Flavonoids and Other Compounds from Ouratea ferruginea (Ochnaceae) as Anticancer and Chemopreventive Agents

Abstract
:1. Introduction
2. Results and Discussion

2.1. Biological Activity

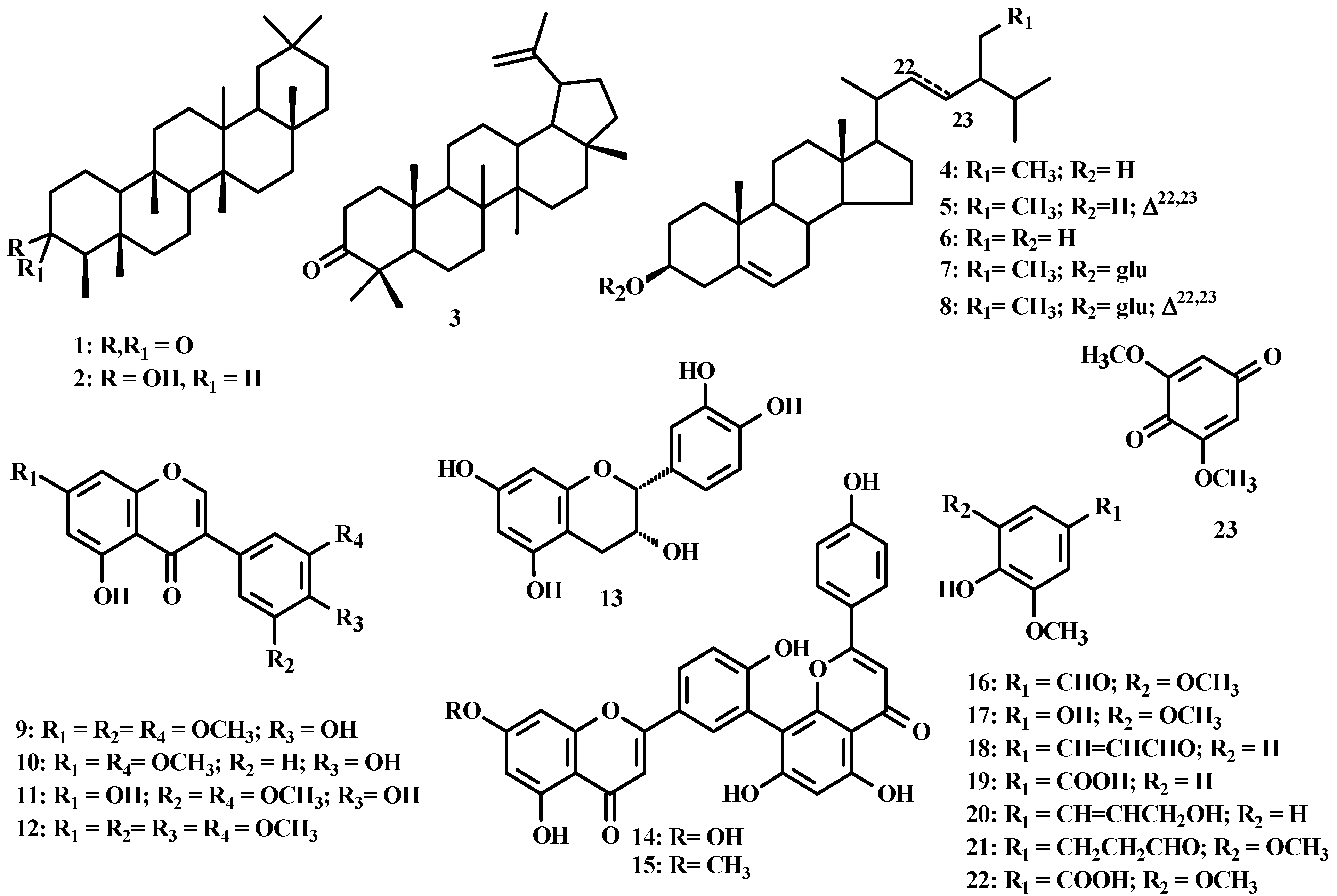{kind=link}
| Compounds [0.08 mg/mL] | ECOD activity | GST activity |
|---|---|---|
| 5,4′-OH-7,3′,5′-OMe-isoflavone(9) | 66.9 ±27.7 | 25.3 ±5.3 |
| 7,3′-Dimethoxy-5,4′-dihydroxyisoflavone (10) | 77.8 ± 11.6 | ND * |
| 5,7,4′-Trihydroxy-3′,5′-dimethoxyisoflavone (11) | 53.5 ± 12.4 | 32.2 ±1.3 |
| Sequoiaflavone (15) | 24.8 ± 1.2 | 77.4 ± 6.7 |
3. Experimental
3.1. Instrumentation and Reagents
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Preparation of Liver Fractions
3.5. Preparation of Samples
3.6. CYP Inhibition Assays
3.7. GST Inhibition Assays
4. Conclusions
Acknowledgments
References
- Salvador, G.S.; Cervi, A.C.; Santos dos, E.P. Flórula do morro dos perdidos, serra de araçatuba, estado do paraná, brasil: Ochnaceae. Estud. Biol. 2005, 27, 13–17. [Google Scholar]
- Suzart, L.R.; Daniel, J.F.S.; Carvalho, M.G.; Kaplan, M.A.C. Biodiversidade flavonoídica e aspectos farmacológicos em espécies dos gêneros Ouratea e Luxemburgia (Ochnaceae). Quim. Nova 2007, 30, 984–987. [Google Scholar] [CrossRef]
- de Carvalho, M.G.; Velandia, J.R.; de Oliveira, J.C.C.; Echevarria, A.; Braz-Filho, R.; Grynberg, N.F. Chemical structure, Cytotoxic Antitumour Activities of Biflavonoids from Brazilian Ouratea (Ochnaceae). In Phytochemical and Pharmacologiy II of the Series “Recent Progress in Medicinal Plants”; Majumdar, D.K., Govil, J.N., Singh, V.K., Eds.; SCI Tech Publishing LLC: Houston, TX, USA, 2002; Volume 8, pp. 77–92. [Google Scholar]
- Moon, Y.J.; Wang, X.; Morris, M.E. Dietary flavonoids: Effects on xenobiotic and carcinogen metabolism. Toxicol. in Vitro 2006, 20, 187–210. [Google Scholar] [CrossRef]
- Wang, J.F.; Zhang, C.C.; Chou, K.C.; Wei, D.Q. Structure of cytochrome P450s and personalized drug. Curr. Med. Chem. 2009, 16, 232–244. [Google Scholar] [CrossRef]
- Hayes, J.D.; Pulford, D.J. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 521–600. [Google Scholar]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar]
- Mahato, S.B.; Kundu, A.P. 13C NMR spectra of pentacyclic triterpenoids-A compilation and some salient features. Phytochemistry 1994, 37, 1517–1575. [Google Scholar]
- Salazar, G.C.M.; Silva, G.D.F.; Duarte, L.P.; Vieira-Filho, S.A.; Lula, I.S. Two epimeric friedelane triterpenes isolated from Maytenus truncata Reiss: 1H and 13C chemical shift assignments. Magn. Reson. Chem. 2000, 38, 977–980. [Google Scholar]
- Rubinstein, I.; Goad, L.J.; Clague, A.D.H.; Mulheirn, L.J. The 220 MHz NMR spectra of phytosterols. Phytochemistry 1976, 15, 195–200. [Google Scholar]
- Benedek, B.; Weniger, B.; Parejo, I.; Bastida, J.; Arango, G.J.; Lobstein, A.; Codina, C. Antioxidant activity of isoflavones and biflavones isolated from Godoya antioquiensis. Arzneim.-Forsch. 2006, 56, 661–664. [Google Scholar]
