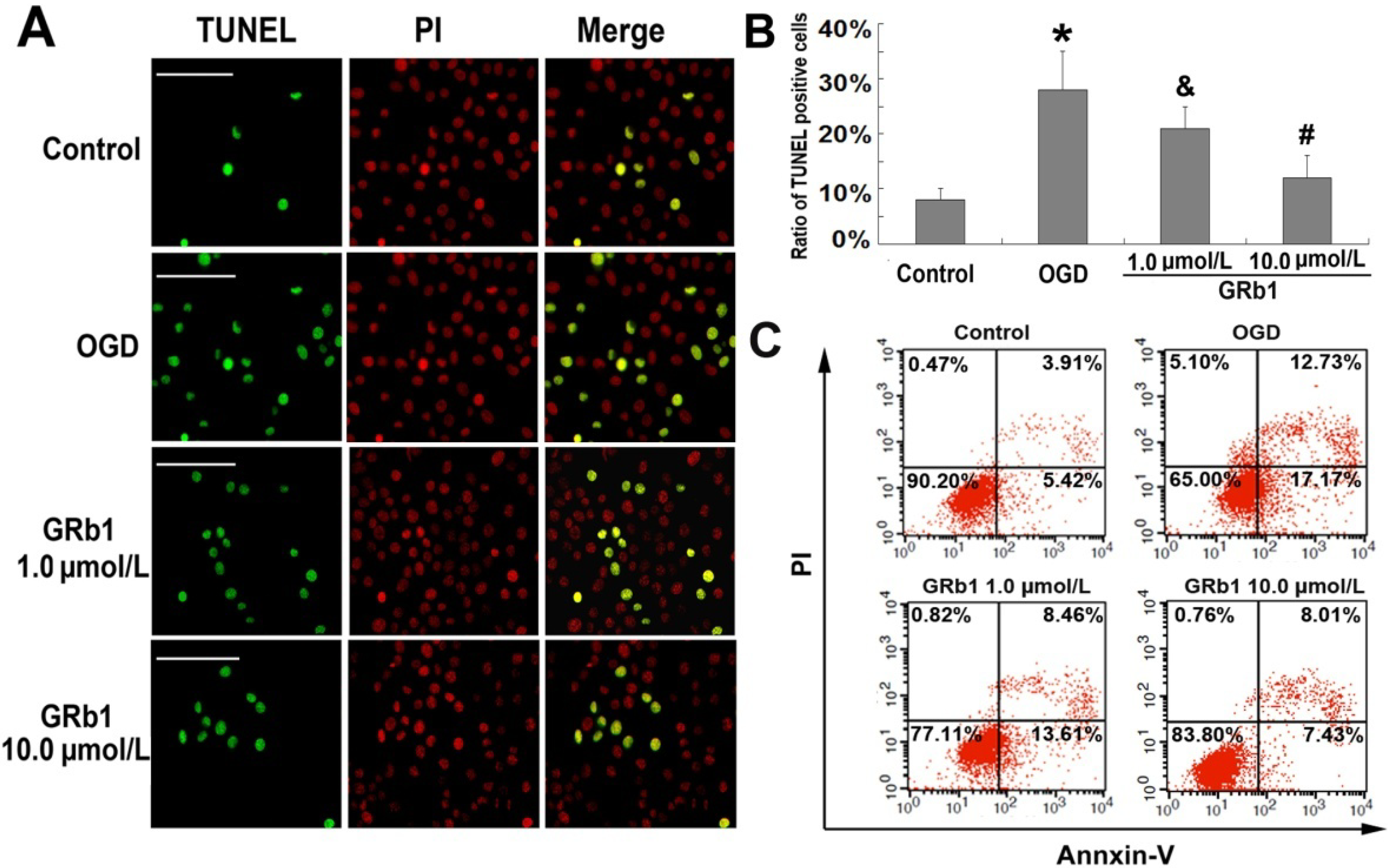
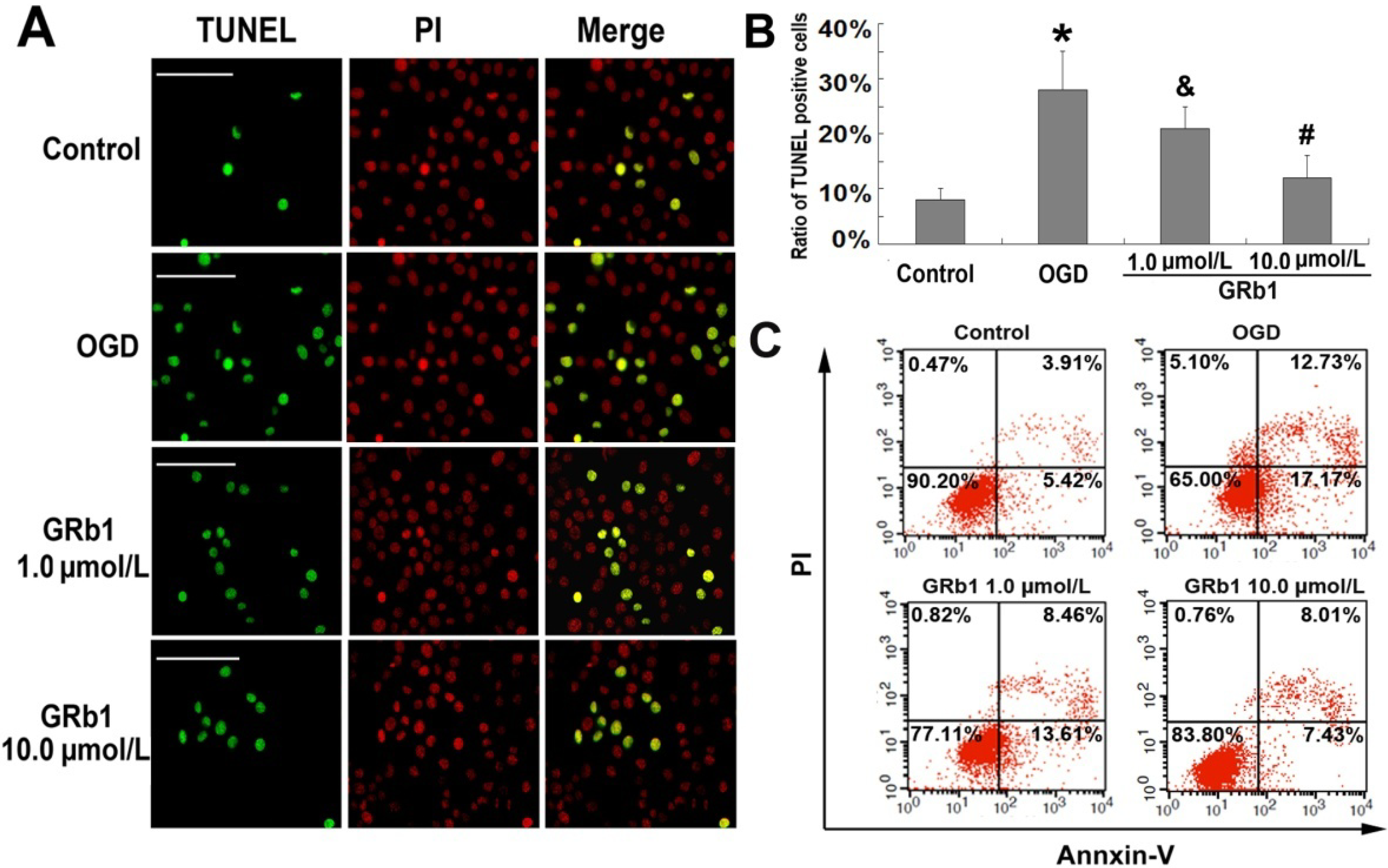
2.2. Ginsenoside Rb1 Inhibited Apoptosis Caused by OGD
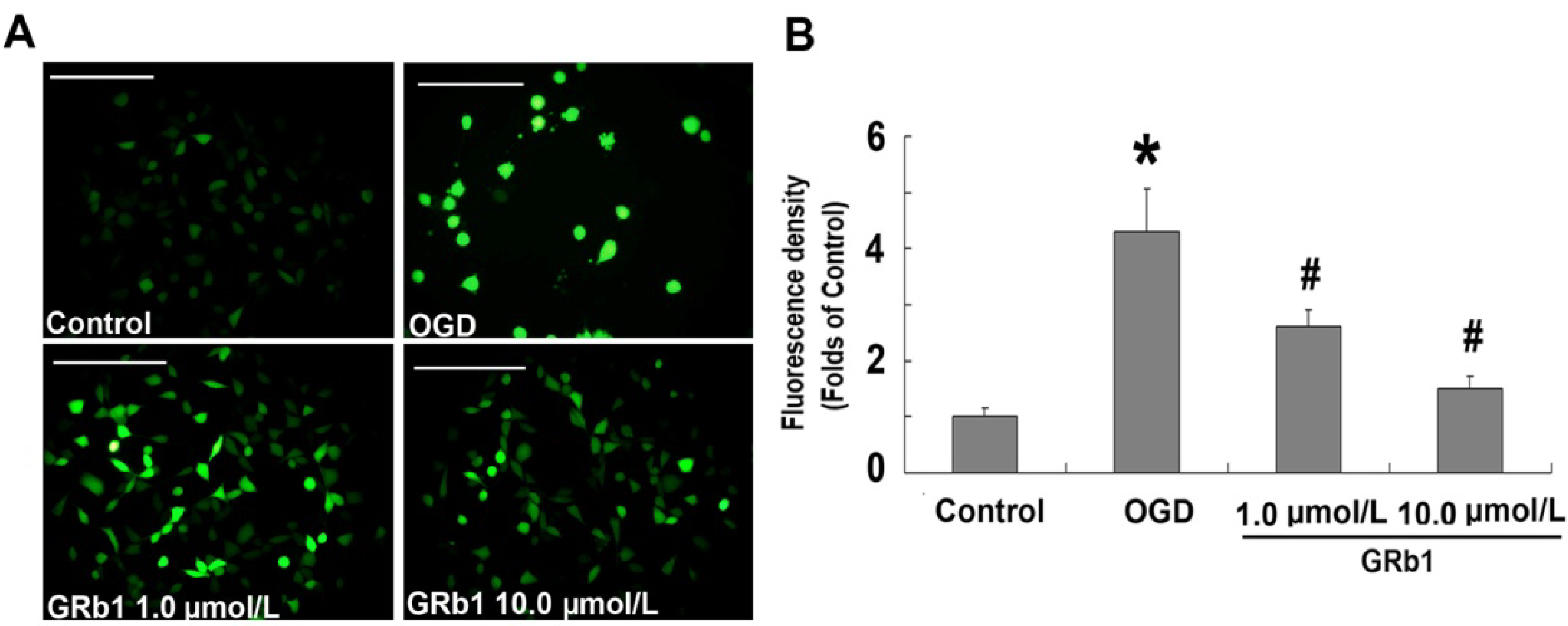
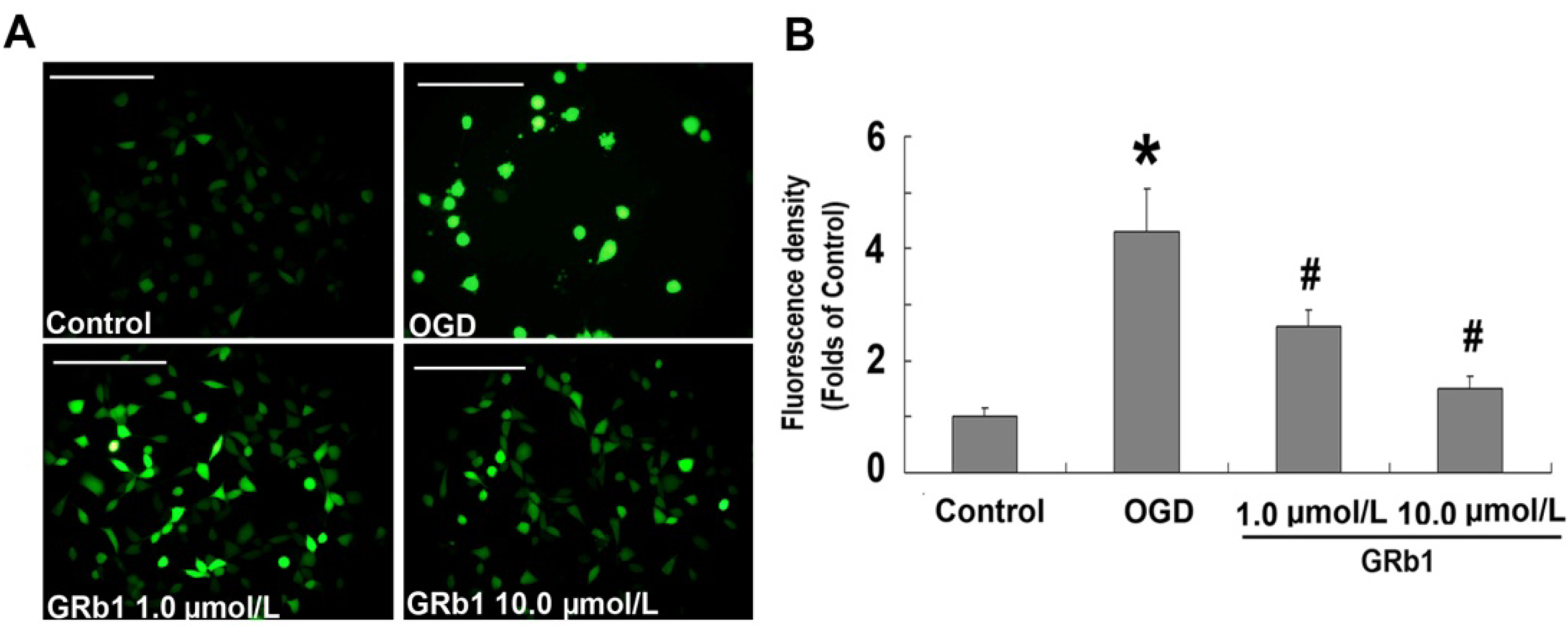
2.3. Ginsenoside Rb1 Attenuated ROS Level
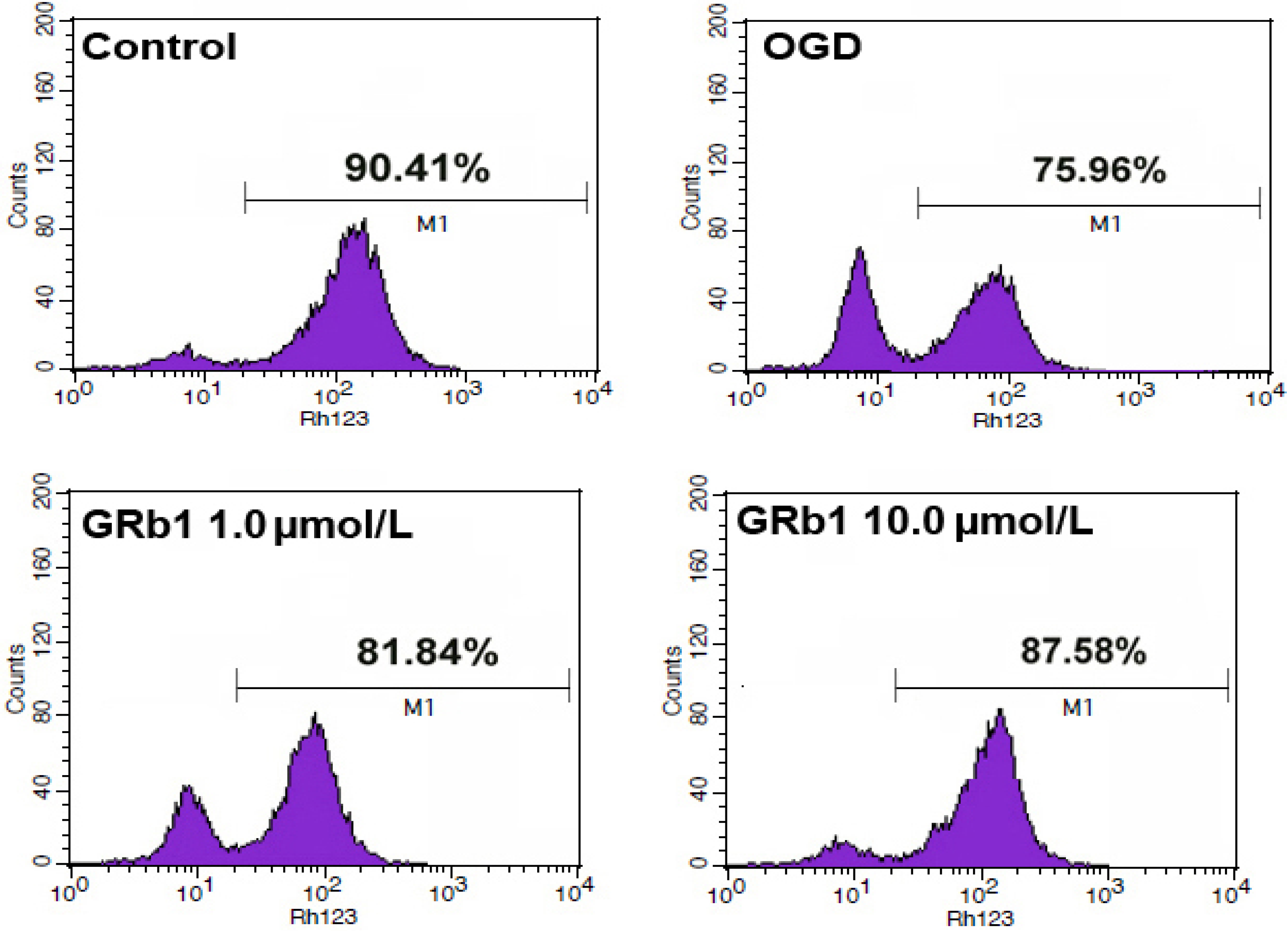
2.4. Ginsenoside Rb1 Sustained Mitochondrial Membrane Potential (ΔΨm)
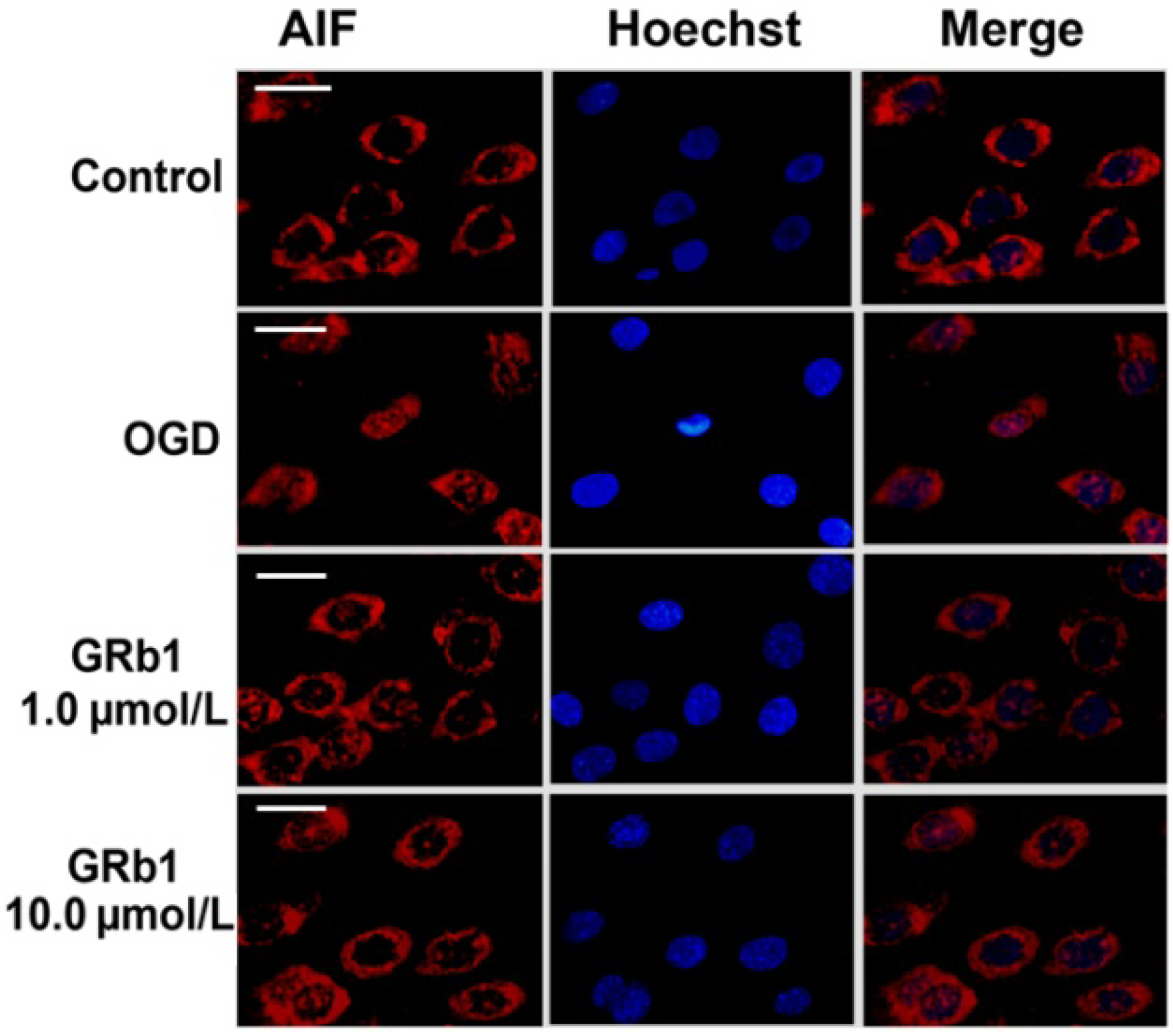
2.5. Ginsenoside Rb1 Inhibited Release of AIF and Cyto c from Mitochondria
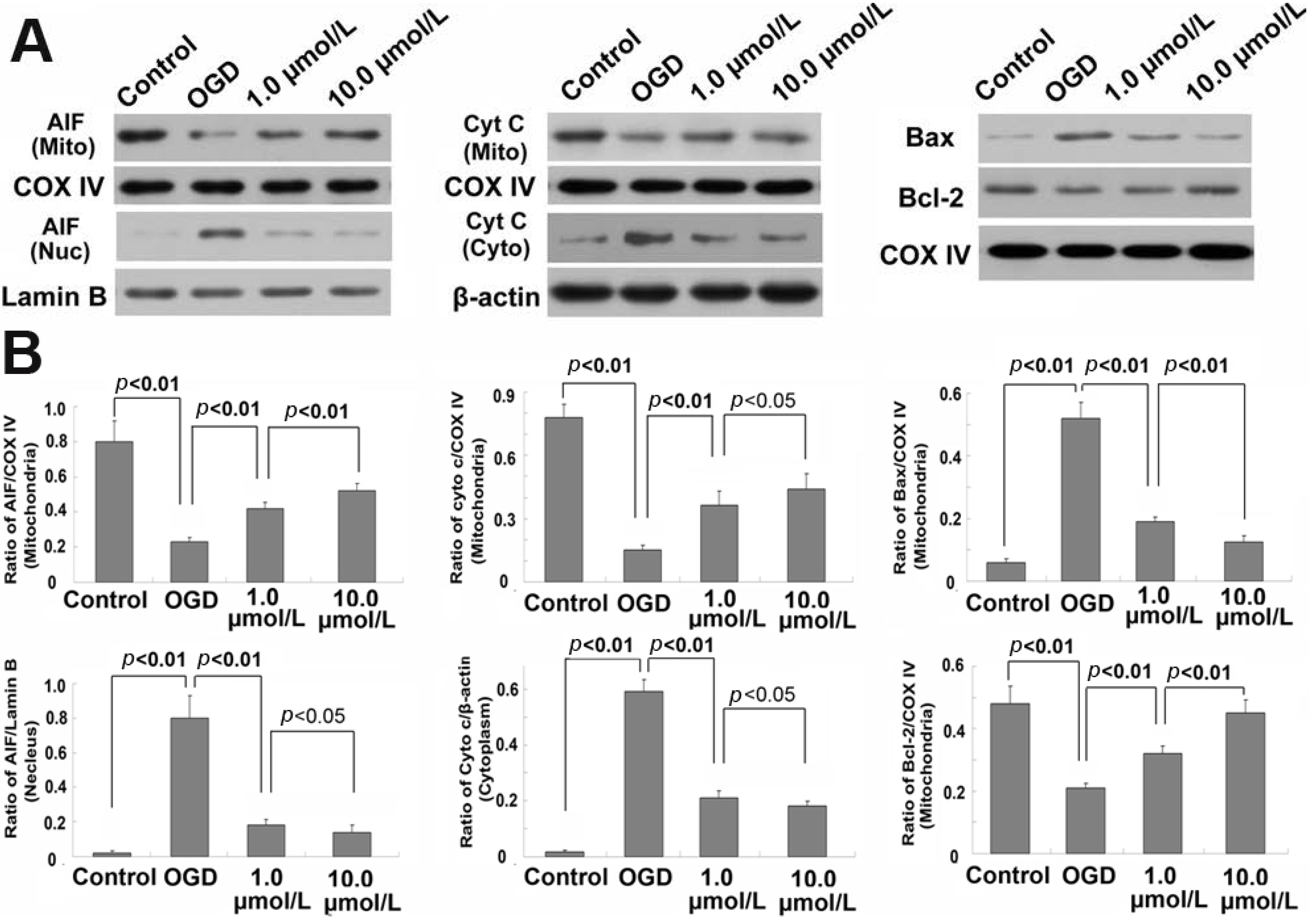
2.6. Ginsenoside Rb1 Improve Bcl-2 Level, but Attenuated Bax Level Within Mitochondria
3. Discussion
4. Experimental
4.1. Reagents
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Oxygen-Glucose Deprivation
4.5. Assessment of Apoptosis by TUNEL Staining
4.6. Detection of Apoptosis by Flow Cytometry
4.7. Measurement of Intracellular ROS Levels
4.8. Mitochondrial Membrane Potential
4.9. Immunofluorescence Staining
4.10. Differential Centrifugation and Cellular Fraction
4.11. Gel Electrophoresis and Western Blotting
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Kirino, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982, 239, 57–69. [Google Scholar] [CrossRef]
- Albers, G.W.; Caplan, L.R.; Coull, B.; Fayad, P.B.; Mohr, J.P.; Saver, J.L.; Sherman, D.G.; TIA Working Group. Transient ischemic attatck―Proposal for a new definition. N. Engl. J. Med. 2002, 347, 1713–1716. [Google Scholar] [CrossRef]
- Li, Y.; Sharov, V.G.; Jiang, N.; Zaloga, C.; Sabbah, H.N.; Chopp, M. Ultrastructural and light microscopic evidence of apoptosis after middle cerebral artery occlusion in the rat. Am. J. Pathol. 1995, 146, 1045–1051. [Google Scholar]
