The Feasibility of Enzyme Targeted Activation for Amino Acid/Dipeptide Monoester Prodrugs of Floxuridine; Cathepsin D as a Potential Targeted Enzyme

Abstract
:1. Introduction
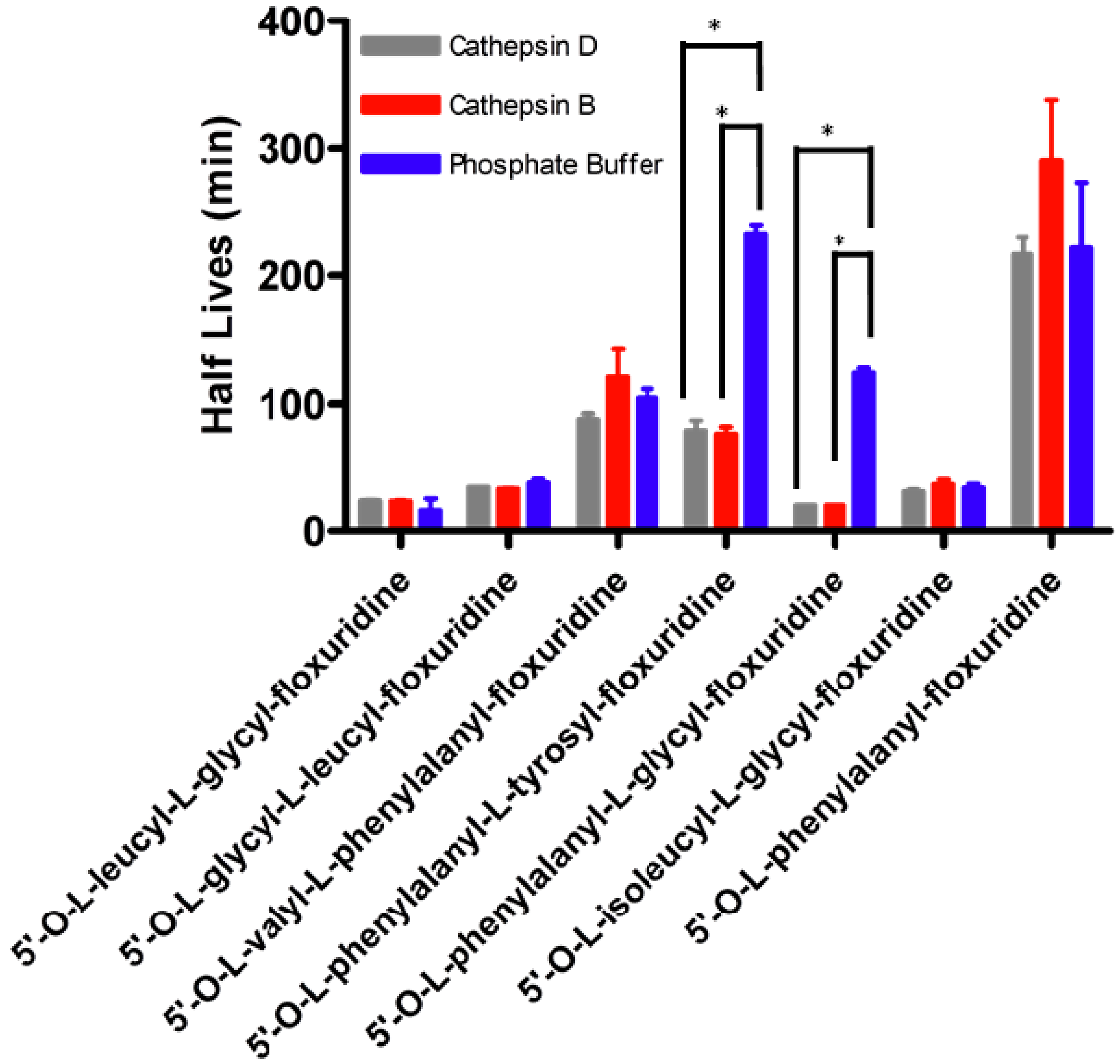
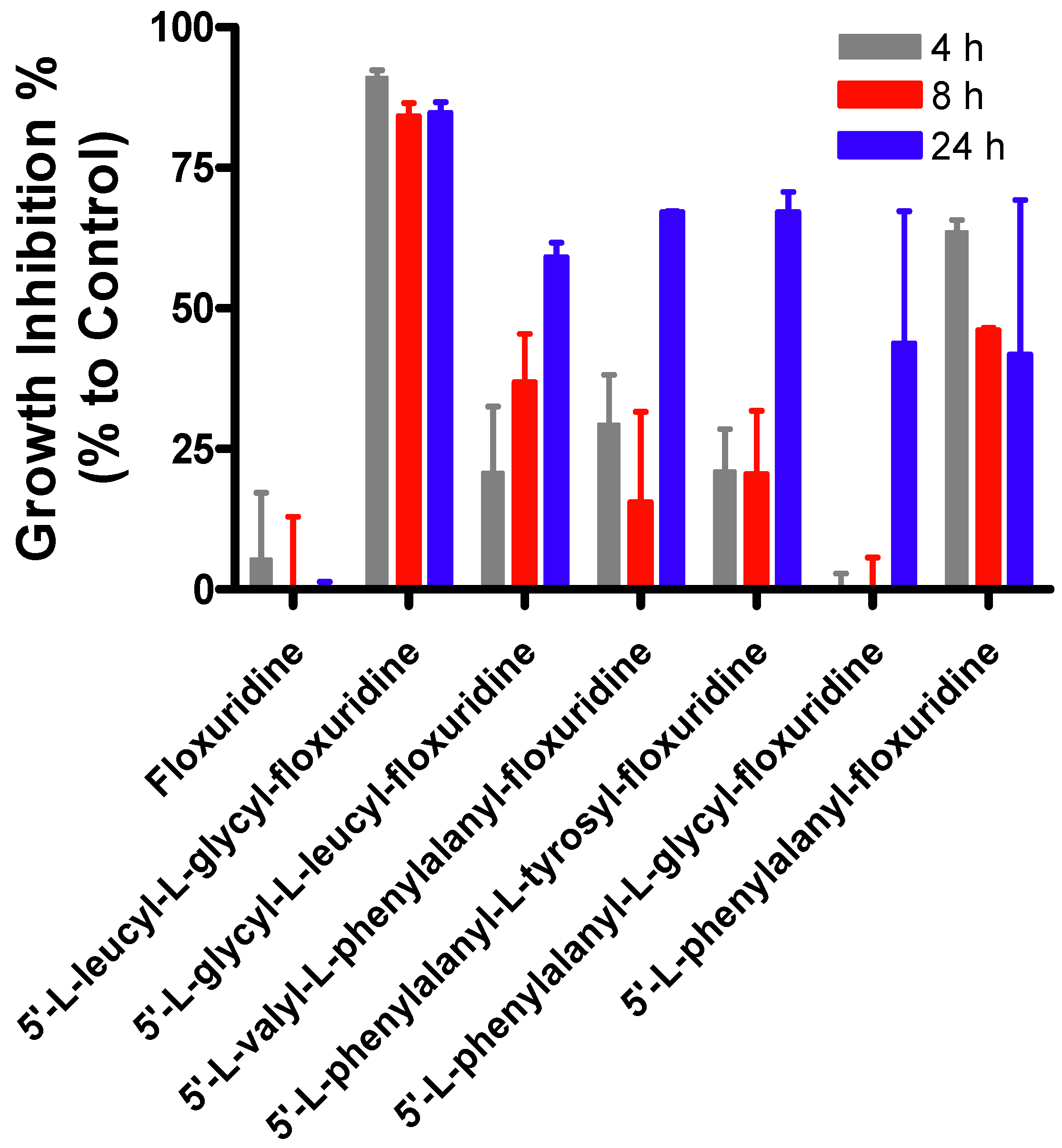
2. Results and Discussion

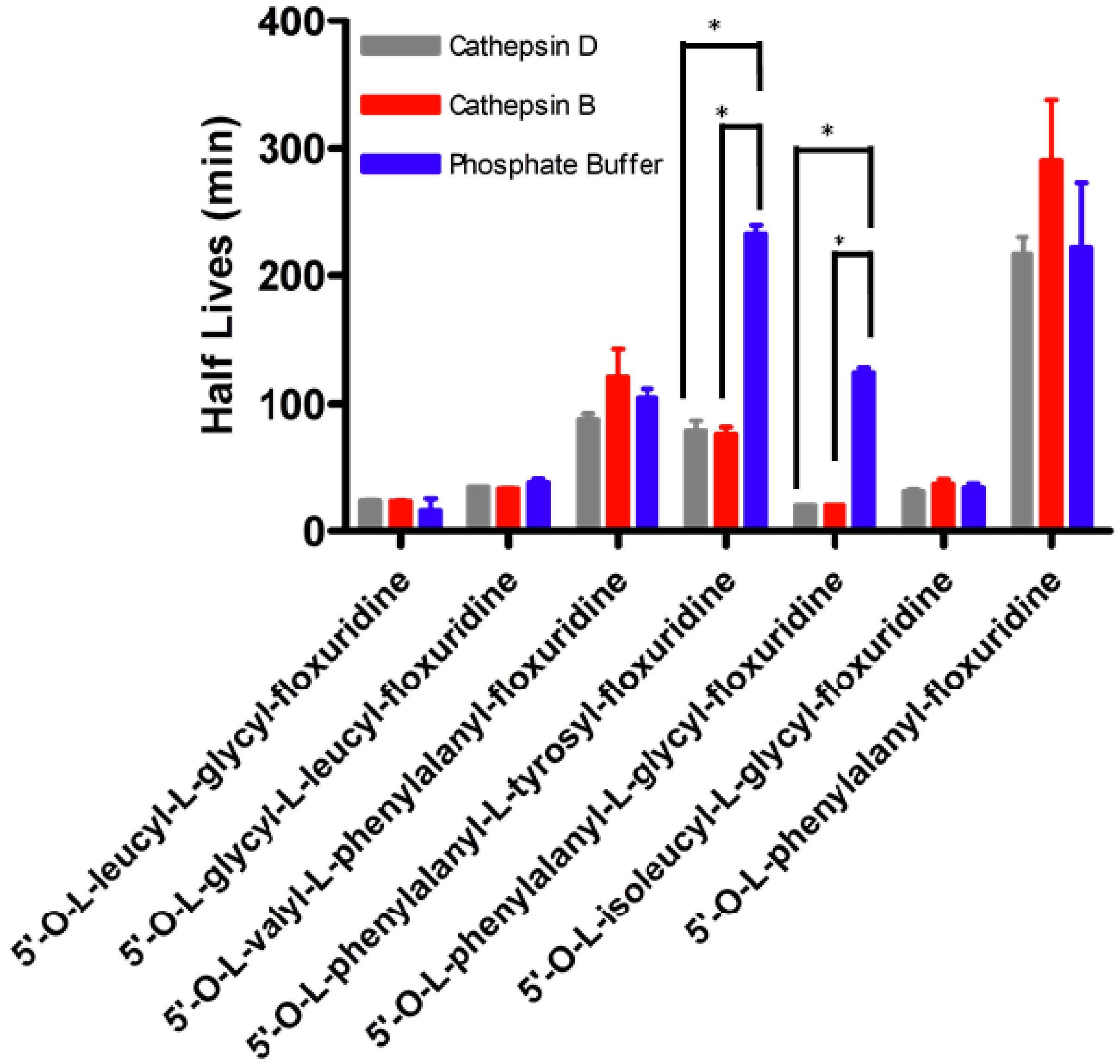{kind=link}
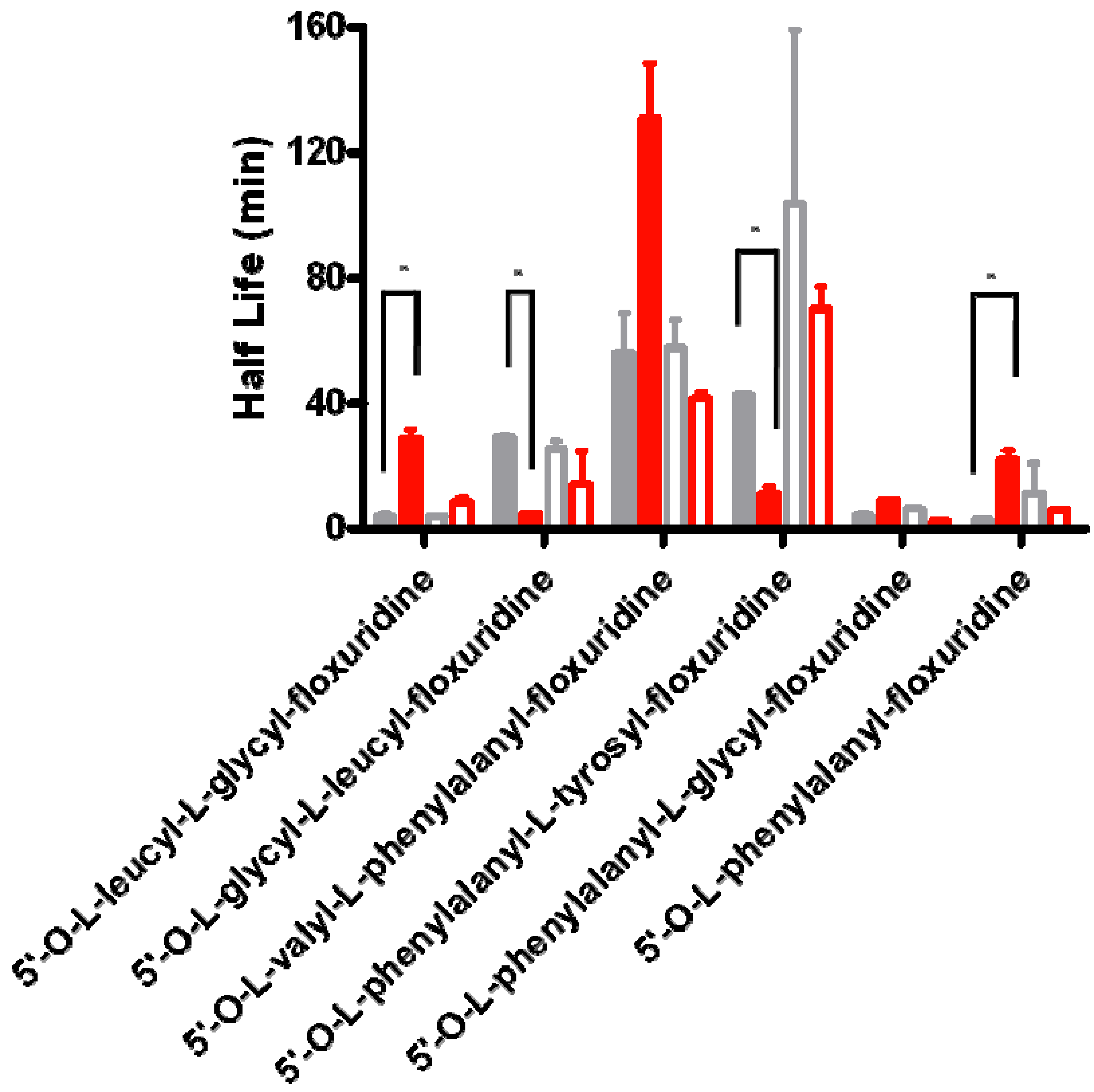{kind=link}
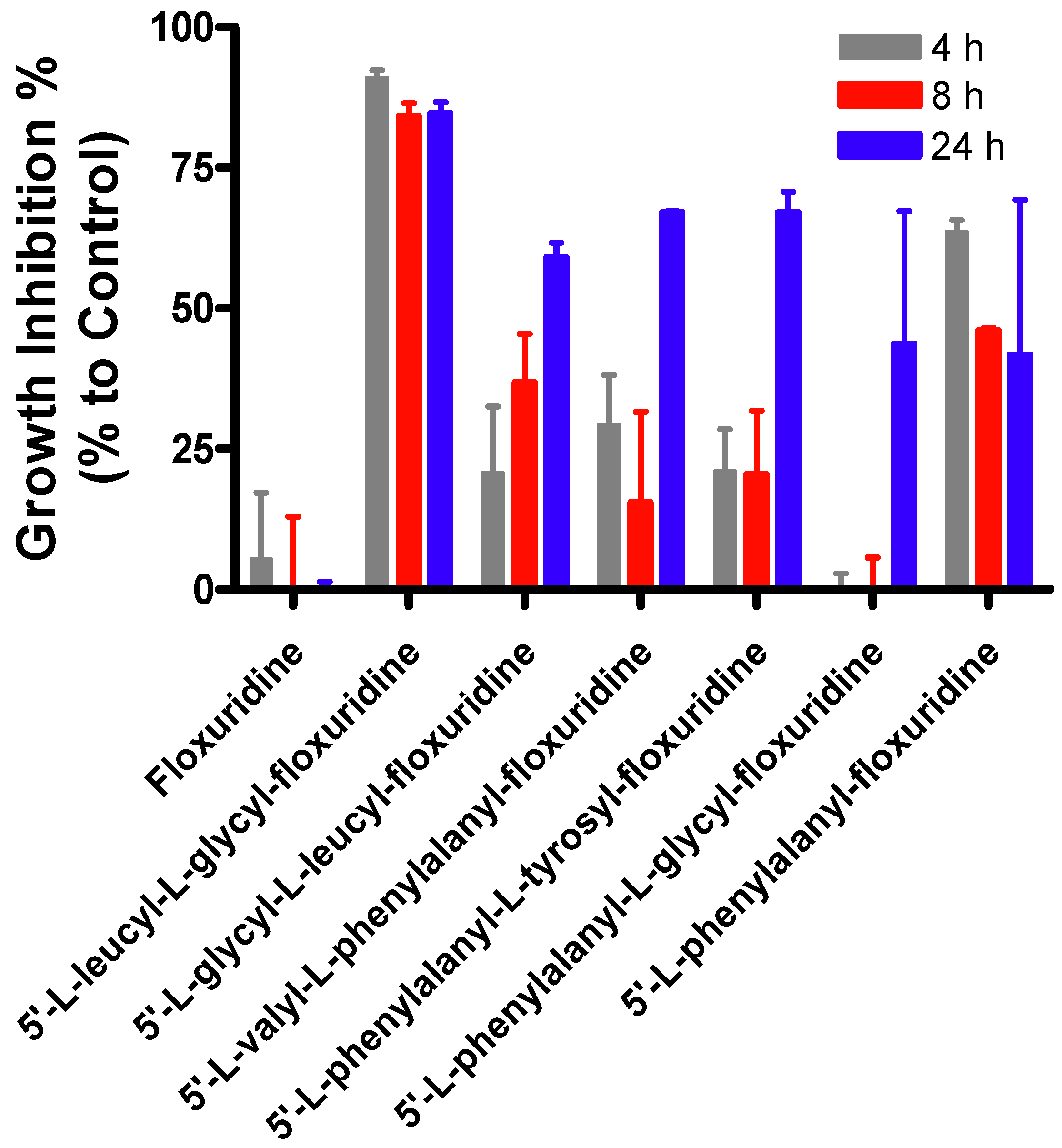{kind=link}
| Enzyme | Capan-2 | AsPC-1 |
|---|---|---|
| Cathepsin B | 1526.6 | 38 |
| Zinc Metalloproteinase | 1057.9 | 48 |
| Cathepsin H | 913.4 | 58.6 |
| Cathepsin D | 891.2 | 3.7 |
| gamma-Glutamyl Hydrolase | 840.6 | 20.3 |
| Calpastatin (CAST) | 746.2 | 28.3 |
| Aminopeptidase B | 726.2 | 14.9 |
| Leucine Aminopeptidase | 700.7 | 15.1 |
| Cathepsin C | 593.4 | 17.6 |
| Puromycin Sensitive Aminopeptidase | 530.2 | 4.9 |
| Prolylcarboxypeptidase | 516.3 | 16.8 |
| Aminoacylase 1 | 511.9 | 16.2 |
| Cytochrome P450 | 432.4 | 13.5 |
| N-Acylaminoacyl-Peptide Hydrolase | 335.5 | 9.7 |
| Tripeptidyl peptidase II (TPP2) | 296.7 | 36.4 |
| Cathepsin U | 288.4 | 7.4 |
| Aminopeptidase P | 264 | 328.2 |
| Dipeptidylpeptidase III (DPP3) | 257.5 | 4.2 |
| Leucine Aminopeptidase | 238 | 20.7 |
| Mitochondrial Intermediate Peptidase (MIPEP) | 206.8 | 35 |
| Aminopeptidase PILS (APPILS) | 195.1 | 2.4 |
| UDP-N-Acetylglucosamine-2-Epimerase (GNE) | 180.4 | 42.3 |
| Caspase 6 | 179 | 0.8 |
| Sentrin-Specific Protease (SENP2) | 176 | 116.5 |
| Glycosylasparaginase | 176 | 130.1 |
| Carboxypeptidase D | 168.2 | 57.3 |
| Transmembrane Protease(TMPRSS4) | 167.8 | 3.9 |
| Cysteine Protease | 167.1 | 17.1 |
| Aspartyl Aminopeptidase (DNPEP) | 163.6 | 24.7 |
| Peptidase D (PEPD) | 163.2 | 3.3 |
| Heparan Sulfate (glucosamine) 3-O-Sulfotransferase 1 (HS3ST1) | 157.1 | 5.3 |
| Aspartylglucosaminidase | 121.5 | 28.6 |
| GPI Transamidase | 119.6 | 112.6 |
| SentrinSUMO-specific protease 3 (SENP3) | 117.8 | 111.9 |
| Caspase 3 | 114.8 | 5.1 |
| Carboxy-Terminal Hydrolase | 111.7 | 56 |
| Caspase 8 | 108.8 | 49.8 |
| Phosphatidylcholine 2-acylhydrolase (cPLA2) | 108.3 | 3.3 |
| Polypeptide 5 (RPS6KA5) | 103.3 | 32.1 |
| beta-Site APP-Cleaving Enzyme | 100.2 | 178.3 |
| Protease 5 (isopeptidase T) (USP5) | 99.9 | 84 |
| Arginyl Aminopeptidase (Aminopeptidase B)-Like 1 (RNPEPL1) | 99.3 | 16.1 |
