Prediction of Acute Mammalian Toxicity Using QSAR Methods: A Case Study of Sulfur Mustard and Its Breakdown Products

Abstract
:1. Introduction
2. Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Name | Chemical Structure | Experimental LD50 (mg/kg) |
|---|---|---|
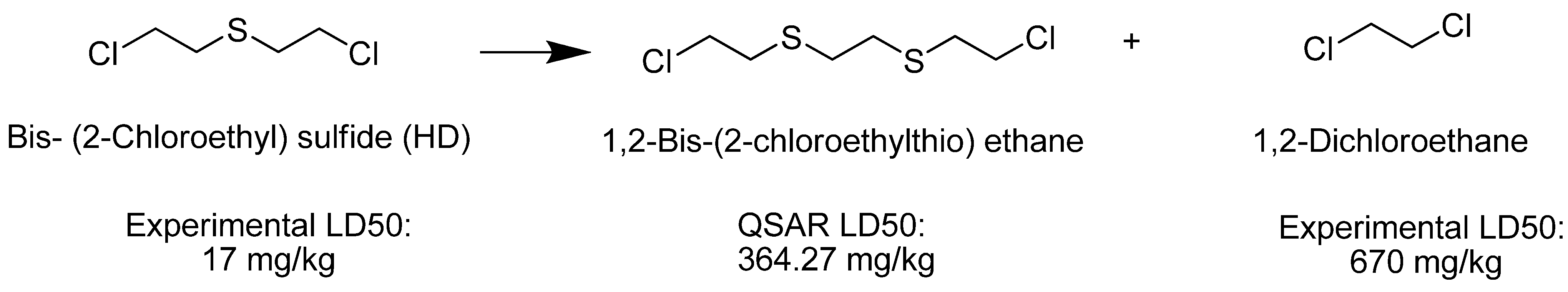
| Bis (2-chloroethyl) sulfide (HD) | 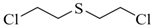 | 17 |
| Bis [2-(2-chloroethylthioethyl) ether] (T) |  | na |
| 1,2-bis(2-chloroethylthio)ethane (Q) |  | na |
| Bis (2-chloroethyl) disulfide | 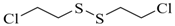 | na |
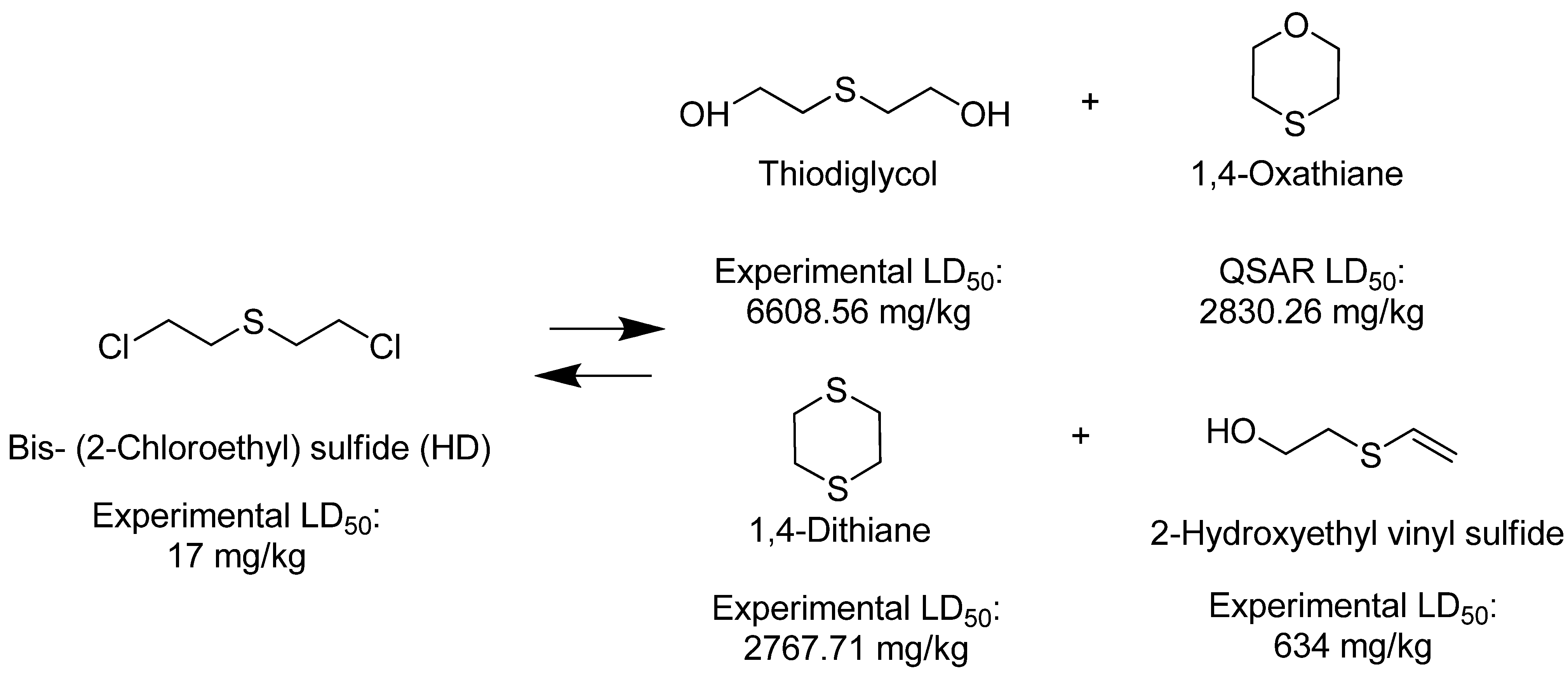
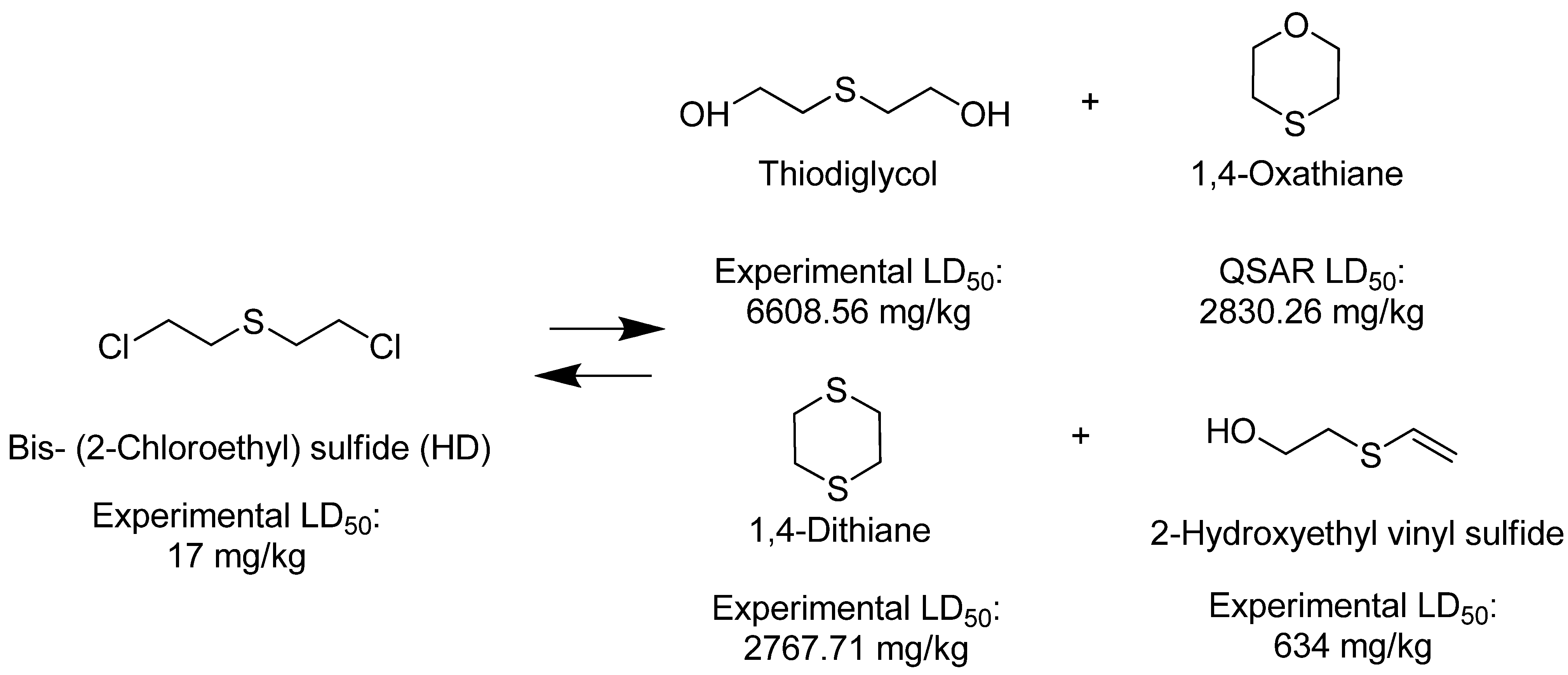
| Thiodiglycol (TDG) | 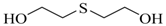 | 6,610 |
| Thiodiglycol sulfoxide |  | na |
| 1,4-Dithiane |  | 2,768 |
| 1,4-Oxathiane | 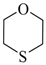 | 2,830 |
| Bis-[2-(2-hydroxyethylthioethyl)]ether (TOH) |  | na |
| Ethanol, 2,2'-[1,2-ethanediylbis(thio)]-bis- (QOH) | 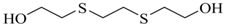 | na |
| 1-(2-hydroxyethylthio)-2-(2-vinylthioethoxy)ethane | 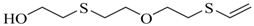 | na |