- Malagelada, C.; Xifró, X.; Miñano, A.; Sabriá, J.; Rodríguez-Alvarez, J. Contribution of caspase-mediated apoptosis to the cell death caused by oxygen-glucose deprivation in cortical cell cultures. Neurobiol. Dis. 2005, 20, 27–37. [Google Scholar] [CrossRef]
- Shah, P.T.; Yoon, K.W.; Xu, X.M.; Broder, L.D. Apoptosis mediates cell death following traumatic injury in rat hippocampal neurons. Neuroscience 1997, 79, 999–1004. [Google Scholar] [CrossRef]
- Friedlander, R.M. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef]
- Genovese, T.; Mazzon, E.; Paterniti, I.; Esposito, E.; Cuzzocrea, S. Neuroprotective effects of olprinone after cerebral ischemia/reperfusion injury in rats. Neurosci. Lett. 2011, 503, 93–99. [Google Scholar] [CrossRef]
- Yao, R.Q.; Qi, D.S.; Yu, H.L.; Liu, J.; Yang, L.H.; Wu, X.X. Quercetin attenuates cell apoptosis in focal cerebral ischemia rat brain via activation of BDNF-TrkB-PI3K/Akt signaling pathway. Neurochem. Res. 2012, 37, 2777–2786. [Google Scholar] [CrossRef]
- Ding, Z.M.; Wu, B.; Zhang, W.Q.; Lu, X.J.; Lin, Y.C.; Geng, Y.J.; Miao, Y.F. Neuroprotective effects of ischemic preconditioning and postconditioning on global brain ischemia in rats through the same effect on inhibition of apoptosis. Int. J. Mol. Sci. 2012, 13, 6089–6101. [Google Scholar] [CrossRef]
- Yuan, Q.L.; Yang, C.X.; Xu, P.; Gao, X.Q.; Deng, L.; Chen, P.; Sun, Z.L.; Chen, Q.Y. Neuroprotective effects of ginsenoside Rb1 on transient cerebral ischemia in rats. Brain Res. 2007, 1167, 1–12. [Google Scholar]