- McCormick, S.; Robson, K.; Bohm, B. Flavonoids of Wyethia angustifolia and W. helenioides. Phytochemistry 1986, 25, 1723–1726. [Google Scholar]
- Tahara, S.; Moriyama, M.; Ingham, J.L.; Mizutani, J. 5,7,3′,4′,5′-pentaoxygenated and 2′,6′-diprenylated isoflavones from Piscidia erythrina. Phytochemistry 1991, 30, 2769–2775. [Google Scholar]
- Pettit, G.R.; Melody, N.; Thornhill, A.; Knight, J.C.; Groy, T.L.; Herald, C.L. Antineoplastic agents. 579. synthesis and cancer cell growth evaluation of E-Stilstatin 3: A resveratrol structural modification. J. Nat. Prod. 2009, 72, 1637–1642. [Google Scholar]
- Harasawa, A.; Tagashira, A. Isolation of 2,6-dimethoxy-1,4-benzoquinone from Hydrangea (Hydrangea macrophylla Seringe var. otaksa Makino) and its deodorant activity against methyl mercaptan. Biosci. Biotech. Biochem. 1994, 58, 2073–2074. [Google Scholar] [CrossRef]
- Kim, H.; Ralph, J. NMR analysis of lignins in CAD-deficient plants. Part 1. Incorporation of hydroxycinnamaldehydes and hydroxybenzaldehydes into lignins. Org. Biomol. Chem. 2003, 1, 268–281. [Google Scholar] [CrossRef]
- Harborne, J.B.; Mabry, T.J. The Flavonoids: Advances in Research; Chapman and Hall: New York, NY, USA, 1982; p. 116. [Google Scholar]
- Markham, K.R.; Sheppard, C.; Geiger, H. 13C NMR Studies of some naturally occurring amentoflavone and hinokiflavone biflavonoids. Phytochemistry 1987, 26, 3335–3337. [Google Scholar] [CrossRef]
- Sivaraman, L.; Leatham, M.P.; Yee, J.; Wilkens, L.R.; Lau, A.F.; Le Marchand, L. CYP1A1 genetic polymorphisms and in situ colorectal cancer. Cancer Res. 1994, 54, 3692–3695. [Google Scholar]
- Raunio, H.; Rautio, A.; Gullstén, H.; Pelkonen, O. Polymorphisms of CYP2A6 and its practical consequences. Br. J. Clin. Pharmacol. 2001, 52, 357–363. [Google Scholar] [CrossRef]
- Udomsuk, L.; Jarukamjorn, K. Factors influencing regulation of CYP2B expression. Thai. Pharm. Health Sci. J. 2009, 4, 524–531. [Google Scholar]
- Guengerich, F.P.; Shimada, T. Oxidation of toxic and carcinogenic chemicals by human cytochrome P-450 enzymes. Chem. Res. Toxicol. 1991, 4, 391–407. [Google Scholar] [CrossRef]
- Moon, J.Y.; Lee, D.W.; Park, K.H. Inhibition of 7-ethoxycoumarin O-deethylase activity in rat liver microsomes by naturally occurring flavonoids: Structure-activity relationships. Xenobiotica 1998, 28, 117–126. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Papakyriakou, A.; Vourloumis, D.; Spandidos, D.A. Comparative CYP1A1 and CYP1B1 substrate and inhibitor profile of dietary flavonoids. Bioorg. Med. Chem. 2011, 19, 2842–2849. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Papakyriakou, A.; Vourloumis, D.; Tsatsakis, A.M.; Spandidos, D.A. Dietary flavonoids in cancer therapy and prevention: Substrates and inhibitors of cytochrome P450 CYP1 enzymes. Pharmacol. Ther. 2010, 126, 9–20. [Google Scholar]
