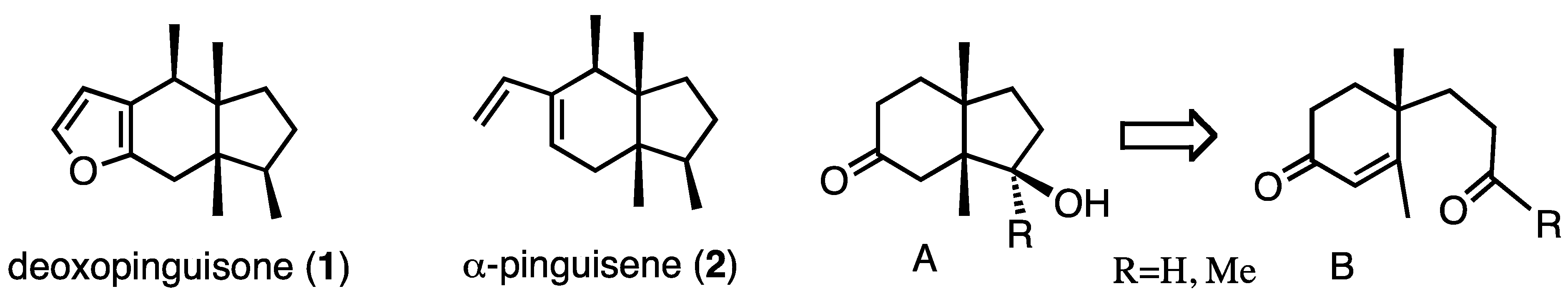
2. Results and Discussion
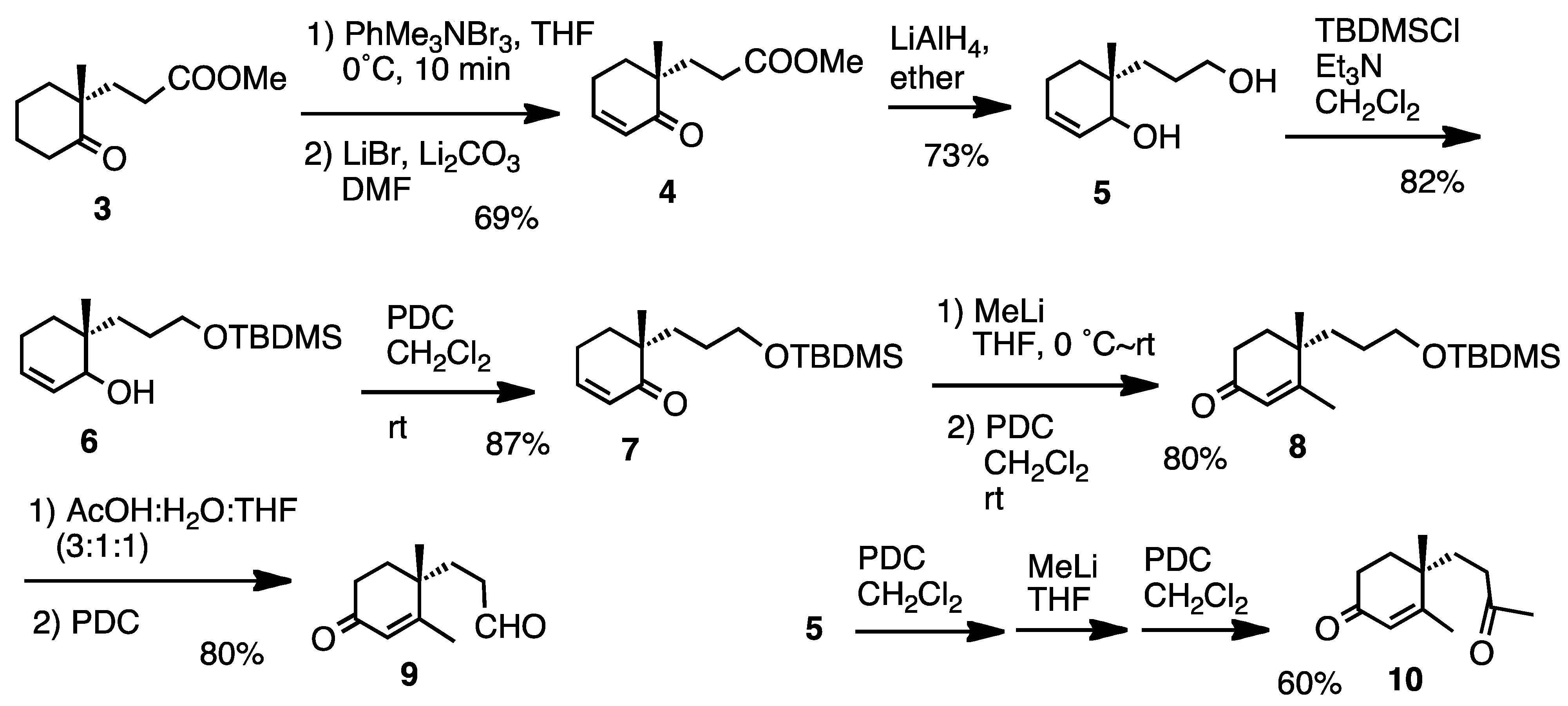
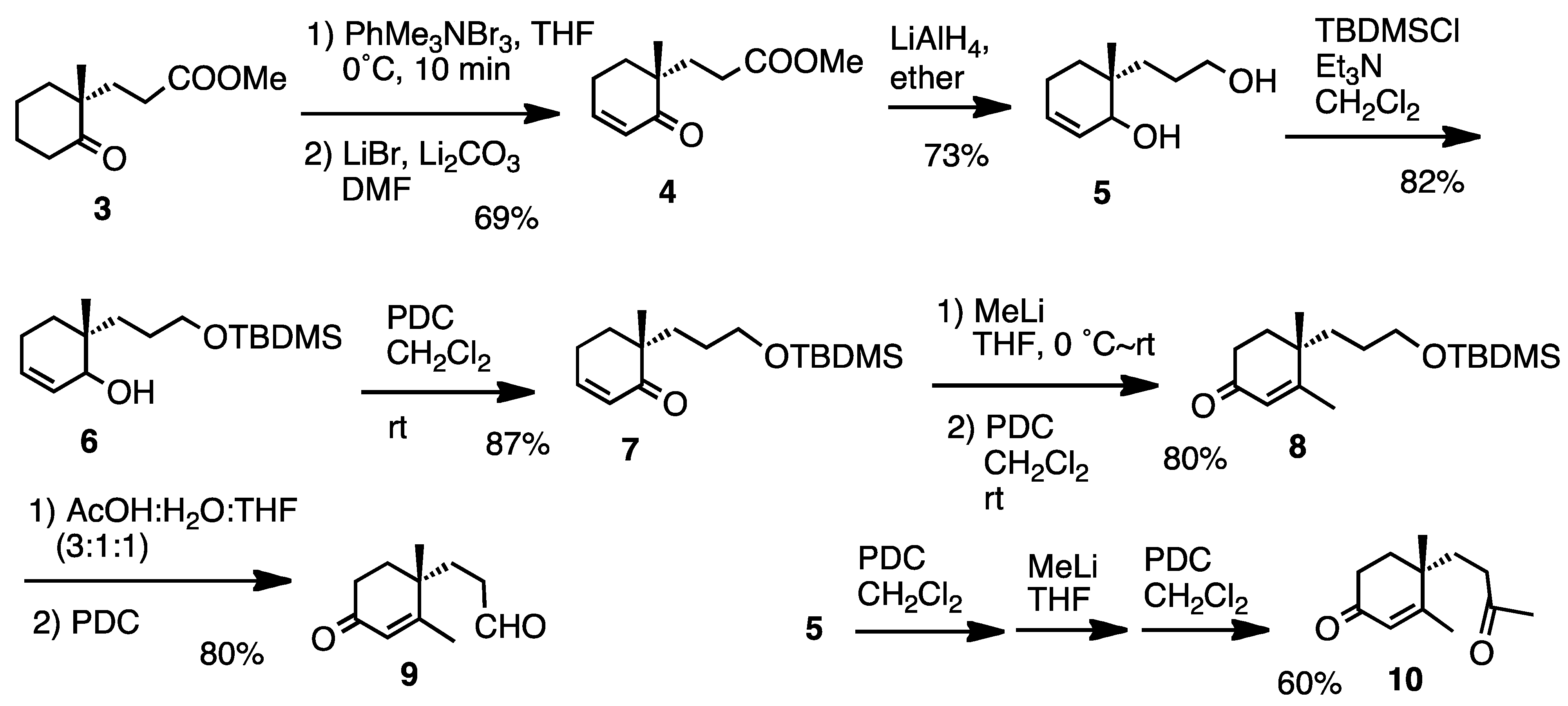
For the synthetic work we chose compounds
9 and
10 (
Scheme 1), which were prepared starting from ketone
3. Several routine reactions afforded aldehyde
9 and ketone
10. This route can also be used in chiral form, because compound
3 is now commercially available and easy to prepare [
21].
Scheme 1.
Preparation of compounds 9 and 10.
Scheme 1.
Preparation of compounds 9 and 10.
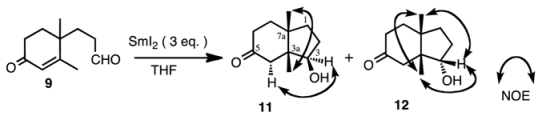
Aldehyde
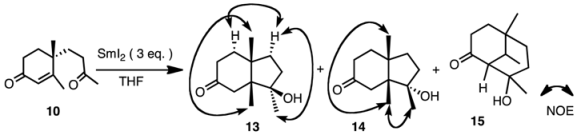
9 was subjected to reaction with SmI
2 (3 equiv.) in THF with or without additives. The results are shown in
Table 1. The products were isomeric keto-alcohols
11 and
12, whose stereochemistries were established from the NOESY spectra. Without an additive, the reactions proceeded smoothly and both compounds were obtained in a ratio of 87:13 in favor of compound
11 at 0 °C (entry 1). When the reaction temperature was raised to rt (entry 2), the ratio of compound
12 increased to 31%. The yields were always good. When MeOH was added as a proton source, the ratio was about 7:3 in favor of compound
11 (entries 3 and 4). The yield varied from 47% to 58%. The reason why the yields were not so high is presumably due to the simple reduction of the double bond to give the corresponding dihydro derivatives, which were not isolated but detected in GC-MS. The ratio of
11 and
12 did not change very much when using HMPA (entries 7 and 8), however, when NiI
2 was added (entries 9 and 10) the ratio of
12 was slightly increased [
22].