| 1-(2-hydroxyethylthio)-2-(2-vinylthio)ethane | 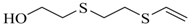 | na |
| 2-Hydroxyethyl vinyl sulfide | 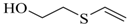 | na |
| 2-Methylnaphthalene | 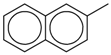 | 1,630 |
| Bis-(2-chloroethyl) ether | 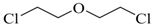 | 75 |
| Bis-(2-ethylexyl)phthalate | 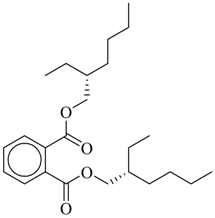 | 30,600 |
| Propanal | 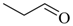 | 1,410 |
| Ethylene Glycol | 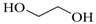 | 4,700 |
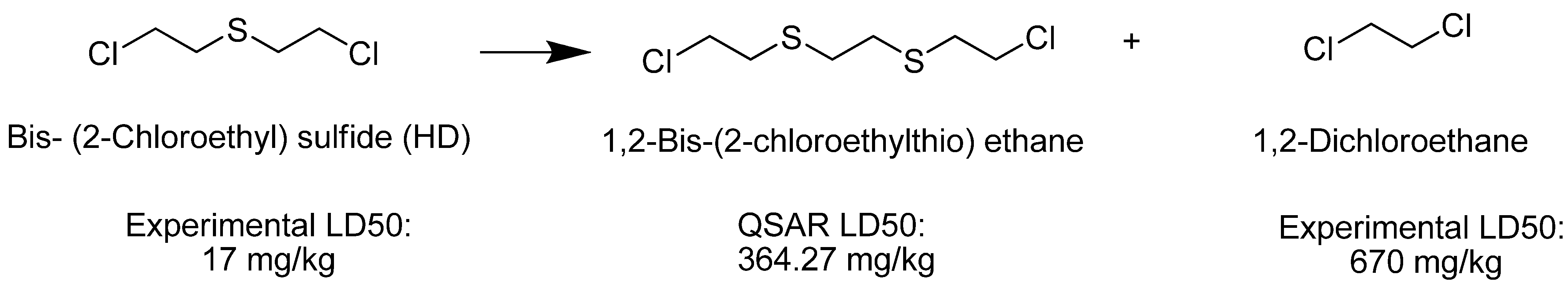
| Ethylene dichloride | 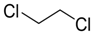 | 670 |
| Bis-(2-Chloroethyl) sulfoxide | 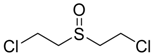 | na |
| Bis-(2-Chloroethyl) sulfone | 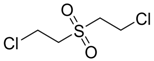 | na |
| Thiodiglycol sulfone |  | na |
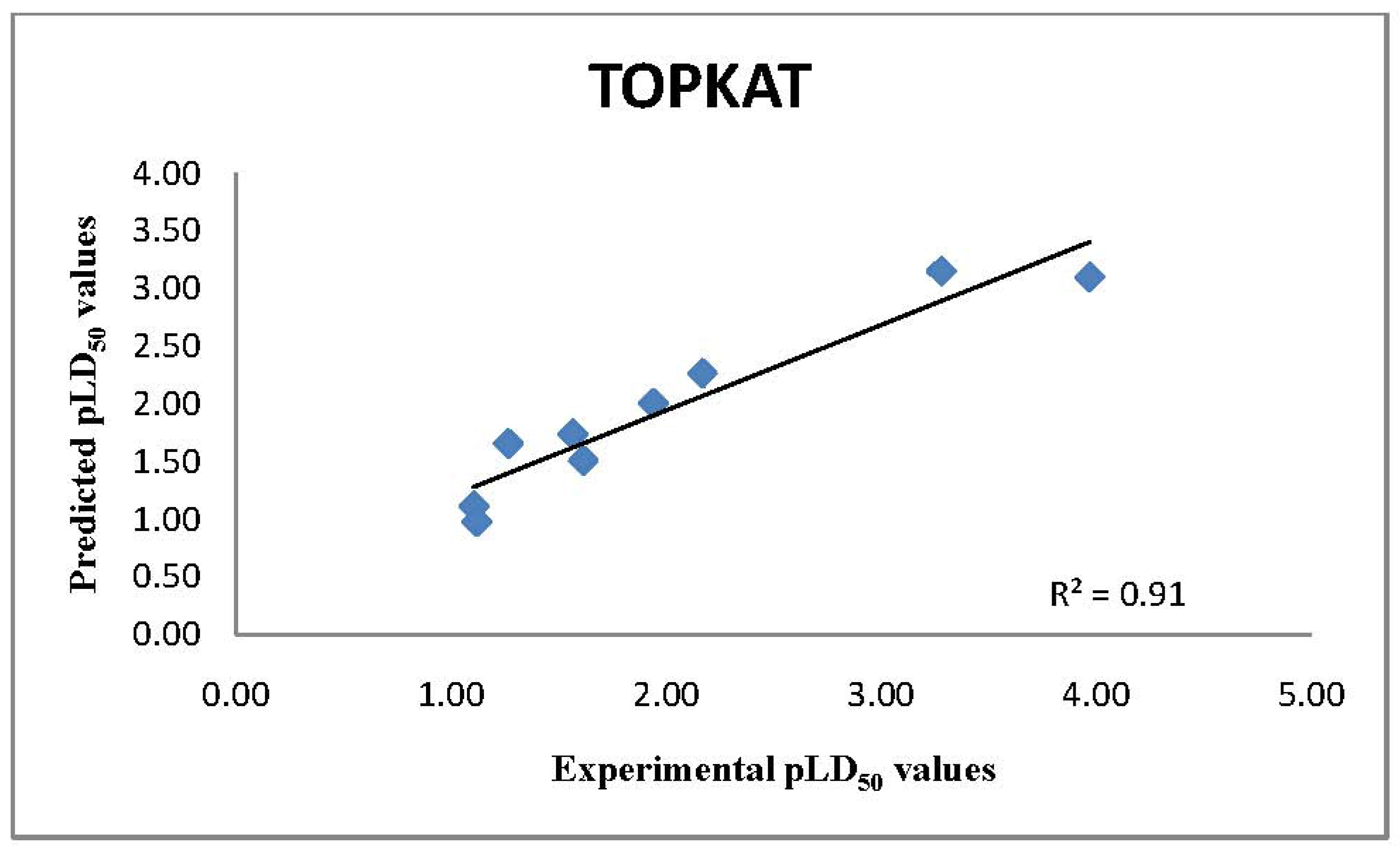
2.1. TOPKAT Predictions of LD50 Values
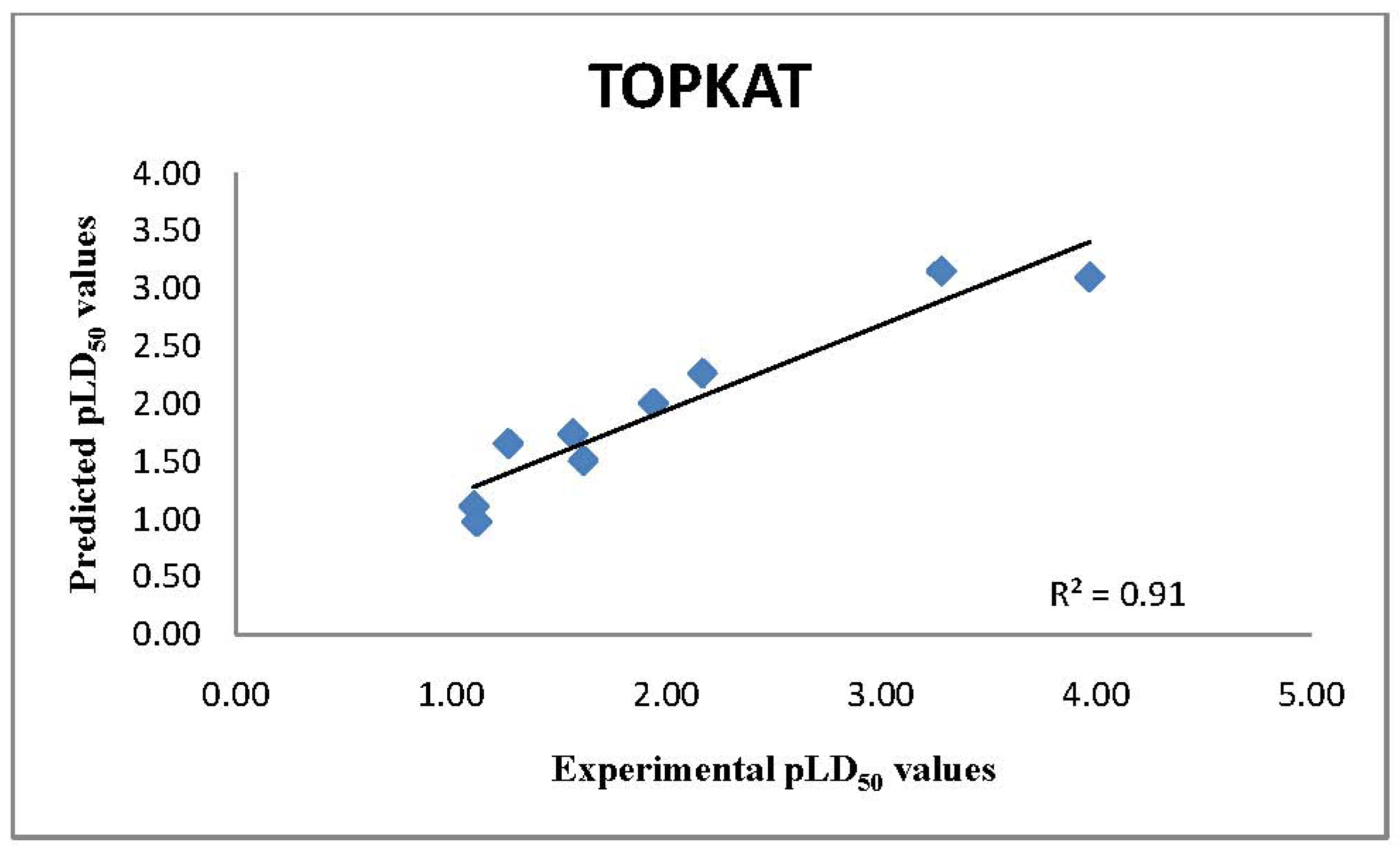
| Chemical Name | Experimental LD50* | TOPKAT | ADMET 2D | ADMET 3D | T.E.S.T Consensus |
|---|---|---|---|---|---|