| SentrinSUMO-Specific Protease (SENP1) | 98.8 | 48.3 |
| Carboxypeptidase M | 97.4 | 7.8 |
| Carboxypeptidase D | 96.9 | 4.9 |
| 3-Phosphoinositide Dependent Protein Kinase-1 (PDPK1) | 94.7 | 53.3 |
| Putative Metalloglycoprotease | 94 | 55.4 |
| Aspartylglucosaminidase (AGA) | 93.2 | 12.5 |
| Carboxypeptidase 1 | 92.8 | 15 |
| Matrix Metalloproteinase 14 (MMP14) | 88.5 | 10.3 |
| Half Life (min) | ||||||
|---|---|---|---|---|---|---|
| Prodrug | Phosphate Buffer pH 7.4 | Capan-2 Cell Homogenate | Cathepsin D Inhibitor | Aminopeptidase Inhibitor | TPP 2 Inhibitor | DPP 3 Inhibitor |
| 5'-O-L-leucyl-L-glycylfloxuridine | 23.1 ± 4.1 § | 3.9 ± 1.1 | 7.9 ± 0.7 | 14.0 ± 4.3 | 5.6 ± 0.6 | 32.2 ± 9.3 |
| 5'-O-L-glycyl-L-leucylfloxuridine | 35.7 ± 0.9 § | 29.2 ± 0.7 | 32.2 ± 0.1 | 33.0 ± 0.4 | 21.0 ± 0.9 | 26.5 ± 17.5 |
| 5'-O-L-valyl-L-phenylalanyl-floxuridine | 104.7 ± 7.0 § | 56.2 ±12.8 | 76.4 ±6.1 | 93.6 ± 29.3 | 46.1 ± 1.9 | 67.7 ± 15.1 |
| 5'-O-L-phenylalanyl-L-tyrosyl-floxuridine | 233.9 ± 6.6 § | 42.8 ± 0.0 | 105.7 ± 12.8 * | 54.7 ± 2.8 | 42.2 ± 1.3 | 47.2 ± 3.6 |
| 5'-O-L-phenylalanyl-L-glycyl-floxuridine | 132.1 ± 10.2 § | 4.3 ± 0.9 | 10.2 ± 3.0 | 25.7 ± 0.4 * | 19.6 ± 0.4 * | 30.1 ± 10.0 |
| 5'-O-L-phenylalanylfloxuridine | 187.0 ± 19.0 § | 3.0 ± 0.1 | 60.6 ± 4.6* | 48.7 ± 0.37 * | 15.3 ± 0.6 * | 28.9 ± 10.7 |

3. Experimental
3.1. Materials
3.2. Floxuridine Prodrug Synthesis
3.3. Cell Culture
3.4. Affymetrix Oligonucleotide Array
3.5. Data Analysis
3.6. HPLC Analysis
3.7. Cell Proliferation Assays
4. Conclusions
Acknowledgements
- Sample Availability: Not available.
References and Notes
- Escalona-Benz, E.; Jockovich, M.E.; Murray, T.G.; Hayden, B.; Hernandez, E.; Feuer, W.; Windle, J.J. Combretastatin A-4 prodrug in the treatment of a murine model of retinoblastoma. Invest. Ophthalmol. Vis. Sci. 2005, 46, 8–11. [Google Scholar]
- Moody, T.W.; Mantey, S.A.; Pradhan, T.K.; Schumann, M.; Nakagawa, T.; Martinez, A.; Fuselier, J.; Coy, D.H.; Jensen, R.T. Development of high affinity camptothecin-bombesin conjugates that have targeted cytotoxicity for bombesin receptor-containing tumor cells. J. Biol. Chem. 2004, 279, 23580–23589. [Google Scholar]
- Senter, P.D.; Beam, K.S.; Mixan, B.; Wahl, A.F. Identification and activities of human carboxylesterases for the activation of CPT-11, a clinically approved anticancer drug. Bioconjug. Chem. 2001, 12, 1074–1080. [Google Scholar]