Table 1.
The reaction of aldehyde
9 with SmI
2.
![Molecules 17 11079 i001]()
Table 1.
The reaction of aldehyde 9 with SmI2. ![Molecules 17 11079 i001]()
| Entry | Additives | Temp (°C) | Yield (%) | Ratio |
|---|
| 11 | 12 |
|---|
| 1 | none | 0 | quant. | 87 | 13 |
| 2 | none | rt | quant. | 69 | 31 |
| 3 | MeOH 2 equiv. | 0 | 58 | 72 | 28 |
| 4 | MeOH 2 equiv. | rt | 47 | 69 | 31 |
| 5 | HMPA 12 equiv. | 0 | 50 | 82 | 18 |
| 6 | HMPA 12 equiv. | rt | 44 | 84 | 16 |
| 7 | NiI2 | 0 | quant. | 75 | 25 |
| 8 | NiI2 | rt | quant. | 56 | 44 |
We next studied the reaction of ketone
10 under various conditions (
Table 2). The products were isomeric keto-alcohols
13 and
14, and a mixture of bicyclic compounds
15, whose structures were determined by spectroscopic analyses. When ketone
10 was subjected to reaction with SmI
2 without additive at 0 °C (entry 1), the products were
13 and
14 in a ratio of 84:16. In this case the major product had the hydroxy group in a β-orientation as determined by the NOESY spectrum (
Table 2).
Table 2.
The reaction of compound
10 with SmI
2.
![Molecules 17 11079 i002]()
Table 2.
The reaction of compound 10 with SmI2. ![Molecules 17 11079 i002]()
| Entry | Additives | Temp. (°C) | Yield (%) | Ratio |
|---|
| 13 | 14 | 15 |
|---|
| 1 | none | 0 | quant. | 84 | 16 | - |
| 2 | none | rt | 84 | 67 | 33 | - |
| 3 | MeOH 2 equiv. | 0 | quant. | 84 | 16 | - |
| 4 | MeOH 2 equiv. | rt | quant. | 55 | 45 | - |
| 5 | HMPA 12 equiv. | 0 | 78 | 61 | 17 | 22 |
| 6 | HMPA 12 equiv. | rt | 71 | 31 | 29 | 40 |
| 7 | NiI2 | 0 | quant. | 53 | 47 | - |
| 8 | NiI2 | rt | 85 | 86 | 14 | - |
When the temperature was raised to rt (entry 2), the ratio of compound
14 increased. This tendency was the same as that of the aldehyde mentioned above. However, when HMPA was added, a third product
15 was also obtained (entries 5 and 6). The ratio of
15 was 40% of the products at rt from a total yield of 71% (entry 6). This product was formed by an aldol type condensation of the samarium enolate of the α,β-unsaturated enone. When NiI
2 was added, the yield was high and the ratio of compound
12 was slightly increased (entries 7 and 8) [
22].
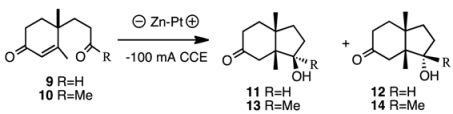
Electrolysis does not use organic solvents and expensive reagents, but rather water and electric power. In order to compare the selectivity, compounds
9 and
10 were subjected to electrolysis conditions as shown in
Table 3. The yields were moderate and the ratio was
11:
12 = 62:38 in the case of aldehyde
9. The results were not very different from those of samarium iodide reduction. However, in the case of ketone
10, only β-alcohol
13 was formed selectively (entry 2).
Table 3.
The electrolysis of
9 and
10.
![Molecules 17 11079 i003]()
Table 3.