| Bis (2-chloroethyl) sulfide | 17 | 125.9 | 536.84 | 349.32 | 38.69 |
| Thiodiglycol (TDG) | 6,608.56 | 2,700 | 1,651.93 | 1,407.24 | 5,426.53 |
| 1,4-Dithiane | 2,767.71 | OPS | 502.47 | 762.79 | 1,602.59 |
| 1,4-Oxathiane | 2,830.26 | 1,900 | 791.98 | 1,185.75 | 1,834.75 |
| 2-Methylnaphthalene | 1,629.3 | 1,400 | 1,884.9 | 1,864.48 | 1,391.74 |
| Bis-(2-chloroethyl) ether | 75 | 100.8 | 215.35 | 130.57 | 44.72 |
| Bis-(2-ethylhexyl)phthalate | 30,600 | >10,000 | 8,781.91 | 6,791.82 | 37,293.36 |
| Propanal | 1,409.62 | 1,800 | 404.01 | 427.17 | 353.02 |
| Ethylene glycol | 4,700 | 6,500 | 2,705.28 | 2,034.17 | 2,587.11 |
| Ethylene dichloride | 670 | 537.5 | 298.37 | 297.55 | 811.49 |
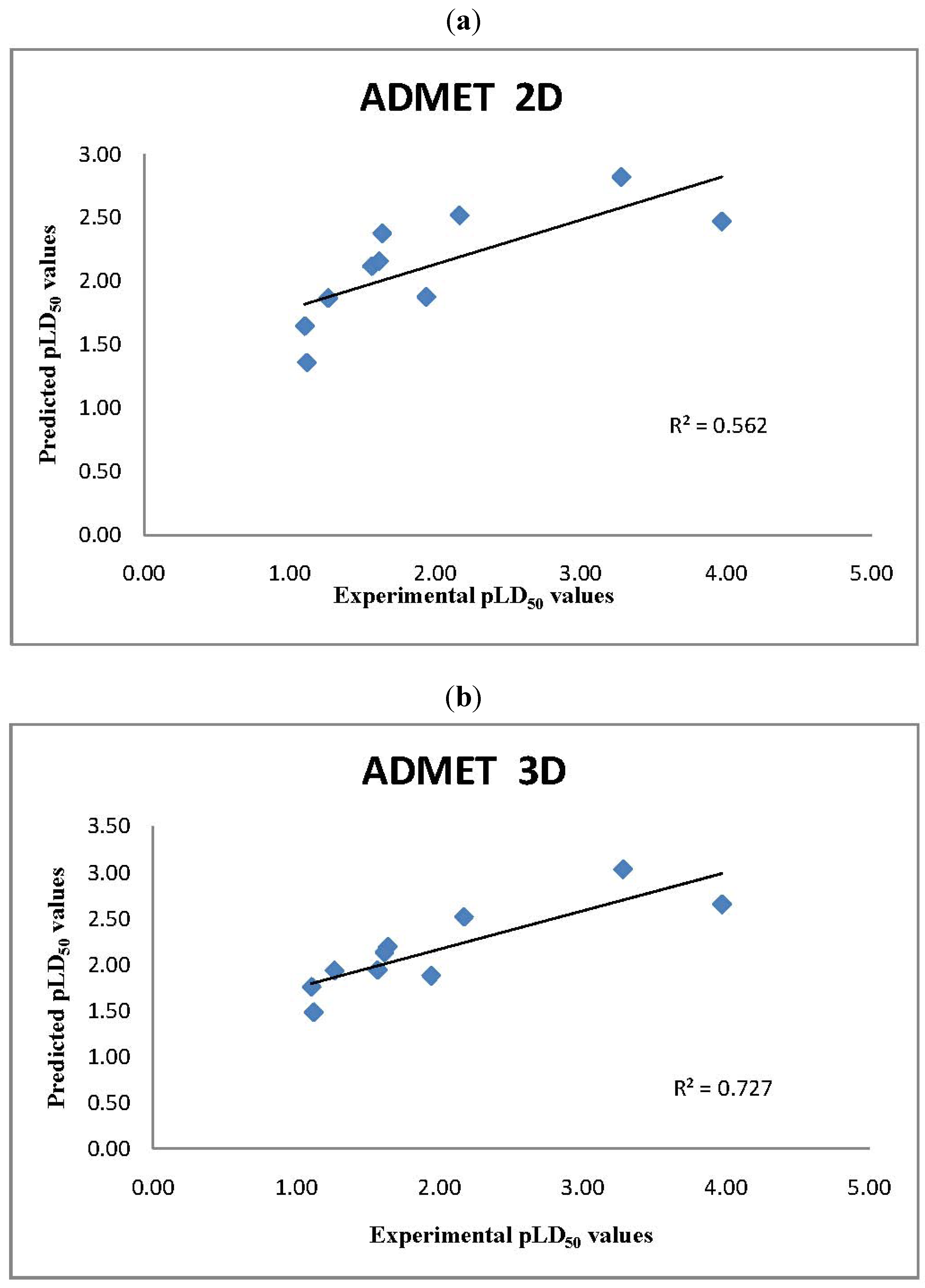
2.2. ADMET Predictor LD50 Values
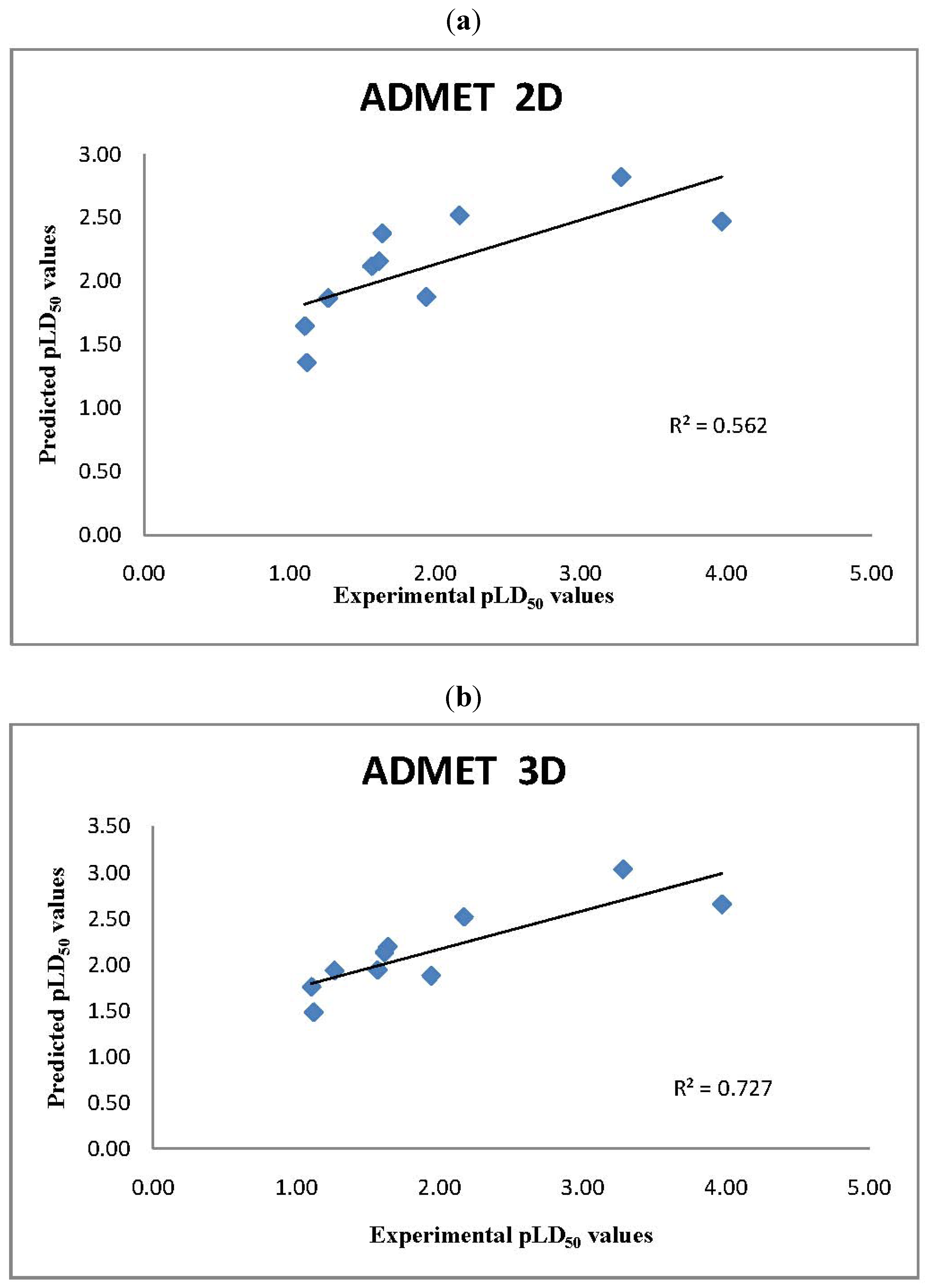
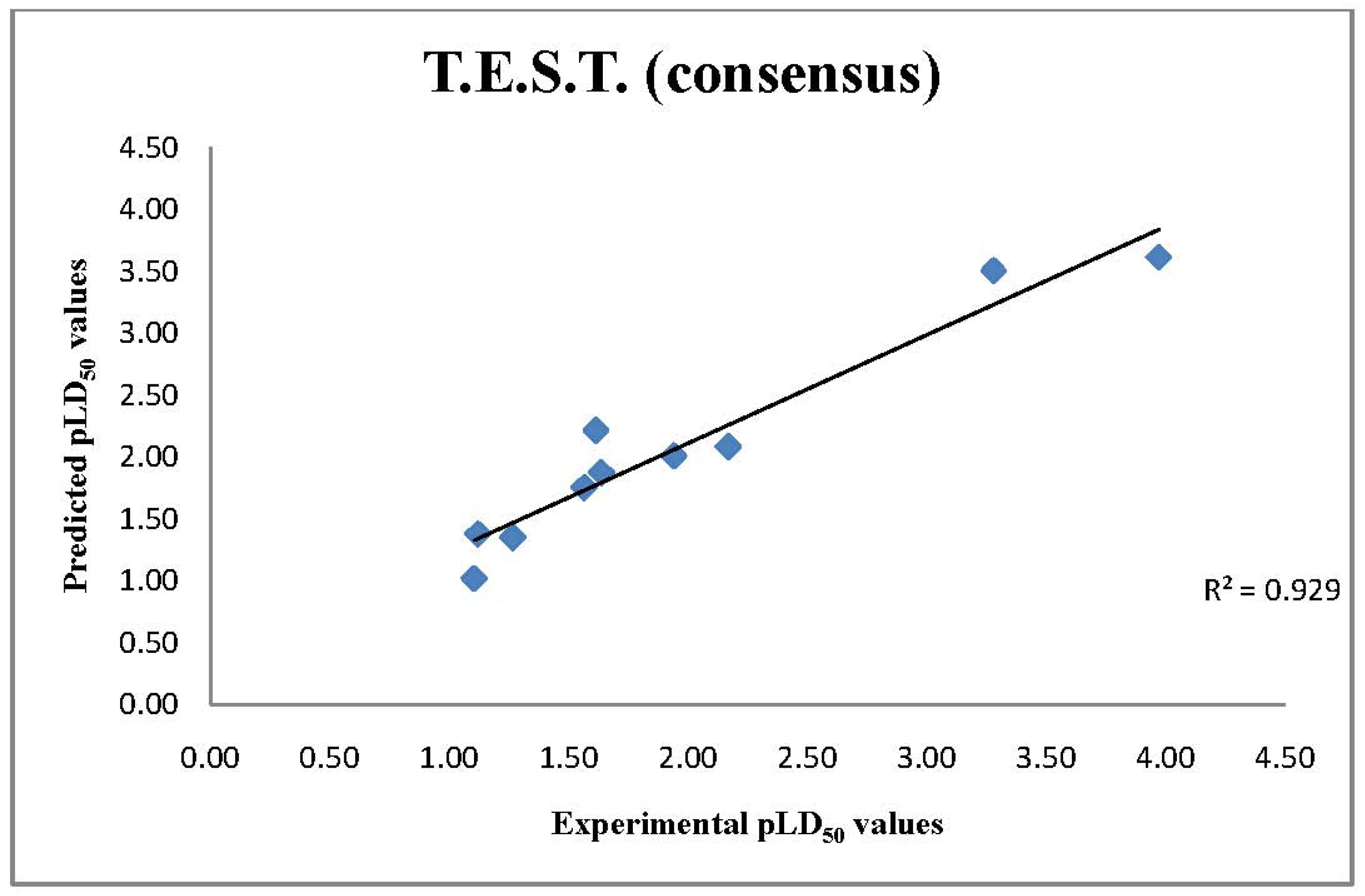
2.3. T.E.S.T. Predictions of LD50 Values
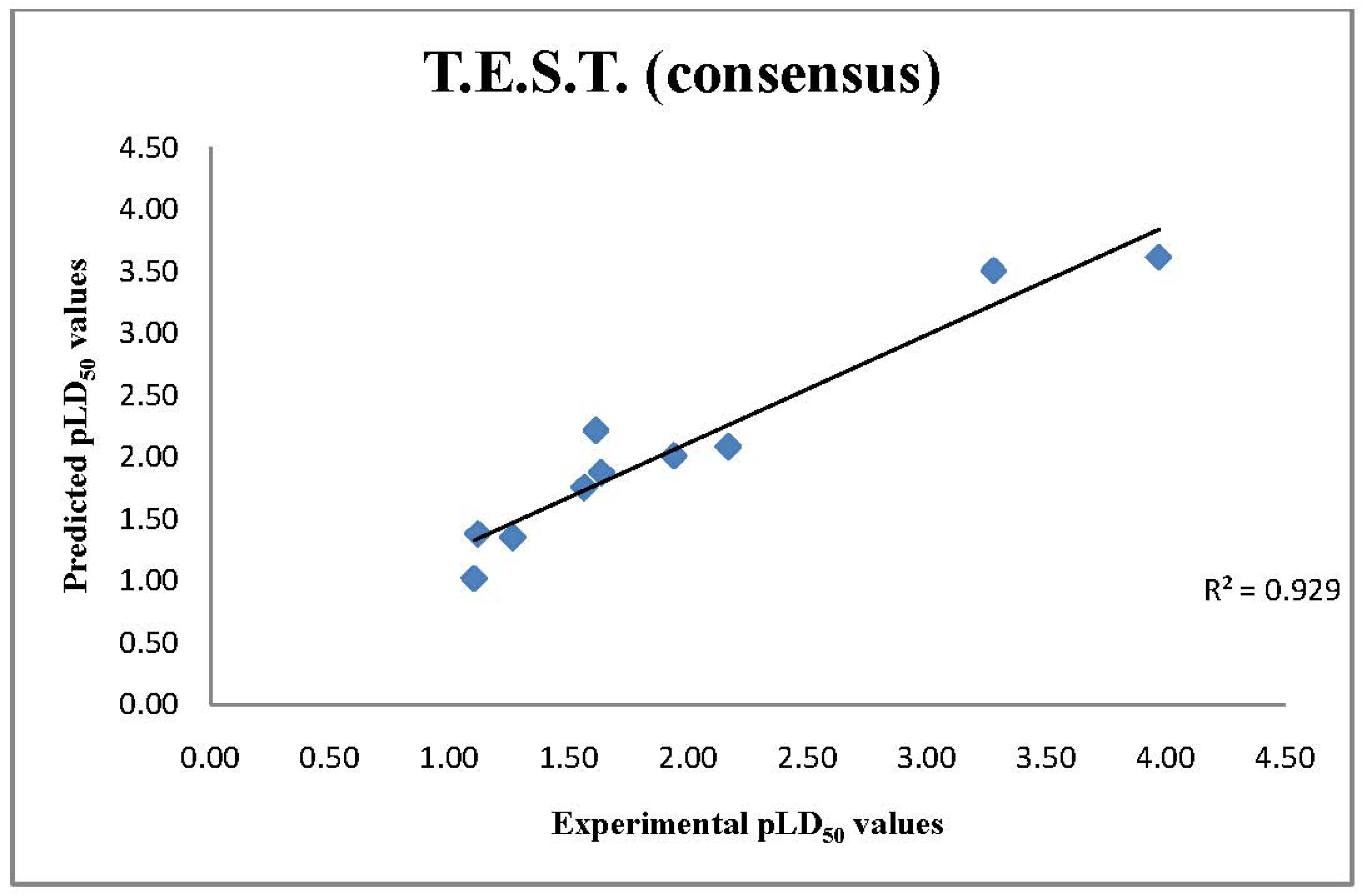
| Chemical Name | TOPKAT | ADMET 2D | ADMET 3D | T.E.S.T (consensus) |
|---|---|---|---|---|
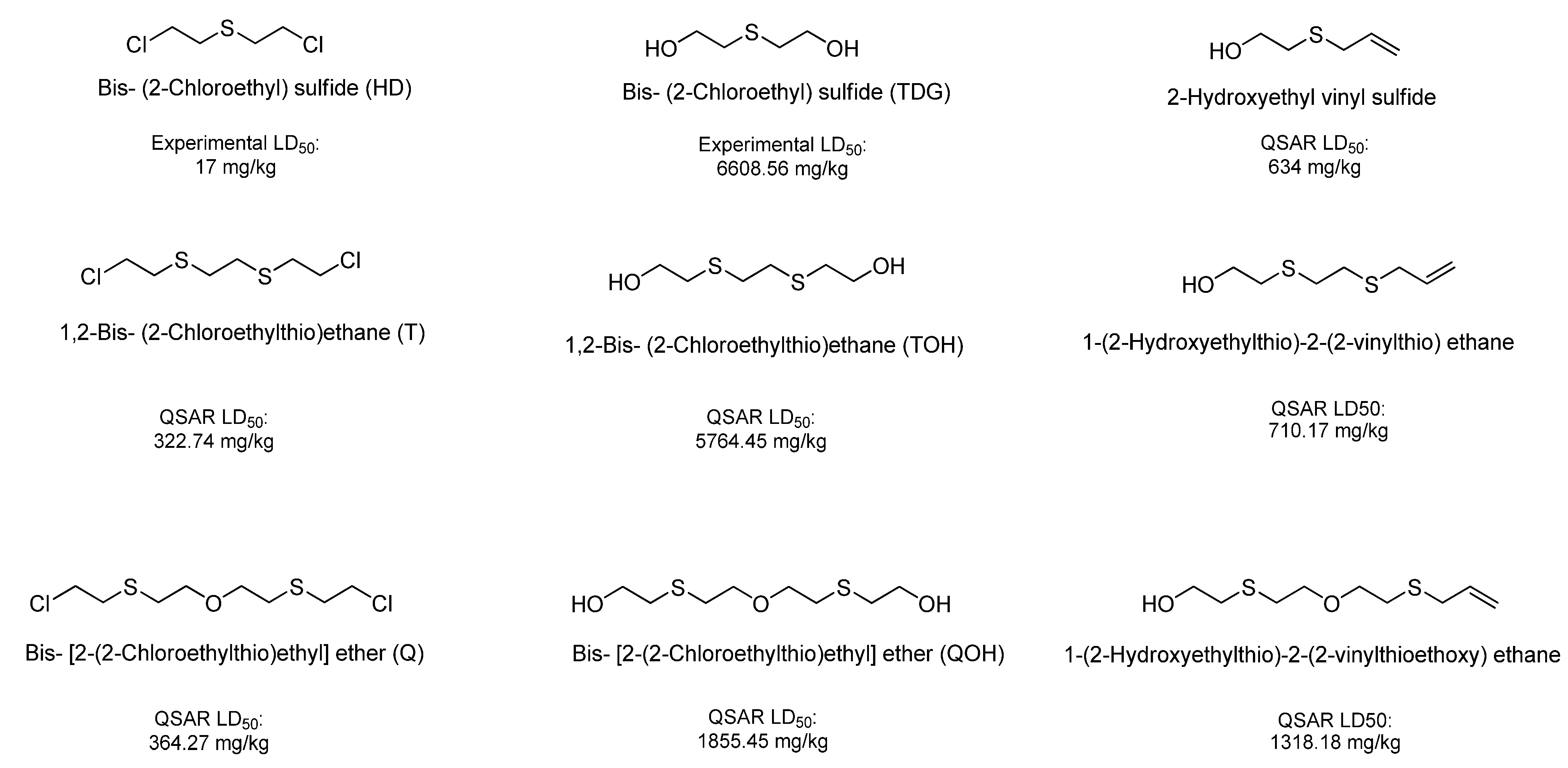
| Bis [2-(2-chloroethylthioethyl) ether] (T) | 816.1 | 197.18 | 110.7 | 166.96 |