- Chan, H.Y.; Wang, H.; Leung, L.K. The red clover (Trifolium pratense) isoflavone biochanin A modulates the biotransformation pathways of 7, 12- dimethylbenz[a]anthracene. Br. J. Nutr. 2003, 90, 87–92. [Google Scholar]
- Leung, H.Y.; Wang, Y.; Chan, H.Y.; Leung, L.K. Developing a high-throughput system for the screening of cytochrome P450 1A1-inhibitory polyphenols. Toxicol. In Vitro 2007, 21, 996–1002. [Google Scholar]
- Wen, X.; Walle, U.K.; Walle, T. 5,7-Dimethoxyflavone downregulates CYP1A1 expression and benzo[a]pyrene-induced DNA binding in Hep G2 cells. Carcinogenesis 2005, 26, 803–809. [Google Scholar]
- Surh, Y.J.; Lee, E.; Lee, J.M. Chemoprotective properties of some pungent ingredients present in red pepper and ginger. Mutat. Res. 1998, 402, 259–267. [Google Scholar] [CrossRef]
- Ghazali, R.; Waring, R.H. Effects of flavonoids on glutathione-S-transferase in human blood platelets, rat liver, rat kidney, and HT-29 colon adenocarcinoma cell-lines: Potential in drug metabolism and chemoprevention. Med. Sci. Res. 1999, 27, 449–451. [Google Scholar]
- Volm, V. Multidrug resistance and its reversal. Anticancer Res. 1998, 18, 2905–2918. [Google Scholar]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Ann. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Greenlee, W.F.; Poland, A. An improved assay of 7-ethoxycoumarin O-deethylase activity: Induction of hepatic enzyme activity in C57BL/6J and DBA/2J mice by phenobarbital, 3-methylcholanthrene and 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Pharmacol. Exp. Ther. 1978, 205, 596–605. [Google Scholar]
- Peterson, G.L. A simplification of the protein assay method of lowry et al. which is more generally applicable. Anal. Biochem. 1977, 83, 346–356. [Google Scholar]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-Transferases: The first enzymatic in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar]
- Sample Availability: Samples of the Ouratea ferruginea are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fidelis, Q.C.; Castro, R.N.; Guilhon, G.M.S.P.; Rodrigues, S.T.; De Salles, C.M.C.; De Salles, J.B.; De Carvalho, M.G. Flavonoids and Other Compounds from Ouratea ferruginea (Ochnaceae) as Anticancer and Chemopreventive Agents. Molecules 2012, 17, 7989-8000. https://doi.org/10.3390/molecules17077989
Fidelis QC, Castro RN, Guilhon GMSP, Rodrigues ST, De Salles CMC, De Salles JB, De Carvalho MG. Flavonoids and Other Compounds from Ouratea ferruginea (Ochnaceae) as Anticancer and Chemopreventive Agents. Molecules. 2012; 17(7):7989-8000. https://doi.org/10.3390/molecules17077989
Chicago/Turabian StyleFidelis, Queli C., Rosane N. Castro, Giselle M. S. P. Guilhon, Silvane T. Rodrigues, Cristiane M. C. De Salles, João B. De Salles, and Mario G. De Carvalho. 2012. "Flavonoids and Other Compounds from Ouratea ferruginea (Ochnaceae) as Anticancer and Chemopreventive Agents" Molecules 17, no. 7: 7989-8000. https://doi.org/10.3390/molecules17077989