The electrolysis of 9 and 10. ![Molecules 17 11079 i003]()
| Entry | Solvent | Additive | Time (h) | SM | Yield (%) | Ratio |
|---|
| 1 | tBuOH-H2O (4:6) | Et4NTsO | 2 | 9 | 53 | 11:12 = 62:38 |
| 2 | tBuOH-H2O (4:6) | Et4NTsO | 2 | 10 | 76 | 13:14 = 100:0 |
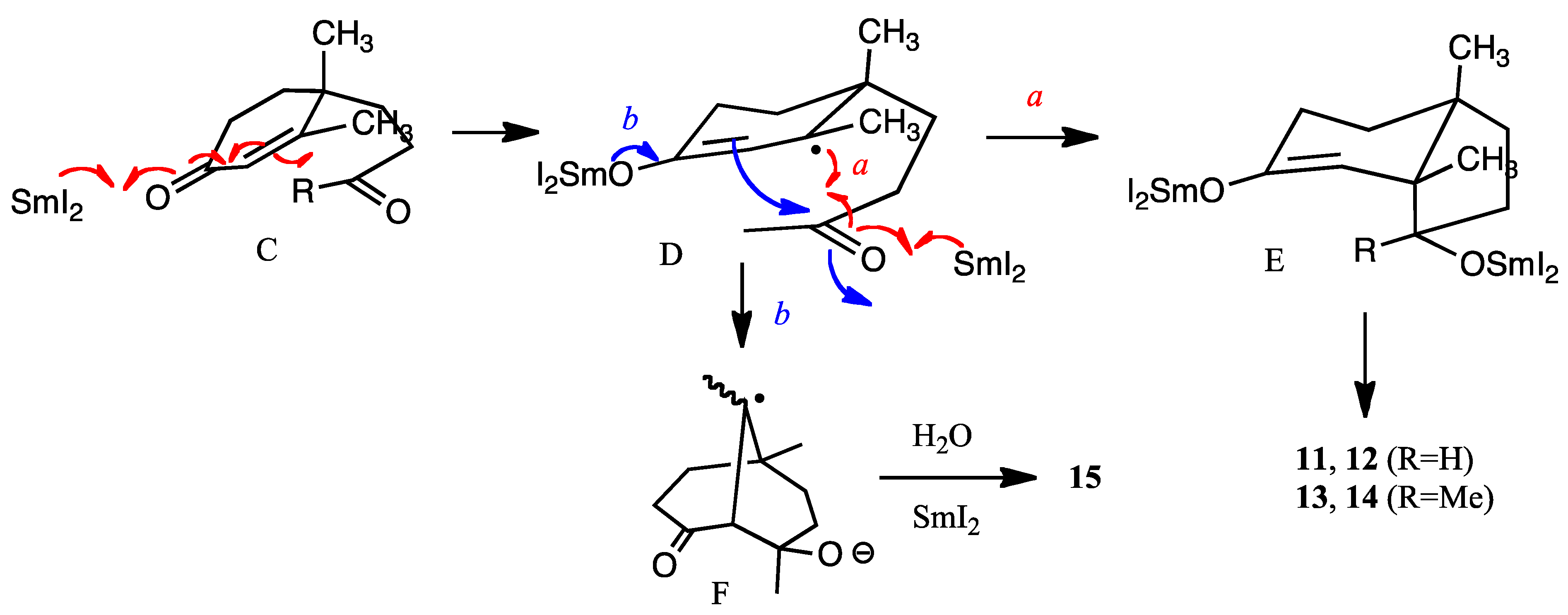
Comparison of the half-wave potentials of α,β-unsaturated carbonyls with those of corresponding saturated carbonyl compounds has been extensively studied in electrochemistry [
23]. The first waves of carbonyl groups, referred to as SCE., are −2.45 V (cyclohexanone), −2.25 V (methyl ethyl ketone), −1.8 V (propionaldehyde), −1.55 V (cyclohex-2-en-1-one), −1.50 V (acrolein), and −1.42 V (methyl vinyl ketone), respectively [
23]. Therefore, the reduction of α,β-unsaturated carbonyl moiety seems easier than that of the isolated ketone carbonyl group with electrochemistry, but the selectivity of the one-electron reduction of the carbonyl moiety using SmI
2 depends on the stereoelectronic properties of the substrate [
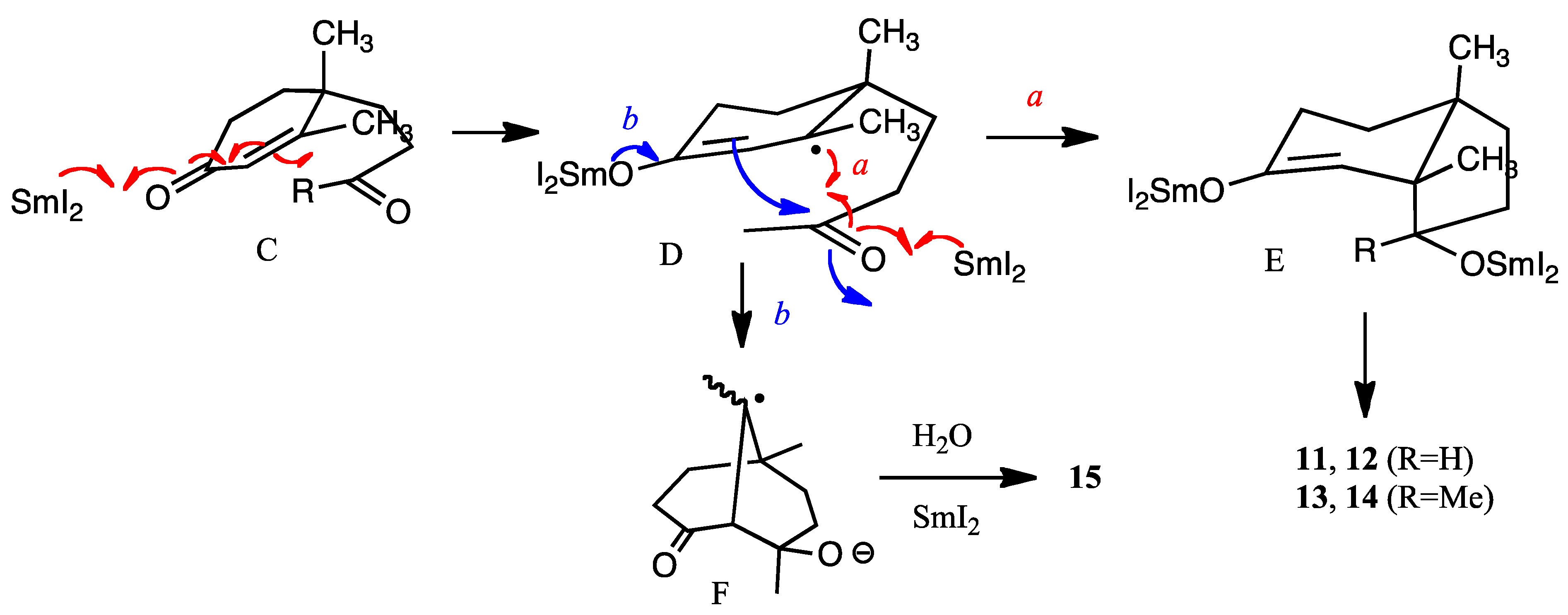
6]. Samarium transfers one-electron to the carbonyl group of the enone moiety to form a radical anion (C to D) (
Figure 2). Then one more samarium atom reduces the ketone carbonyl group and the carbon-carbon bond is formed as shown by arrow
a to afford E (alkene first mechanism [
2]). The hydroxy group is outside the bicyclic ring formed, because the samarium ion radical is large and the outside position is more energetically favored than inside of the ring. Thus, this configuration is more or less predominant. However, with HMPA as the additive, the reducing power must be higher than that with samarium alone [
24], and the enone moiety is susceptible to reduction to afford a samarium enolate D. Then the aldol-type cyclization occurs to afford bicyclic anion radical F from D (shown by arrow
b in
Figure 2). Further reduction of this anion radical F and protonation afford product
15. Electrolysis also creates a similar transition state leading to similar results. The reason why compound
10 produces
13 much more selectively is not clear at this stage. However, it is assumed that solvent molecules surround the methyl ketone carbonyl group resulting in the bulkier CO (solvent) moiety with consequent protrusion outside of the ring leading to the β-alcohol
13 [
24].
Figure 2.
Mechanisms of reductive cyclization (R = H, Me).
Figure 2.
Mechanisms of reductive cyclization (R = H, Me).
3. Experimental