| 1,2-Bis(2-chloroethylthio)ethane (Q) | OPS | 630.39 | 369.39 | 93.02 |
| Bis (2-chloroethyl) disulfide | 153.6 | 512.3 | 443.37 | 351.90 |
| Thiodiglycol sulfoxide | 1,400 | 3,524.73 | 2,912.39 | 4,094.75 |
| Bis-[2-(2-hydroxyethylthioethyl)]ether (TOH) | OPS | 1,355.55 | 1,665.45 | 14,272.34 |
| Ethanol, 2,2'-[1,2-ethanediylbis(thio)]-bis- (QOH) | OPS | 1,014.26 | 1,045.92 | 3,506.17 |
| 1-(2-Hydroxyethylthio)-2-(2-vinylthioethoxy)-ethane | OPS | 574.96 | 649.36 | 2,730.21 |
| 1-(2-Hydroxyethylthio)-2-(2-vinylthio)ethane | OPS | 391.58 | 399 | 1,339.93 |
| 2-Hydroxyethyl vinyl sulfide | OPS | 474.79 | 448.1 | 979.11 |
| Bis-(2-chloroethyl) sulfoxide | 67.9 | 449.01 | 265.64 | 577.05 |
| Bis-(2-chloroethyl) sulfone | 227 | 462.07 | 260.21 | 249.89 |
| Thiodiglycol sulfone | 2,800 | 2,715.91 | 1,805.24 | 12,190.02 |
3. Discussion
| Chemical Name | QSAR estimated LD50 (average of TOPKAT, 2D, 3D ADMET predictor and T.E.S.T.) | Toxicity Class * |
|---|---|---|
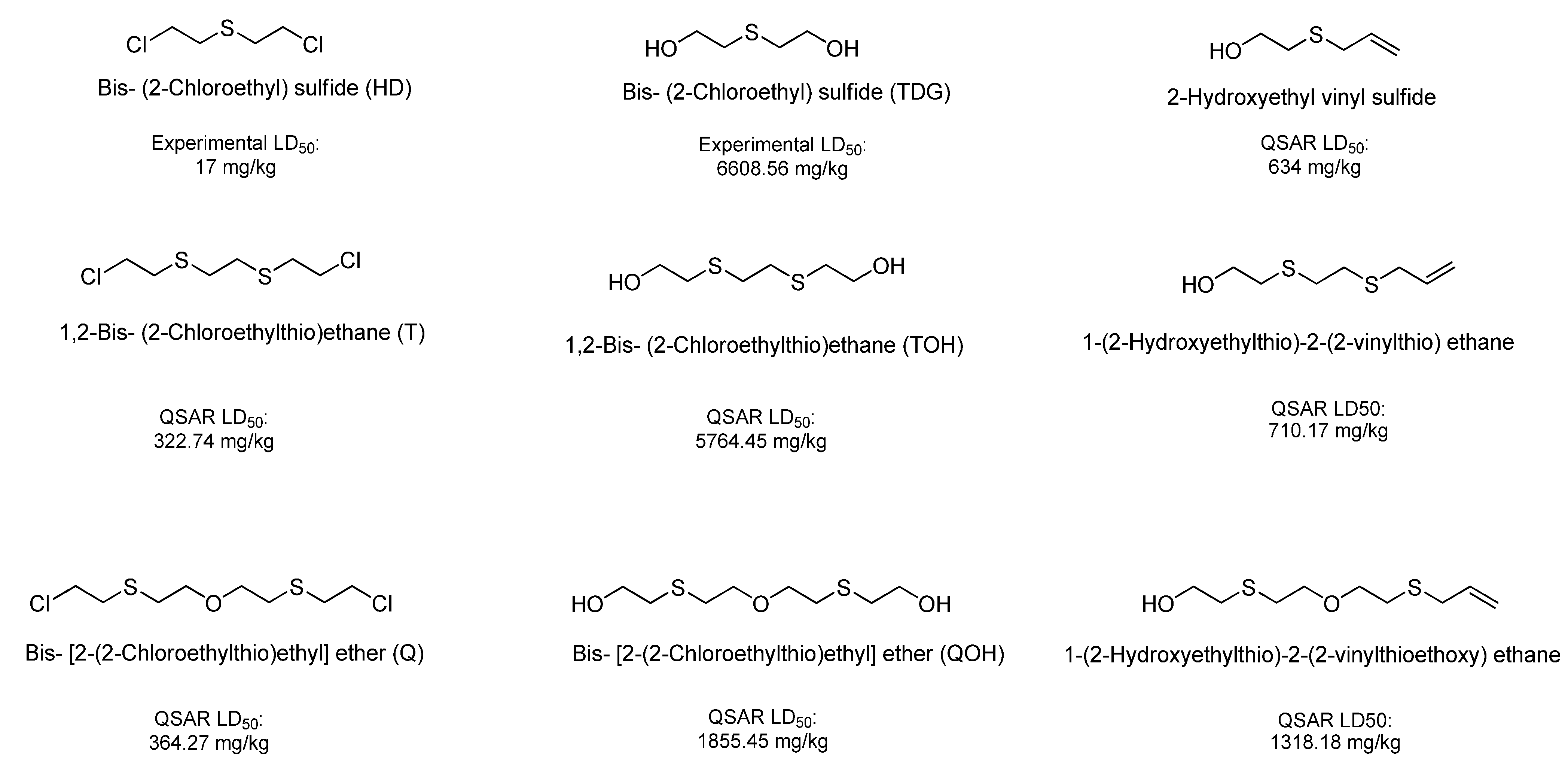
| Bis [2-(2-chloroethylthioethyl) ether] (T) | 322.74 | Very Toxic |