- Bras, A.P.; Sitar, D.S.; Aoki, F.Y. Comparative bioavailability of acyclovir from oral valacyclovir and acyclovir in patients treated for recurrent genital herpes simplex virus infection. Can. J. Clin. Pharmacol. 2001, 8, 207–211. [Google Scholar]
- Curran, M.; Noble, S. Valganciclovi. Drugs 2001, 61, 1145–1150, discussion 1151-1152.. [Google Scholar]
- Linden, K.; Zhou, X.X.; Stable, L. Validation of microdialysis sampling for oral availability studies by means of a new ganciclovir prodrug. Pharmacol. Toxicol. 2002, 90, 297–302. [Google Scholar]
- Tolle-Sander, S.; Lentz, K.A.; Maeda, D.Y.; Coop, A.; Polli, J.E. Increased acyclovir oral bioavailability via a bile acid conjugate. Mol. Pharm. 2004, 1, 40–48. [Google Scholar]
- Song, X.; Lorenzi, P.L.; Landowski, C.P.; Vig, B.S.; Hilfinger, J.M.; Amidon, G.L. Amino acid ester prodrugs of the anticancer agent gemcitabine: Synthesis, bioconversion, metabolic bioevasion, and hPEPT1-mediated transport. Mol. Pharm. 2005, 2, 157–167. [Google Scholar]
- Nielsen, C.U.; Andersen, R.; Brodin, B.; Frokjaer, S.; Taub, M.E.; Steffansen, B. Dipeptide model prodrugs for the intestinal oligopeptide transporter. Affinity for and transport via hPepT1 in the human intestinal Caco-2 cell line. J. Control. Release 2001, 76, 129–138. [Google Scholar] [CrossRef]
- Thomsen, A.E.; Friedrichsen, G.M.; Sorensen, A.H.; Andersen, R.; Nielsen, C.U.; Brodin, B.; Begtrup, M.; Frokjaer, S.; Steffansen, B. Prodrugs of purine and pyrimidine analogues for the intestinal di/tri-peptide transporter PepT1: affinity for hPepT1 in Caco-2 cells, drug release in aqueous media and in vitro metabolism. J. Control. Release 2003, 86, 279–292. [Google Scholar]
- Vabeno, J.; Nielsen, C.U.; Ingebrigtsen, T.; Lejon, T.; Steffansen, B.; Luthman, K. Dipeptidomimetic ketomethylene isosteres as pro-moieties for drug transport via the human intestinal di-/tripeptide transporter hPEPT1: Design, synthesis, stability, and biological investigations. J. Med. Chem. 2004, 47, 4755–4765. [Google Scholar]
- Grem, J.L. 5-Fluorouracil: Forty-plus and still ticking. A review of its preclinical and clinical development. Invest. New Drugs 2000, 18, 299–313. [Google Scholar] [CrossRef]