3.1. General
IR spectra were measured on a JASCO FT/IR-5300 spectrophotometer. The 1H and 13C-NMR spectra were taken using a Varian Unity 600 (at 600 MHz and 150 MHz, respectively) and a Varian Unity 200 (200 MHz and 50 MHz, respectively) spectrometer. Mass spectra including high-resolution mass spectra were recorded on a JEOL JMS-700 MStation. A Chemcopak Nucleosil 50-5 column (4.8 × 250 mm) was used for HPLC (JASCO pump system). For GC-MS an Agilent GC 6890 system equipped with a MS detector 5973 was used and the product ratios were determined by the area %. Silica gel 60 (70–230 mesh, Fuji Silysia) was used for column chromatography and silica gel 60 F254 plates (Merck) were used for TLC.
3.1.1. General Procedure for Smi2 Reduction
A solution of substrate in dry THF was introduced into a solution of SmI2 at a certain temperature. Saturated solution of sodium potassium tartrate was added and the solvent was evaporated. The mixture was extracted with ether and worked up as usual. The residue was purified by silica-gel column chromatography.
3.1.2. General Procedure for Electrolysis
Zn and Pt were used for the cathode and anode, respectively, with Et4NOTs in tBuOH-H2O (2:3) (20 mL), CCE at 100 mA (from −1.5 to −2.0 V vs. SCE) at rt. Work-up: benzene was added and most of the water was removed under reduced pressure. The residue was extracted with EtOAc and the organic layer was washed with Sat. NaCl solution. The organic layer was dried (MgSO4), and the filtrate was evaporated to give a residue, which was purified by silica-gel column chromatography or HPLC.
3.2. Preparation of Methyl 3-(1-Methyl-2-oxocyclohex-3-enyl)propanoate (4)
Ketone (3, 1 g, 0.5 mmol) was treated with phenyltrimethylammonium tribromide (1.9 g, 0.88 mmol) in THF (5 mL) at 0 °C for 10 min. After the usual work-up, the residue (925 mg) was successively treated with LiBr (585 mg, 5.6 mmol) and Li2CO3 (148 mg, 3.3 mmol) in DMF (4 mL) at 150 °C for 18 h. Usual work-up and purification afforded enone 4 (684 mg, 69%). Oil; IR (FT): 1730, 1660, 1630 cm−1; 1H-NMR (200 MHz, CDCl3): δ 1.09 (3H, s), 1.73–1.99 (4H, m), 2.03–2.47 (4H, m), 3.65 (3H, s), 5.91 (1H, dt, J = 10.2, 1.8 Hz), 6.89 (1H, dt, J = 10.2, 4.0 Hz); 13C-NMR (50 MHz, CDCl3): δ 21.4 (CH3), 22.9 (CH2), 28.9 (CH2), 31.1 (CH2), 33.4 (CH2), 43.7 (C), 51.4 (CH3), 128.2 (CH), 148.6 (CH), 173.9 (C), 203.1 (C); MS m/z 196 (M)+, 181, 165, 136, 110, 68 (base), 55, HRMS Found m/z 196.1078. Calcd for C11H16O3 196.1100.