| 1,2-Bis(2-chloroethylthio)ethane (Q) | 364.27 | Very Toxic |
| Bis (2-chloroethyl) disulfide | 365.29 | Very Toxic |
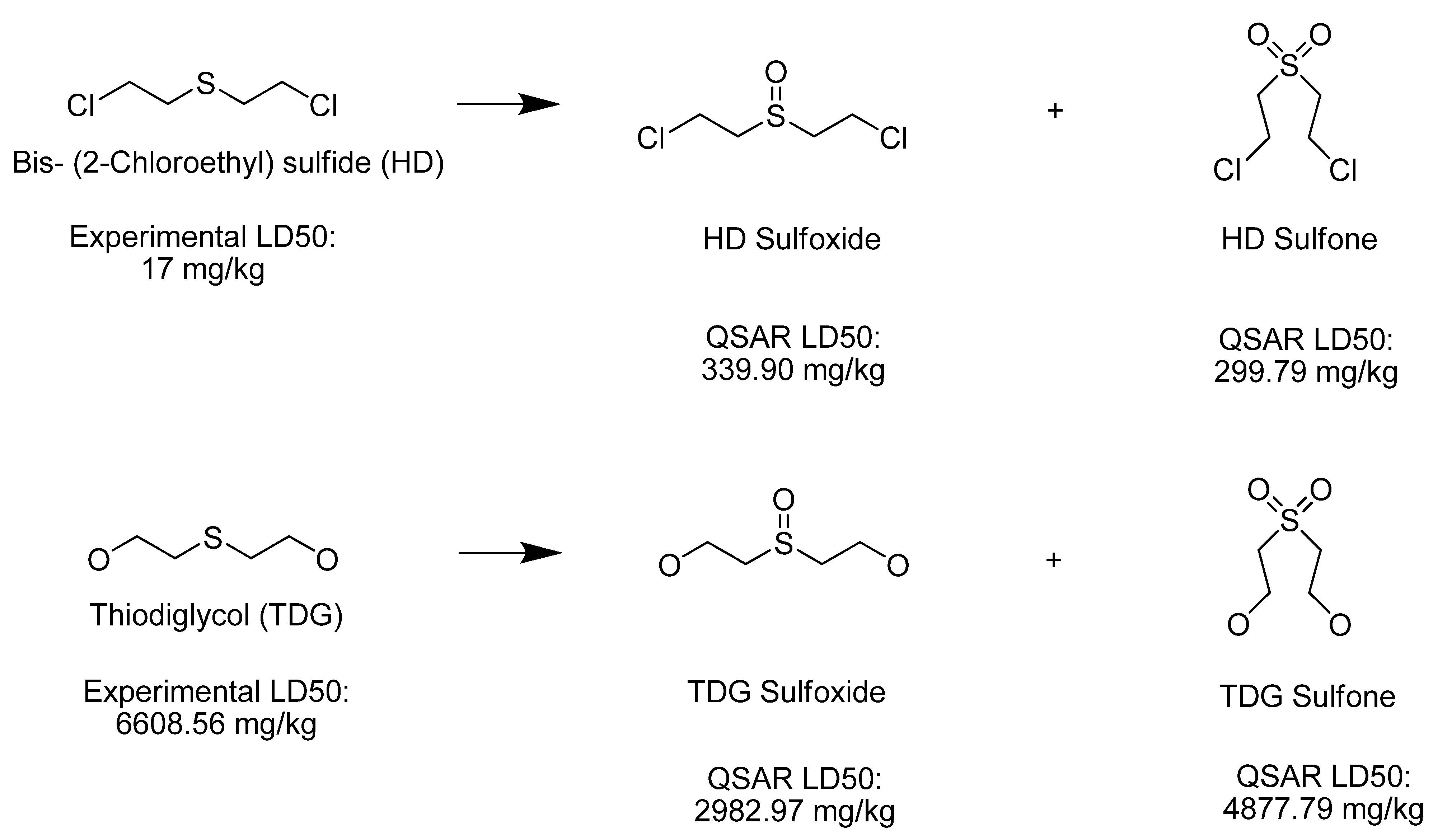
| Thiodiglycol sulfoxide | 2,982.97 | Moderately Toxic |
| Bis-[2-(2-hydroxyethylthioethyl)]ether (TOH) | 5,764.45 | Slightly Toxic |
| Ethanol, 2,2'-[1,2-ethanediylbis(thio)]-bis- (QOH) | 1,855.45 | Moderately Toxic |
| 1-(2-hydroxyethylthio)-2-(2-vinylthioethoxy)-ethane | 1,318.18 | Moderately Toxic |
| 1-(2-Hydroxyethylthio)-2-(2-vinylthio)ethane | 710.17 | Moderately Toxic |
| 2-Hydroxyethyl vinyl sulfide | 634.00 | Moderately Toxic |
| Bis-(2-Chloroethyl) sulfoxide | 339.90 | Very Toxic |
| Bis-(2-Chloroethyl) sulfone | 299.79 | Very Toxic |
| Thiodiglycol sulfone | 4,877.79 | Moderately Toxic |
4. Materials and Methods
4.1. Toxicity Prediction by Komputer Assisted Technology (TOPKAT)
TOPKAT Rat Oral LD50 Model
4.2. ADMET Predictor Rat Oral LD50 Model
4.3. Toxicity Estimation Software Tool (T.E.S.T.)
4.3.1. The Hierarchical Clustering Method
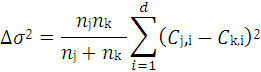
4.3.2. The FDA MDL QSAR Method

4.3.3. The Nearest Neighbor Method
4.4. Applicability Domains (ADs)
4.4.1. AD of the TOPKAT Model
4.4.2. AD of the ADMET Predictor Model
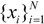 be a descriptor set of model X. For each descriptor, xi(i= 1,…, N), its minimum and maximum values,
be a descriptor set of model X. For each descriptor, xi(i= 1,…, N), its minimum and maximum values, 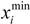 and
and 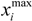 , are determined over the training set of X. A new compound C is said to be within the scope of X if the value of each relevant descriptor ci(i= 1,…, N) calculated for C is contained within the corresponding interval
, are determined over the training set of X. A new compound C is said to be within the scope of X if the value of each relevant descriptor ci(i= 1,…, N) calculated for C is contained within the corresponding interval 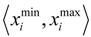 with tolerance equal to 10% of the interval length. Such a compound has its X value typed in black bold font. Otherwise, the compound in question is outside the scope of X and its X value is marked by magenta font (Simulations Plus Inc.).