- Zhu, J.; Jiang, Y.; Wu, L.; Lu, T.; Xu, G.; Liu, X. Suppression of local inflammation contributes to the neuroprotective effect of ginsenoside Rb1 in rats with cerebral ischemia. Neuroscience 2012, 202, 342–351. [Google Scholar] [CrossRef]
- Liang, Y.Y.; Wang, B.; Qian, D.M.; Li, L.; Wang, Z.H.; Hu, M.; Song, X.X. Inhibitory effects of Ginsenoside Rb1 on apoptosis caused by HSV-1 in human glioma cells. Virol. Sin. 2012, 27, 19–25. [Google Scholar]
- Gao, X.Q.; Yang, C.X.; Chen, G.J.; Wang, G.Y.; Chen, B.; Tan, S.K.; Liu, J.; Yuan, Q.L. Ginsenoside Rb1 regulates the expressions of brain-derived neurotrophic factor and caspase-3 and induces neurogenesis in rats with experimental cerebral ischemia. J. Ethnopharmacol. 2010, 132, 393–399. [Google Scholar] [CrossRef]
- Wu, Y.; Xia, Z.Y.; Dou, J.; Zhang, L.; Xu, J.J.; Zhao, B.; Lei, S.; Liu, H.M. Protective effect of ginsenoside Rb1 against myocardial ischemia/reperfusion injury in streptozotocin-induced diabetic rats. Mol. Biol. Rep. 2011, 38, 4327–4335. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, T.; Lu, J.; Li, S.; Wan, L.; Long, D.; Li, Q.; Feng, L.; Li, Y. Rb1 postconditioning attenuates liver warm ischemia-reperfusion injury through ROS-NO-HIF pathway. Life Sci. 2011, 88, 598–605. [Google Scholar] [CrossRef]
- Lim, J.H.; Wen, T.C.; Matsuda, S.; Tanaka, J.; Maeda, N.; Peng, H.; Aburaya, J.; Ishihara, K.; Sakanaka, M. Protection of ischemic hippocampal neurons by ginsenoside Rb1, a main ingredient of ginseng root. Neurosci. Res. 1997, 28, 191–200. [Google Scholar] [CrossRef]