3.3. Preparation of 6-(3-Hydroxypropyl)-6-methylcyclohex-2-en-1-ol (5)
A solution of enone 4 (172 mg, 0.88 mmol) in ether (20 mL) was treated with LiAlH4 (101 mg, 2.64 mmol) at 0 °C for 2 h. Usual work-up afforded the diol 5 (109 mg, 73%) after purification as a mixture of diatereomers. Oil; IR (FT): 3330, 3020, 1660 cm−1; 1H-NMR (200 MHz, CDCl3): δ 0.87 (3H, s), 0.88 (3H, s), 1.2–1.7 (12H, m), 1.92–2.08 (2H, m), 3.03 (2H, br s), 3.5–4.0 (6H, m), 5.5–6.0 (4H, m); 13C-NMR (50 MHz, CDCl3): δ 18.3 (CH3), 21.3 (CH3), 22.7 (CH2), 22.8 (CH2), 26.2 (CH2), 26.6 (CH2), 29.6 (CH2), 30.3 (CH2), 32.1 (CH2), 35.1 (CH2), 35.6 (C), 35.8 (C), 63.3 (CH2), 71.7 (CH), 72.2 (CH), 128.1 (CH), 128.6 (CH), 129.7 (CH), 129.8 (CH); MS (CI) m/z 169 [M-2+H]+, 153, 135 (base), 109; HRMS (CI) Found m/z 169.1223 [M-2+H]+. Calcd for C10H17O2 169.1229.
3.4. Preparation of 6-(3-t-Butyldimethylsilyloxypropyl)-6-methylcyclohex-2-en-1-ol (6)
A solution of diol (5, 480 mg, 2.6 mmol) in CH2Cl2 (6 mL) was treated with Et3N (0.5 mL, 3.4 mmol) and TBDMSCl (473 mg, 3.1 mmol) at rt for 18 h. Usual work-up and purification afforded silyl ether 6 (658 mg, 82%). Oil; IR (FT): 3400, 1660 cm−1; 1H-NMR (200 MHz, CDCl3): δ 0.04 (6H, s), 0.88 (9H, s), 0.89 (3H, s), 1.16–1.75 (6H, m), 1.92–2.04 (2H, m), 3.58 (1H, d, J = 6.6 Hz), 3.60 (2H, t, J = 7.0 Hz), 3.74 (1H, br s), 5.62–5.74 (1H, m), 5.79 (1H, dt, J = 9.2, 2.6 Hz); 13C-NMR (50 MHz, CDCl3): δ −5.3 (CH3), 18.4 (C), 21.4 (CH3), 22.9 (CH2), 26.0 (CH3), 26.5 (CH2), 29.7 (CH2), 31.5 (CH2), 35.7 (C), 64.0 (CH2), 72.0 (CH), 128.3 (CH), 130.0 (CH); MS (CI) m/z 284 (M)+, 267, 227, 135 (base), 93, 83, 75; HRMS (CI) Found m/z 284.2155 (M)+. Calcd for C16H32O2Si 284.2172.
3.5. Preparation of 6-(3-t-Butyldimethylsilyloxypropyl)-6-methylcyclohex-2-en-1-one (7)
A solution of alcohol 6 (68 mg, 0.24 mmol) was oxidized with PDC (270 mg, 0.72 mmol) in CH2Cl2 in the presence of molecular sieves 3A (201 mg) at rt for 18 h. Usual work-up afforded enone 7 (59 mg, 87%) after purification. Oil; IR (FT): 1680 cm−1; 1H-NMR (200 MHz, CDCl3): δ 0.03 (6H, s), 0.87 (9H, s), 1.07 (3H, s), 1.32–1.63 (4H, m), 1.69–1.97 (2H, m), 2.19–2.50 (2H, m), 3.56 (2H, t, J = 7.0 Hz), 5.88 (1H, dt, J = 10.2, 2.0 Hz), 6.83 (1H, dt, J = 10.2, 4.0 Hz); 13C-NMR (50 MHz, CDCl3): δ −5.4 (CH3X2), 18.2 (C), 21.7 (CH3), 23.1 (CH2), 25.9 (CH3X3), 27.4 (CH2), 32.3 (CH2), 33.5 (CH2), 44.2 (C), 63.5 (CH2), 128.6 (CH), 148.5 (CH), 204.4 (C); MS (CI) m/z 283 [M+H]+, 267, 225, 151 (base); CI-HRMS Found m/z 283.2097 [M+H]+. Calcd for C16H31O2Si 283.2093.
3.6. Preparation of 4-(3-t-Butyldimethylsilyloxypropyl)-3,4-dimethylcyclohex-2-en-1-one (8)
MeLi (0.88 mL, 1 mmol) was added to a stirred solution of enone 7 (39 mg, 0.22 mmol) and the mixture was stirred at 0°C for 22 h. Usual work-up afforded a residue 36 mg), which was successively treated with PDC (140 mg, 0.36 mmol) in CH2Cl2 (4 mL) at rt for 4 h. Usual work-up and purification afforded enone 8 (33 mg, 80%). Oil; IR (FT): 1680, 1620 cm−1; 1H-NMR (200 MHz, CDCl3): δ 0.04 (6H, s), 0.89 (9H, s), 1.14 (3H, s), 1.18–1.76 (6H, m), 1.90 (3H, d, J = 1.2 Hz), 2.39 (1H, J = 6.4 Hz), 2.43 (1H, dd, J = 6.4, 2.1 Hz), 3.5–3.7 (2H, m), 5.80 (1H, d, J = 1.2 Hz); 13C-NMR (50 MHz, CDCl3): δ −5.3 (CH3X2), 18.3 (C), 20.0 (CH3), 24.2 (CH3), 25.9 (CH3X3), 27.7 (CH2), 33.4 (CH2), 34.1 (CH2), 34.8 (CH2), 38.2 (C), 63.2 (CH2), 127.2 (CH), 168.9 (C), 199.3 (C); MS (CI) m/z 297 [M+H]+, 281, 239 (base), 57, CI-HRMS Found m/z 297.2226. Calcd for C17H33O2Si 297.2250.