- Parker, W.B.; Cheng, Y.C. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol. Ther. 1990, 48, 381–395. [Google Scholar]
- Willmore, E.; Durkacz, B.W. Cytotoxic mechanisms of 5-fluoropyrimidines. Relationships with poly(ADP-ribose) polymerase activity, DNA strand breakage and incorporation into nucleic acids. Biochem. Pharmacol. 1993, 46, 205–211. [Google Scholar]
- Laskin, J.D.; Evans, R.M.; Slocum, H.K.; Burke, D.; Hakala, M.T. Basis for natural variation in sensitivity to 5-fluorouracil in mouse and human cells in culture. Cancer Res. 979, 39, 383–390. [Google Scholar]
- Yamada, M.; Nakagawa, H.; Fukushima, M.; Shimizu, K.; Hayakawa, T.; Ikenaka, K. In vitro study on intrathecal use of 5-fluoro-2'-deoxyuridine (FdUrd) for meningeal dissemination of malignant brain tumors. J. Neurooncol. 1998, 37, 115–121. [Google Scholar]
- Birnie, G.D.; Heidelberger, C. In vitro synthesis of acidsoluble thymine compounds by human neoplastic tissues. Cancer Res. 1963, 23, 420–430. [Google Scholar]
- Bencharit, S.; Morton, C.L.; Howard-Williams, E.L.; Danks, M.K.; Potter, P.M.; Redinbo, M.R. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nat. Struct. Biol. 2002, 9, 337–342. [Google Scholar]
- Oosterhoff, D.; Pinedo, H.M.; van der Meulen, I.H.; de Graaf, M.; Sone, T.; Kruyt, F.A.; van Beusechem, V.W.; Haisma, H.J.; Gerritsen, W.R. Secreted and tumour targeted human carboxylesterase for activation of irinotecan. Br. J. Cancer 2002, 87, 659–664. [Google Scholar]
- Wu, M.H.; Yan, B.; Humerickhouse, R.; Dolan, M.E. Irinotecan activation by human carboxylesterases in colorectal adenocarcinoma cells. Clin. Cancer Res. 2002, 8, 2696–2700. [Google Scholar]
- Xu, G.; Zhang, W.; Ma, M.K.; McLeod, H.L. Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin. Cancer Res. 2002, 8, 2605–2611. [Google Scholar]
- Harel, M.; Hyatt, J.L.; Brumshtein, B.; Morton, C.L.; Wadkins, R.M.; Silman, I.; Sussman, J.L.; Potter, P.M. The 3D structure of the anticancer prodrug CPT-11 with Torpedo californica acetylcholinesterase rationalizes its inhibitory action on AChE and its hydrolysis by butyrylcholinesterase and carboxylesterase. Chem. Biol. Interact. 2005, 157–158, 153–157. [Google Scholar]