- Manzanero, S.; Santro, T.; Arumugam, T.V. Neuronal oxidative stress in acute ischemic stroke: Sources and contribution to cell injury. Neurochem. Int. 2013, 62, 712–718. [Google Scholar] [CrossRef]
- Choi, A.Y.; Choi, J.H.; Lee, J.Y.; Yoon, K.S.; Choe, W.; Ha, J.; Yeo, E.J.; Kang, I. Apigenin protects HT22 murine hippocampal neuronal cells against endoplasmic reticulum stress-induced apoptosis. Neurochem. Int. 2010, 57, 143–152. [Google Scholar] [CrossRef]
- Guo, R.B.; Wang, G.F.; Zhao, A.P.; Gu, J.; Sun, X.L.; Hu, G. Paeoniflorin protects against ischemia-induced brain damages in rats via inhibiting MAPKs/NF-κB-mediated inflammatory responses. PLoS One 2012, 7, e49701. [Google Scholar]
- Zhang, T.L.; Fu, J.L.; Geng, Z.; Yang, J.J.; Sun, X.J. The neuroprotective effect of losartan through inhibiting AT1/ASK1/MKK4/JNK3 pathway following cerebral I/R in rat hippocampal CA1 region. CNS Neurosci. Ther. 2012, 18, 981–987. [Google Scholar] [CrossRef]
- Jordan, J.; de Groot, P.W.; Galindo, M.F. Mitochondria: The headquarters in ischemia-induced neuronal death. Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 98–106. [Google Scholar] [CrossRef]
- Lee, H.J.; Lyu da, H.; Koo, U.; Lee, S.J.; Hong, S.S.; Kim, K.; Kim, K.H.; Lee, D.; Mar, W. Inhibitory effect of 2-arylbenzofurans from the Mori Cortex Radicis (Moraceae) on oxygen glucose deprivation (OGD)-induced cell death of SH-SY5Y cells. Arch. Pharm. Res. 2011, 34, 1373–1380. [Google Scholar] [CrossRef]
- Zhao, L.P.; Ji, C.; Lu, P.H.; Li, C.; Xu, B.; Gao, H. Oxygen glucose deprivation (OGD)/re-oxygenation-induced in vitro neuronal cell death involves mitochondrial cyclophilin-D/P53 signaling axis. Neurochem. Res. 2013, 38, 705–713. [Google Scholar] [CrossRef]