3.7. Preparation of 3-(1,2-Dimethyl-4-oxocyclohex-2-enyl)propanal (9)
Enone 8 (67 mg, 0.23 mmol) was treated with AcOH:H2O:THF (3:1:1) (6 mL) at rt overnight. Usual work-up afforded a residue (25 mg), which was subjected to PDC (978 mg, 2.6 mmol) oxidation in CH2Cl2 (30 mL in the presence of molecular sieves 3A (903 mg) at rt for 2 h. Usual work-up afforded enone 9 (33 mg, 80%) after purification. Oil; IR (FT): 1720, 1670, 1620 cm−1; 1H-NMR (200 MHz, CDCl3): δ 1.16 (3H, s), 1.65–1.97 (4H, m), 1.88 (3H, d, J = 1.3 Hz), 2.19–2.60 (2H, m), 2.40 (2H, t, J = 5.8 Hz), 5.82 (1H, d, J = 1.3 Hz), 9.80 (1H, t, J = 1.3 Hz); 13C-NMR (50 MHz, CDCl3): δ 19.9 (CH3), 23.8 (CH3), 29.8 (CH2), 33.3 (CH2), 33.9 (CH2), 37.7 (C), 39.1 (CH2), 127.9 (CH), 167.3 (C), 198.9 (C), 201.3 (CH); MS m/z 180 (M+), 162, 152, 124, 109, 95 (base), 81, 67, 55; HRMS Found m/z 180.1151 (M)+. Calcd for C11H16O2 180.1151.
3.8. Preparation of 4-(3-Oxobutyl)-3,4-dimethylcyclohex-2-en-1-one (10)
Diol 5 (509 mg, 3 mmol) was oxidized with PDC (3.4 g, 9 mmol) in CH2Cl2 at rt overnight. Usual work-up afforded a residue (97 mg), which was treated with MeLi (9.3 mL, 10 mmol) in THF (30 mL) at rt for 16 h. The residue after usual work-up was further treated with PDC (2.6 g, 7 mmol) in CH2Cl2 at rt overnight. Usual work-up afforded enone 5 (349 mg, 60%) after purification. Oil; IR (FT): 1710, 1670, 1610 cm−1; 1H-NMR (200 MHz, CDCl3): δ 1.16 (3H, s), 1.64–1.97 (4H, m), 1.89 (3H, d, J = 1.1 Hz), 2.17 (3H, s), 2.23–2.58 (4H, m), 5 .83 (1H, d, J = 1.1 Hz); 13C-NMR (50 MHz, CDCl3): δ 19.9 (CH3), 24.0 (CH3), 30.1 (CH3), 31.6 (CH2), 33.3 (CH2), 34.0 (CH2), 37.8 (C), 38.5 (CH2), 127.7 (CH), 167.7 (C), 198.9 (C), 207.9 (C); MS m/z 194 (M+), 176, 124 (base), 109, 95, 79, 67, 55; HRMS Found m/z 194.1295 (M)+. Calcd for C12H18O2 194.1307.
3.9. (3R*,3aR*,7aS*)-3-Hydroxy-3a,7a-dimethylhexahydro-1H-inden-5(6H)-one (11))
Oil; IR (FT): 3440, 1710 cm−1; 1H-NMR (600 MHz, CDCl3): δ 0.90 (3H, s, 3a-CH3), 1.16 (3H, s, 7a-CH3), 1.55-1.74 (4H, m, H-1,1,2β,7β), 1.84 (1H, ddd, J = 15.5, 11.8, 4.9 Hz, H-7α), 2.12 (1H, dd, J = 14.4, 1.9 Hz, H-4β), 2.13-2.19 (1H, m, H-2α), 2.25 (1H, dtd, J = 14.4, 4.9, 1.9 Hz, H-6α), 2.29 (1H, dd, J = 14.4, 1.9 Hz, H-4α), 2.39 (1H, dddd, J = 14.4, 11.8, 6.0, 1.1 Hz, H-6β), 3.96 (1H, dd, J = 8.5, 6.6 Hz, H-3); 13C-NMR (150 MHz, CDCl3): δ 16.9 (3a-CH3), 22.3 (7a-CH3), 29.4 (C-2), 34.8 (C-1), 36.8 (C-7), 37.8 (C-6), 41.5 (C-7a), 47.5 (C-4), 50.7 (C-3a), 78.0 (C-3) , 212.5 (C-5); MS m/z 182 (M+), 164, 139, 124, 111 (base), 96, 84, 79, 69, 55; HRMS Found m/z 182.1298 (M)+. Calcd for C11H18O2 182.1307.