- Harel, M.; Hyatt, J.L.; Brumshtein, B.; Morton, C.L.; Yoon, K.J.; Wadkins, R.M.; Silman, I.; Sussman, J.L.; Potter, P.M. The crystal structure of the complex of the anticancer prodrug 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothecin (CPT-11) with Torpedo californica acetylcholinesterase provides a molecular explanation for its cholinergic action. Mol. Pharmacol. 2005, 67, 1874–1881. [Google Scholar]
- Kondoh, K.; Tsuji, N.; Kamagata, C.; Sasaki, M.; Kobayashi, D.; Yagihashi, A.; Watanabe, N. A novel aspartic protease gene, ALP56, is up-regulated in human breast cancer independently from the cathepsin D gene. Breast Cancer Res. Treat. 2003, 78, 37–44. [Google Scholar]
- Steinfeld, S.; Maho, A.; Chaboteaux, C.; Daelemans, P.; Pochet, R.; Appelboom, T.; Kiss, R. Prolactin up-regulates cathepsin B and D expression in minor salivary glands of patients with Sjogren’s syndrome. Lab. Invest. 2000, 80, 1711–1720. [Google Scholar]
- Skrzydlewska, E.; Sulkowska, M.; Wincewicz, A.; Koda, M.; Sulkowski, S. Evaluation of serum cathepsin B and D in relation to clinicopathological staging of colorectal cancer. World J. Gastroenterol. 2005, 11, 4225–4229. [Google Scholar]
- Dumartin, L.; Whiteman, H.J.; Weeks, M.E.; Hariharan, D.; Dmitrovic, B.; Iacobuzio-Donahue, C.A.; Brentnall, T.A.; Bronner, M.P.; Feakins, R.M.; Timms, J.F.; et al. AGR2 is a novel surface antigen that promotes the dissemination of pancreatic cancer cells through regulation of cathepsins B and D. Cancer Res. 2011, 71, 7091–7102. [Google Scholar]
- Whiteman, H.J.; Weeks, M.E.; Dowen, S.E.; Barry, S.; Timms, J.F.; Lemoine, N.R.; Crnogorac-Jurcevic, T. The role of S100P in the invasion of pancreatic cancer cells is mediated through cytoskeletal changes and regulation of cathepsin D. Cancer Res. 2007, 67, 8633–8642. [Google Scholar]
- Chauhan, S.S.; Goldstein, L.J.; Gottesman, M.M. Expression of cathepsin L in human tumors. Cancer Res. 1991, 51, 1478–1481. [Google Scholar]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135. [Google Scholar]
- Rochefort, H.; Liaudet-Coopman, E. Cathepsin D in cancer metastasis: A protease and a ligand. Apmis 1999, 107, 86–95. [Google Scholar]
- Turk, B.; Stoka, V.; Rozman-Pungercar, J.; Cirman, T.; Droga-Mazovec, G.; Oresic, K.; Turk, V. Apoptotic pathways: involvement of lysosomal proteases. Biol. Chem. 2002, 383, 1035–1044. [Google Scholar]