- Du, C.P.; Tan, R.; Hou, X.Y. Fyn kinases play a critical role in neuronal apoptosis induced by oxygen and glucose deprivation or amyloid-β peptide treatment. CNS Neurosci. Ther. 2012, 18, 754–761. [Google Scholar]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Rau, T.F.; Lu, Q.; Sharma, S.; Sun, X.; Leary, G.; Beckman, M.L.; Hou, Y.; Wainwright, M.S.; Kavanaugh, M.; Poulsen, D.J.; et al. Oxygen glucose deprivation in rat hippocampal slice cultures results in alterations in carnitine homeostasis and mitochondrial dysfunction. PLoS One 2012, 7, e40881. [Google Scholar] [CrossRef]
- James, D.; Parone, P.A.; Terradillos, O.; Lucken-Ardjomande, S.; Montessuit, S.; Martinou, J.C. Mechanisms of mitochondrial outer membrane permeabilization. Novartis Found. Symp. 2007, 287, 170–176. [Google Scholar] [CrossRef]
- Marutani, E.; Kosugi, S.; Tokuda, K.; Khatri, A.; Nguyen, R.; Atochin, D.N.; Kida, K.; van Leyen, K.; Arai, K.; Ichinose, F. A novel hydrogen sulfide-releasing N-methyl-D-aspartate receptor antagonist prevents ischemic neuronal death. J. Biol. Chem. 2012, 287, 32124–32135. [Google Scholar] [CrossRef]
- Li, Z.; Cui, G.; Wang, J.; Yu, Z.; Zhao, L.; Lv, Z. Nemo-like kinase (NLK) involves in neuronal apoptosis after traumatic brain injury. Cell. Mol. Neurobiol. 2012, 32, 381–389. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Suzuki, H.; Sozen, T.; Altay, O.; Zhang, J.H. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir. Suppl. 2011, 110, 43–48. [Google Scholar]