with tolerance equal to 10% of the interval length. Such a compound has its X value typed in black bold font. Otherwise, the compound in question is outside the scope of X and its X value is marked by magenta font (Simulations Plus Inc.).4.4.3. AD of the Hierarchical Clustering Method
4.4.4. AD of the FDA MDL QSAR Method
4.4.5. AD of the Nearest Neighbor Method
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Agency for Toxic Substances and Disease Registry (ATSDR), Toxicological Profile for Sulfur Mustard; ATSDR: Atlanta, GA, USA, 2003.
- International Agency for Research on Cancer (IARC), Monographs; IARC: Lyon, France, 1987; pp. 403–405.
- Lu, F.; Kacew, S. Basic toxicology. In Fundamentals, Target Organs, and Risk Assessment; Taylor & Francis: New York, NY, USA, 2002. [Google Scholar]
- Balls, M. Why modification of the LD50 test will not be enough. Lab. Anim. 1991, 25, 198–206. [Google Scholar] [CrossRef]
- Fiedler, H.; Hutzinger, O.; Giesy, J.P. Utility of the QSAR modeling system for predicting the toxicity of substances on the European inventory of existing commercial chemicals. Toxicol. Environ. Chem. 1990, 28, 167–188. [Google Scholar] [CrossRef]
- Cronin, M.T.; Dearden, J.C. QSAR in toxicology. 2. Prediction of acute mammalian toxicity and interspecies correlations. QSAR 1995, 14, 117–120. [Google Scholar]
- Greene, N. Computer systems for the prediction of toxicity: An update. Adv. Drug Deliv. Rev. 2002, 54, 417–431. [Google Scholar] [CrossRef]
- Tsakovska, I.; Lessigiarska, I.; Netzeva, T.; Worth, A.P. A mini review of mammalian toxicity (Q)SAR models. QSAR Comb. Sci. 2008, 27, 41–48. [Google Scholar] [CrossRef]
- Demchuk, E.; Ruiz, P.; Chou, S.; Fowler, B.A. SAR/QSAR methods in public health practice. Toxicol. Appl. Pharmacol. 2011, 254, 192–197. [Google Scholar] [CrossRef]
- Schultz, T.W.; Seward, J.R. Health-effects related structure-toxicity relationships: A paradigm for the first decade of the new millennium. Sci. Total Environ. 2000, 249, 73–84. [Google Scholar] [CrossRef]
- Simon-Hettich, B.; Rothfuss, A.; Steger-Hartmann, T. Use of computer-assisted prediction of toxic effects of chemical substances. Toxicology 2006, 224, 156–162. [Google Scholar] [CrossRef]
- Jaworska, J.S.; Comber, M.A.C.; van Leeuwen, C.J. Summary of a workshop on regulatory acceptance of (Q)SARs for human health and environmental endpoints. Environ. Health Perspect. 2003, 111, 1358–1360. [Google Scholar] [CrossRef]
- Ruiz, P.; Mumtaz, M.; Gombar, V. Assessing the toxic effects of ethylene glycol ethers using Quantitative Structure Toxicity Relationship models. Toxicol. Appl. Pharmacol. 2011, 254, 198–205. [Google Scholar] [CrossRef]
- Gombar, V.K. Quantiative structure-activity relationships in toxicology: From fundamentals to applications. Adv. Mol. Toxicol. 1997, 125–139. [Google Scholar]
- Klopman, G.; Zhu, H.; Fuller, M.A.; Saiakhov, R.D. Searching for an enhanced predictive tool for mutagenicity. SAR QSAR Environ. Res. 2004, 15, 251–263. [Google Scholar] [CrossRef]
- Richard, A.M.; Benigni, R. AI and SAR approaches for predicting chemical carcinogenicity: Survey and status report. SAR QSAR Environ. Res. 2002, 13, 1–19. [Google Scholar] [CrossRef]
- Enslein, K. QSTR applications in acute, chronic, and developmental toxicity, and carcinogenicity. Adv. Mol. Toxicol. 1998, 141–164. [Google Scholar]
- Cronin, M.T.; Walker, J.D.; Jaworska, J.S.; Comber, M.H.; Watts, C.D.; Worth, A.P. Use of QSARs in international decision-making frameworks to predict ecologic effects and environmental fate of chemical substances. Environ. Health Perspect. 2003, 111, 1376–1390. [Google Scholar] [CrossRef]
- McKinney, J.D.; Richard, A.; Waller, C.; Newman, M.C.; Gerberick, F. The Practice of Structure Activity Relationships (SAR) in Toxicology. Toxicol. Sci. 2000, 56, 8–17. [Google Scholar] [CrossRef]
- Richard, A.M. Commercial toxicology prediction systems: A regulatory perspective. Toxicol. Lett. 1998, 102–103, 611–616. [Google Scholar] [CrossRef]
- Gombar, V.K.; Jain, D.V.S. Quantification of molecular shape and its correlation with physico-chemical properties. Indian J. Chem. 1987, 24A, 554–555. [Google Scholar]
- Enslein, K. The future of toxicity prediction with QSAR. In Vitro Toxicol. 1993, 6, 163–169. [Google Scholar]
- Ruiz, P.; Faroon, O.; Moudgal, C.J.; Hansen, H.; De Rosa, C.T.; Mumtaz, M. Prediction of the health effects of polychlorinated biphenyls (PCBs) and their metabolites using quantitative structure-activity relationship (QSAR). Toxicol. Lett. 2008, 181, 53–65. [Google Scholar] [CrossRef]
- Gombar, V.K.; Enslein, K. Quantitative Structure-Activity Relationship (QSAR) studies using electronic descriptors calculated from topological and Molecular Orbital (MO) methods. QSAR 1990, 9, 321–325. [Google Scholar]
- Hall, L.H.; Mohney, B.; Kier, L.B. The electrotopological state: Structure information at the atomic level for molecular graphs. J. Chem. Inf. Comput. Sci. 1991, 31, 76–82. [Google Scholar] [CrossRef]
- Zheng, W.; Tropsha, A. Novel Variable Selection Quantitative Structure−Property relationship approach based on the k-Nearest-Neighbor principle. J. Chem. Inf. Comput. Sci. 1999, 40, 185–194. [Google Scholar]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.; McDowell, R.M.; Gramatica, P. Methods for reliability, uncertainty assessment, and applicability evaluations of classifications and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef]
- Martin, T.M.; Harten, P.; Venkatapathy, R.; Das, S.; Young, D.M. A hierarchical clustering methodology for the estimation of toxicity. Toxicol. Mech. Methods 2008, 18, 251–266. [Google Scholar] [CrossRef]
- Tropsha, A.; Golbraikh, A. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 2007, 13, 3494–3504. [Google Scholar] [CrossRef]
- Zhu, H.; Martin, T.M.; Ye, L.; Sedykh, A.; Young, D.M.; Tropsha, A. Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure. Chem. Res. Toxicol. 2009, 22, 1913–1921. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.-D.; Lee, K.-H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Papa, E.; Kovarich, S.; Gramatica, P. On the use of local and global QSPRs for the prediction of physico-chemical properties of polybrominated diphenyl ethers. Mol. Inform. 2011, 30, 232–240. [Google Scholar] [CrossRef]
- Li, J.Z.; Gramatica, P. Classification and virtual screening of androgen receptor antagonists. J. Chem. Inf. Model. 2010, 50, 861–874. [Google Scholar] [CrossRef]
- Sazonovas, A.; Japertas, P.; Didziapetris, R. Estimation of reliability of predictions and model applicability domain evaluation in the analysis of acute toxicity (LD 50). SAR QSAR Environ. Res. 2010, 21, 127–148. [Google Scholar] [CrossRef]
- TOPKAT User Guide Version 6.2; Accelrys: San Diego, CA, USA, 2004.