- Shen, J.; Person, M.D.; Zhu, J.; Abbruzzese, J.L.; Li, D. Protein expression profiles in pancreatic adenocarcinoma compared with normal pancreatic tissue and tissue affected by pancreatitis as detected by two-dimensional gel electrophoresis and mass spectrometry. Cancer Res. 2004, 64, 9018–9026. [Google Scholar]
- Abbott, D.E.; Margaryan, N.V.; Jeruss, J.S.; Khan, S.; Kaklamani, V.; Winchester, D.J.; Hansen, N.; Rademaker, A.; Khalkhali-Ellis, Z.; Hendrix, M.J. Reevaluating cathepsin D as a biomarker for breast cancer: serum activity levels versus histopathology. Cancer Biol. Ther. 2010, 9, 23–30. [Google Scholar]
- Baurain, R.; Masquelier, M.; Deprez-De Campeneere, D.; Trouet, A. Amino acid and dipeptide derivatives of daunorubicin. 2. Cellular pharmacology and antitumor activity on L1210 leukemic cells in vitro and in vivo. J. Med. Chem. 1980, 23, 1171–1174. [Google Scholar] [CrossRef]
- Briozzo, P.; Morisset, M.; Capony, F.; Rougeot, C.; Rochefort, H. In vitro degradation of extracellular matrix with Mr 52,000 cathepsin D secreted by breast cancer cells. Cancer Res. 1988, 48, 3688–3692. [Google Scholar]
- Keppler, D.; Fondaneche, M.C.; Dalet-Fumeron, V.; Pagano, M.; Burtin, P. Immunohistochemical and biochemical study of a cathepsin B-like proteinase in human colonic cancers. Cancer Res. 1988, 48, 6855–6862. [Google Scholar]
- Maciewicz, R.A.; Wardale, R.J.; Etherington, D.J.; Paraskeva, C. Immunodetection of cathepsins B and L present in and secreted from human pre-malignant and malignant colorectal tumour cell lines. Int. J. Cancer 1989, 43, 478–486. [Google Scholar]
- Masquelier, M.; Baurain, R.; Trouet, A. Amino acid and dipeptide derivatives of daunorubicin. 1. Synthesis, physicochemical properties, and lysosomal digestion. J. Med. Chem. 1980, 23, 1166–1170. [Google Scholar] [CrossRef]
- Breistol, K.; Hendriks, H.R.; Berger, D.P.; Langdon, S.P.; Fiebig, H.H.; Fodstad, O. The antitumour activity of the prodrug N-L--leucyl-doxorubicin and its parent compound doxorubicin in human tumour xenografts. Eur. J. Cancer 1998, 34, 1602–1606. [Google Scholar]
- Quinney, S.K.; Sanghani, S.P.; Davis, W.I.; Hurley, T.D.; Sun, Z.; Murry, D.J.; Bosron, W.F. Hydrolysis of capecitabine to 5'-deoxy-5-fluorocytidine by human carboxylesterases and inhibition by loperamide. J. Pharmacol. Exp. Ther. 2005, 313, 1011–1016. [Google Scholar]