- Nakka, V.P.; Gusain, A.; Mehta, S.L.; Raghubir, R. Molecular mechanisms of apoptosis in cerebral ischemia: Multiple neuroprotective opportunities. Mol. Neurobiol. 2008, 37, 7–38. [Google Scholar] [CrossRef]
- Chen, F.; Chen, Y.; Kang, X.; Zhou, Z.; Zhang, Z.; Liu, D. Anti-apoptotic function and mechanism of ginseng saponins in Rattus pancreatic β-cells. Biol. Pharm. Bull. 2012, 35, 1568–1573. [Google Scholar]
- Cai, B.X.; Jin, S.L.; Luo, D.; Lin, X.F.; Gao, J. Ginsenoside Rb1 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Biol. Pharm. Bull. 2009, 32, 837–841. [Google Scholar] [CrossRef]
- Liang, J.M.; Xu, H.Y.; Zhang, X.J.; Li, X.; Zhang, H.B.; Ge, P.F. Role of mitochondrial function in the protective effects of ischaemic postconditioning on ischaemia/reperfusion cerebral damage. J. Int. Med. Res. 2013, 41, 618–627. [Google Scholar] [CrossRef]
- Jiang, J.; Jiang, J.; Zuo, Y.; Gu, Z. Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson’s disease. Int. J. Mol. Med. 2013, 31, 825–832. [Google Scholar]
- Agudo-López, A.; Miguel, B.G.; Fernández, I.; Martínez, A.M. Involvement of mitochondria on neuroprotective effect of sphingosine-1-phosphate in cell death in an in vitro model of brain ischemia. Neurosci. Lett. 2010, 470, 130–133. [Google Scholar] [CrossRef]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [CrossRef]
- Lü, J.M.; Weakley, S.M.; Yang, Z.; Hu, M.; Yao, Q.; Chen, C. Ginsenoside Rb1 directly scavenges hydroxyl radical and hypochlorous acid. Curr. Pharm. Des. 2012, 18, 6339–6347. [Google Scholar] [CrossRef]