3.10. (3S*,3aR*,7aS*)-3-Hydroxy-3a,7a-dimethylhexahydro-1H-inden-5(6H)-one (12)
Oil; IR (FT): 3320, 1710 cm−1; 1H-NMR (600 MHz, CDCl3): δ 0.89 (3H, s, 3a-CH3), 1.04 (3H, s, 7a-CH3), 1.53 (1H, ddd, J = 12.9, 10.2, 6.5 Hz, H-1β), 1.60–1.71 (2H, m, H-2α,7α), 1.91–1.95 (1H, m, H-1α), 2.07 (1H, br d, J = 14.3 Hz, H-4β), 2.07–2.14 (1H, m, H-7β), 2.16–2.23 (1H, m, H-2β), 2.25-2.30 (1H, m, H-6β), 2.38 (1H, br d, J = 14.3 Hz, H-4α), 2.37–2.42 (1H, m, H-6α), 3.96 (1H, dd, J = 7.7, 4.5 Hz, H-3); 13C-NMR (150 MHz, CDCl3): δ 21.6 (3a-CH3), 23.4 (7a-CH3), 30.3 (C-2), 34.9 (C-1), 35.8 (C-7), 37.6 (C-6), 41.6 (C-7a), 45.5 (C-4), 51.3 (C-3a), 81.9 (C-3), 213.0 (C-5); MS (EI) m/z 182 (M+), 164, 139, 124, 111, 95, 84 (base), 69, 55; HRMS Found m/z 182.1308 (M)+. Calcd for C11H18O2 182.1307.
3.11. (3R*,3aR*,7aS*)-3-Hydroxy-3,3a,7a-trimethylhexahydro-1H-inden-5(6H)-one (13)
Oil; IR (FT): 3480, 1710 cm−1; 1H-NMR (600 MHz, CDCl3): δ 0.89 (3H, s, 3a-CH3), 1.15 (3H, s, 7a-CH3), 1.16 (3H, s, 3-CH3), 1.69 (1H, ddd, J = 14.2, 7.4, 4.9 Hz, H-7β), 1.77–1.81 (3H, m, H-1,2,2), 1.84 (1H, m, H-7α), 1.85–1.89 (1H, m, H-1), 1.96 (1H, dd J = 14.0, 1.1 Hz, H-4β), 2.20 (1H, dddd, J = 17.3, 9.3, 4.9, 1.1 Hz, H-6β), 2.28 (1H, d, J = 14.0 Hz, H-4α), 2.34 (1H, ddd, J = 17.3, 7.4, 4.9 Hz, H-6α); 13C-NMR (150 MHz, CDCl3): δ 16.7 (3a-CH3), 23.5 (3-CH3), 26.6 (7a-CH3), 36.1 (C-6), 36.3 (C-7), 37.1 (C-2), 37.6 (C-1), 42.7 (C-7a), 49.7 (C-4), 50.9 (C-3a), 84.1 (C-3), 213.7 (C-5); MS m/z 196 (M)+, 178, 150, 139, 123, 111, 84 (base), 69, 55; HRMS Found m/z 196.1452 (M)+. Calcd for C12H20O2 196.1464.
3.12. (3S*,3aR*,7aS*)-3-Hydroxy-3,3a,7a-trimethylhexahydro-1H-inden-5(6H)-one (14)
Oil; IR (FT): 3400, 1700 cm−1; 1H-NMR (200 MHz, CDCl3): δ 0.86 (3H, s), 0.98 (3H, s), 1.18 (3H, s), 1.40–1.50 (3H, m), 1.54-1.61 (2H, m), 1.71–1.79 (2H, m), 1.81–1.85 (1H, m), 1.88 (1H, d, J = 11.8 Hz), 1.88–1.94 (1H, m); 13C-NMR (50 MHz, CDCl3): δ 17.2 (CH3), 22.9 (CH3), 26.8 (CH3), 35.1 (CH2), 35.4 (CH2), 35.7 (CH2), 36.6 (CH2), 42.6 (CH2), 44.2 (C), 53.4 (C), 92.3 (C), 213.5 (C); MS m/z 196 (M+), 178, 151, 139, 125, 109, 95, 84 (base), 69, 55; HRMS Found m/z 196.1460 (M)+. Calcd for C12H20O2 196.1463.
3.13. 8-Hydroxy-5,8,9-trimethylbicyclo[3.3.1]nonan-2-one (Mixture of Diatereoisomers) (15)
Oil; IR (FT): 3420, 1700 cm−1; 1H-NMR (200 MHz, CDCl3): δ 0.90 (0.25H, s), 0.93 (0.75H, d, J = 7.1 Hz), 0.96 (0.75H, s), 1.08 (0.25H, s), 1.40 (0.75H, s), 1.42 (0.25H, s); 13C-NMR (150 MHz, CDCl3): δ 28.0, 28.7, 31.4, 35.8, 38.2, 39.5, 40.5, 65.5, 66.6, 69.9, 71.8, 214.1 (CO); HRMS Found m/z 196.1465 (M)+. Calcd for C12H20O2 196.1463.


{kind=link}
{kind=link}
{kind=link}