- Schuller, J.; Cassidy, J.; Dumont, E.; Roos, B.; Durston, S.; Banken, L.; Utoh, M.; Mori, K.; Weidekamm, E.; Reigner, B. Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother. Pharmacol. 2000, 45, 291–297. [Google Scholar]
- Tsukamoto, Y.; Kato, Y.; Ura, M.; Horii, I.; Ishikawa, T.; Ishitsuka, H.; Sugiyama, Y. Investigation of 5-FU disposition after oral administration of capecitabine, a triple-prodrug of 5-FU, using a physiologically based pharmacokinetic model in a human cancer xenograft model: Comparison of the simulated 5-FU exposures in the tumour tissue between human and xenograft model. Biopharm. Drug Dispos. 2001, 22, 1–14. [Google Scholar]
- Hu, M.; Subramanian, P.; Mosberg, H.I.; Amidon, G.L. Use of the peptide carrier system to improve the intestinal absorption of L-alpha-methyldopa: Carrier kinetics, intestinal permeabilities,and in vitro hydrolysis of dipeptidyl derivatives of L--alpha-methyldopa. Pharm. Res. 1989, 6, 66–70. [Google Scholar]
- Han, H.; de Vrueh, R.L.; Rhie, J.K.; Covitz, K.M.; Smith, P.L.; Lee, C.P.; Oh, D.M.; Sadee, W.; Amidon, G.L. 5'-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm. Res. 1998, 15, 1154–1159. [Google Scholar]
- Landowski, C.P.; Song, X.; Lorenzi, P.L.; Hilfinger, J.M.; Amidon, G.L. Floxuridine amino acid ester prodrugs: enhancing Caco-2 permeability and resistance to glycosidic bond metabolism. Pharm. Res. 2005, 22, 1510–1518. [Google Scholar]
- Landowski, C.P.; Vig, B.S.; Song, X.; Amidon, G.L. Targeted delivery to PEPT1-overexpressing cells: Acidic, basic, and secondary floxuridine amino acid ester prodrugs. Mol. Cancer Ther. 2005, 4, 659–667. [Google Scholar]
- Lorenzi, P.L.; Landowski, C.P.; Song, X.; Borysko, K.Z.; Breitenbach, J.M.; Kim, J.S.; Hilfinger, J.M.; Townsend, L.B.; Drach, J.C.; Amidon, G.L. Amino acid ester prodrugs of 2-bromo-5,6-dichloro-1-(beta-D-ribofuranosyl)benzimidazole enhance metabolic stability in vitro and in vivo. J. Pharmacol. Exp. Ther. 2005, 314, 883–890. [Google Scholar]
- Tsume, Y.; Hilfinger, J.M.; Amidon, G.L. Enhanced cancer cell growth inhibition by dipeptide prodrugs of floxuridine: Increased transporter affinity and metabolic stability. Mol. Pharm. 2008, 5, 717–727. [Google Scholar]