- Li, J.; Shao, Z.H.; Xie, J.T.; Wang, C.Z.; Ramachandran, S.; Yin, J.J.; Aung, H.; Li, C.Q.; Qin, G.; Hoek, T.V.; et al. The effects of ginsenoside Rb1 on JNK in oxidative injury in cardiomyocytes. Arch. Pharm. Res. 2012, 35, 1259–1267. [Google Scholar] [CrossRef]
- Kong, H.L.; Li, Z.Q.; Zhao, Y.J.; Zhao, S.M.; Zhu, L.; Li, T.; Fu, Y.; Li, H.J. Ginsenoside Rb1 protects cardiomyocytes against CoCl2-induced apoptosis in neonatal rats by inhibiting mitochondria permeability transition pore opening. Acta Pharmacol. Sin. 2010, 31, 687–695. [Google Scholar] [CrossRef]
- Kim, G.S.; Jung, J.E.; Narasimhan, P.; Sakata, H.; Yoshioka, H.; Song, Y.S.; Okami, N.; Chan, P.H. Release of mitochondrial apoptogenic factors and cell death are mediated by CK2 and NADPH oxidase. J. Cereb. Blood Flow Metab. 2012, 32, 720–730. [Google Scholar] [CrossRef]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. Neurosci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef]
- Hu, G.; Wu, Z.; Yang, F.; Zhao, H.; Liu, X.; Deng, Y.; Shi, M.; Zhao, G. Ginsenoside Rd blocks AIF mitochondrio-nuclear translocation and NF-κB nuclear accumulation by inhibiting poly(ADP-ribose) polymerase-1 after focal cerebral ischemia in rats. Neurol. Sci. 2013. [Google Scholar] [CrossRef]
- Yang, X.; Wang, S.; Lin, Y.; Han, Y.; Qiu, X.; Zhao, X.; Cao, L.; Wang, X.; Chi, Z. Poly (ADP-ribose)polymerase inhibition protects epileptic hippocampal neurons from apoptosis via suppressing Akt-mediated apoptosis-inducing factor translocation in vitro. Neuroscience 2013, 231, 353–362. [Google Scholar] [CrossRef]
- Zemlyak, I.; Brooke, S.M.; Singh, M.H.; Sapolsky, R.M. Effects of overexpression of antioxidants on the release of cytochrome c and apoptosis-inducing factor in the model of ischemia. Neurosci. Lett. 2009, 453, 182–185. [Google Scholar] [CrossRef]