- T. E. S.T Tool, User’s Guide for T.E.S.T, Version 4.0.; US EPA: Cincinatti, OH, USA, 2011.
- User Manual Version 5.5; Simulation Plus Inc, S. P.: Lancaster, CA, USA, 2011.
- Stedeford, T.; Zhao, Q.J.; Dourson, M.L.; Banasik, M.; Hsu, C.H. The application of non-default uncertainty factors in the US EPA’s Integrated Risk Information System (IRIS). Part I: UFL, UFS, and “Other uncertainty factors”. J. Environ. Sci. Heal. C 2007, 25, 245–279. [Google Scholar] [CrossRef]
- Votano, J.R.; Parham, M.; Hall, L.H.; Kier, L.B.; Oloff, S.; Tropsha, A.; Xie, Q.; Tong, W. Three new consensus QSAR models for the prediction of Ames genotoxicity. Mutagenesis 2004, 19, 365–377. [Google Scholar] [CrossRef]
- Moore, D.R.J.; Breton, R.L.; MacDonald, D.B. A comparison of model performance for six quantitative structure-activity relationship packages that predict acute toxicity to fish. Environ. Toxicol. Chem. 2003, 22, 1799–1809. [Google Scholar] [CrossRef]
- Tong, W.; Hong, H.; Fang, H.; Xie, Q.; Perkins, R. Decision forest: Combining the predictions of multiple independent decision tree models. J. Chem. Inf. Comput. Sci. 2003, 43, 525–531. [Google Scholar] [CrossRef]
- Tunkel, J.; Mayo, K.; Austin, C.; Hickerson, A.; Howard, P. Practical considerations on the use of predictive models for regulatory purposes. Environ. Sci. Technol. 2005, 39, 2188–2199. [Google Scholar] [CrossRef]
- Devillers, J.; Mombelli, E. Evaluation of the OECD QSAR Application Toolbox and Toxtree for estimating the mutagenicity of chemicals. Part 2. alpha-beta unsaturated aliphatic aldehydes. SAR QSAR Environ. Res. 2010, 21, 771–783. [Google Scholar] [CrossRef]
- Fjodorova, N.; Novich, M.; Vrachko, M.; Smirnov, V.; Kharchevnikova, N.; Zholdakova, Z.; Novikov, S.; Skvortsova, N.; Filimonov, D.; Poroikov, V.; et al. Directions in QSAR modeling for regulatory uses in OECD member countries, EU and in Russia. J. Environ. Sci. Heal. C 2008, 26, 201–236. [Google Scholar] [CrossRef]
- Liu, H.; Papa, E.; Gramatica, P. QSAR prediction of estrogen activity for a large set of diverse chemicals under the guidance of OECD principles. Chem. Res. Toxicol. 2006, 19, 1540–1548. [Google Scholar] [CrossRef]
- Canadian Center for Occupational Health & Safety. What is an LD50 and LC50. Available online: http://www.ccohs.ca/oshanswers/chemicals/LD50.html#_1_6 (accessed on 26 July 2012).
- Barratt, M.D. Integrating computer prediction systems with in vitro methods towards a better understanding of toxicology. Toxicol. Lett. 1998, 102–103, 617–621. [Google Scholar] [CrossRef]
- National Research Council (U.S.), Committee on Toxicity Testing and Assessment of Environmental Agents. In Toxicity Testing in the 21st Century: A Vision and a Strategy; National Academies Press: Washington, DC, USA, 2007; pp. xvii, 196.
- Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Series on Testing and Assessment; OECD: Paris, France, 2005; p. 96.
- National Research Council (U.S.), Committee on Applications of Toxicogenomic Technologies to Predictive Toxicology. In Applications of Toxicogenomic Technologies to Predictive Toxicology and Risk Assessment; National Academies Press: Washington, DC, USA, 2007; pp. xxii, 275.
- Schrage, A.; Hempel, K.; Schulz, M.; Kolle, S.N.; van Ravenzwaay, B.; Landsiedel, R. Refinement and reduction of acute oral toxicity testing: A critical review of the use of cytotoxicity data. Atla.-Altern. Lab. Anim. 2011, 39, 273–295. [Google Scholar]
- Worth, A.P.; Bassan, A.; De Bruijn, J.; Gallegos Saliner, A.; Netzeva, T.; Pavan, M.; Patlewicz, G.; Tsakovska, I.; Eisenreich, S. The role of the European Chemicals Bureau in promoting the regulatory use of (Q)SAR methods. SAR QSAR Environ. Res. 2007, 18, 111–125. [Google Scholar]
- Organisation for Economic Co-operation and Development (OECD), Report on the Regulatory Uses and Applications in OECD Member Countries of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models in the Assessment of New and Existing Chemicals, Series on Testing and Assessment; OECD: Paris, France, 2006; p. 79.
- Moudgal, C.J.; Lipscomb, J.C.; Bruce, R.M. Potential health effects of drinking water disinfection by-products using quantitative structure toxicity relationship. Toxicology 2000, 147, 109–131. [Google Scholar] [CrossRef]
- Venkatapathy, R.; Wang, C.Y.; Bruce, R.M.; Moudgal, C. Development of quantitative structure-activity relationship (QSAR) models to predict the carcinogenic potency of chemicals: I. Alternative toxicity measures as an estimator of carcinogenic potency. Toxicol. Appl. Pharmacol. 2009, 234, 209–221. [Google Scholar] [CrossRef]
- Romesburg, H.C. Cluster Analysis for Researchers; LULU Press: North Carolina, NC, USA, 1984. [Google Scholar]
- Contrera, J.F.; Matthews, E.J.; Daniel Benz, R. Predicting the carcinogenic potential of pharmaceuticals in rodents using molecular structural similarity and E-state indices. Regul. Toxicol. Pharmacol. 2003, 38, 243–259. [Google Scholar] [CrossRef]
- Netzeva, T.; Worth, A.P.; Aldenberg, T.; Benigni, R.; Cronin, M.T.; Gramatica, P.; Jaworska, J.; Kahn, S.; Klopman, G.; Marchant, C.A.; et al. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. The report and recommendations of ECVAM Workshop 52. Altern. Lab. Anim. 2005, 33, 1–19. [Google Scholar]
- Schultz, T.W.; Hewitt, M.; Netzeva, T.I.; Cronin, M.T.D. Assessing Applicability Domains of Toxicological QSARs: Definition, Confidence in Predicted Values, and the Role of Mechanisms of Action. QSAR Comb. Sci. 2007, 26, 238–254. [Google Scholar] [CrossRef]
- Tetko, I.V.; Sushko, I.; Pandey, A.K.; Zhu, H.; Tropsha, A.; Papa, E.; Oberg, T.; Todeschini, R.; Fourches, D.; Varnek, A. Critical Assessment of QSAR Models of Environmental Toxicity against Tetrahymenapyriformis: Focusing on Applicability Domain and Overfitting by Variable Selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [Google Scholar] [CrossRef] [Green Version]
- Roy, P.P.; Kovarich, S.; Gramatica, P. QSAR Model Reproducibility and Applicability: A Case Study of Rate Constants of Hydroxyl Radical Reaction Models Applied to PolybrominatedDiphenyl Ethers and (Benzo-)Triazoles. J. Comput. Chem. 2011, 32, 2386–2396. [Google Scholar] [CrossRef]
- Pohl, H.R.; Chou, C.H.; Ruiz, P.; Holler, J.S. Chemical risk assessment and uncertainty associated with extrapolation across exposure duration. Regul.Toxicol. Pharmacol. 2010, 57, 18–23. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ruiz, P.; Begluitti, G.; Tincher, T.; Wheeler, J.; Mumtaz, M. Prediction of Acute Mammalian Toxicity Using QSAR Methods: A Case Study of Sulfur Mustard and Its Breakdown Products. Molecules 2012, 17, 8982-9001. https://doi.org/10.3390/molecules17088982
Ruiz P, Begluitti G, Tincher T, Wheeler J, Mumtaz M. Prediction of Acute Mammalian Toxicity Using QSAR Methods: A Case Study of Sulfur Mustard and Its Breakdown Products. Molecules. 2012; 17(8):8982-9001. https://doi.org/10.3390/molecules17088982
Chicago/Turabian StyleRuiz, Patricia, Gino Begluitti, Terry Tincher, John Wheeler, and Moiz Mumtaz. 2012. "Prediction of Acute Mammalian Toxicity Using QSAR Methods: A Case Study of Sulfur Mustard and Its Breakdown Products" Molecules 17, no. 8: 8982-9001. https://doi.org/10.3390/molecules17088982