- Hashimoto, R.; Yu, J.; Koizumi, H.; Ouchi, Y.; Okabe, T. Ginsenoside Rb1 prevents MPP(+)-induced apoptosis in PC12 cells by stimulating estrogen receptors with consequent activation of ERK1/2, Akt and inhibition of SAPK/JNK, p38 MAPK. Evid. Based Complement. Alternat. Med. 2012, 2012, e693717. [Google Scholar]
- Er, E.; Oliver, L.; Cartron, P.F.; Juin, P.; Manon, S.; Vallette, F.M. Mitochondria as the target of the pro-apoptotic protein Bax. Biochim. Biophys. Acta 2006, 1757, 1301–1311. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liang, J.; Yu, Y.; Wang, B.; Lu, B.; Zhang, J.; Zhang, H.; Ge, P. Ginsenoside Rb1 Attenuates Oxygen-Glucose Deprivation-Induced Apoptosis in SH-SY5Y Cells via Protection of Mitochondria and Inhibition of AIF and Cytochrome c Release. Molecules 2013, 18, 12777-12792. https://doi.org/10.3390/molecules181012777
Liang J, Yu Y, Wang B, Lu B, Zhang J, Zhang H, Ge P. Ginsenoside Rb1 Attenuates Oxygen-Glucose Deprivation-Induced Apoptosis in SH-SY5Y Cells via Protection of Mitochondria and Inhibition of AIF and Cytochrome c Release. Molecules. 2013; 18(10):12777-12792. https://doi.org/10.3390/molecules181012777
Chicago/Turabian StyleLiang, Jianmin, Ying Yu, Boyu Wang, Bin Lu, Jizhou Zhang, Hongbo Zhang, and Pengfei Ge. 2013. "Ginsenoside Rb1 Attenuates Oxygen-Glucose Deprivation-Induced Apoptosis in SH-SY5Y Cells via Protection of Mitochondria and Inhibition of AIF and Cytochrome c Release" Molecules 18, no. 10: 12777-12792. https://doi.org/10.3390/molecules181012777