- Tsume, Y.; Vig, B.S.; Sun, J.; Landowski, C.P.; Hilfinger, J.M.; Ramachandran, C.; Amidon, G.L. Enhanced absorption and growth inhibition with amino acid monoester prodrugs of floxuridine by targeting hPEPT1 transporters. Molecules 2008, 13, 1441–1454. [Google Scholar]
- Vig, B.S.; Lorenzi, P.J.; Mittal, S.; Landowski, C.P.; Shin, H.C.; Mosberg, H.I.; Hilfinger, J.M.; Amidon, G.L. Amino acid ester prodrugs of floxuridine: Synthesis and effects of structure, stereochemistry, and site of esterification on the rate of hydrolysis. Pharm. Res. 2003, 20, 1381–1388. [Google Scholar]
- Berg, T.; Gjoen, T.; Bakke, O. Physiological functions of endosomal proteolysis. Biochem. J. 1995, 307, 313–326. [Google Scholar]
- Claus, V.; Jahraus, A.; Tjelle, T.; Berg, T.; Kirschke, H.; Faulstich, H.; Griffiths, G. Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J. Biol. Chem. 1998, 273, 9842–9851. [Google Scholar]
- Kageshita, T.; Yoshii, A.; Kimura, T.; Maruo, K.; Ono, T.; Himeno, M.; Nishimura, Y. Biochemical and immunohistochemical analysis of cathepsins B, H, L and D in human melanocytic tumour. Arch. Dermatol. Res. 1995, 287, 266–272. [Google Scholar]
- Li, W.; Yuan, X.M. Increased expression and translocation of lysosomal cathepsins contribute to macrophage apoptosis in atherogenesis. Ann. NY Acad. Sci. 2004, 1030, 427–433. [Google Scholar]
- Roberg, K.; Ollinger, K. Oxidative stress causes relocation of the lysosomal enzyme cathepsin D with ensuing apoptosis in neonatal rat cardiomyocytes. Am. J. Pathol. 1998, 152, 1151–1156. [Google Scholar]
- Sameni, M.; Elliott, E.; Ziegler, G.; Fortgens, P.H.; Dennison, C.; Sloane, B.F. Cathepsin B and D are localized at the surface of human breast cancer cells. Pathol. Oncol. Res. 1995, 1, 43–53. [Google Scholar]
- Gronborg, M.; Kristiansen, T.Z.; Iwahori, A.; Chang, R.; Reddy, R.; Sato, N.; Molina, H.; Jensen, O.N.; Hruban, R.H.; Goggins, M.G.; et al. Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol. Cell. Proteomics 2006, 5, 157–171. [Google Scholar]
- Reid, W.A.; Valler, M.J.; Kay, J. Immunolocalization of cathepsin D in normal and neoplastic human tissues. J. Clin. Pathol. 1986, 39, 1323–1330. [Google Scholar]
- Deville-Bonne, D.; El Amri, C.; Meyer, P.; Chen, Y.; Agrofoglio, L.A.; Janin, J. Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: Structural and catalytic properties. Antivir. Res. 2010, 86, 101–120. [Google Scholar]
- Kohchi, Y.; Hattori, K.; Oikawa, N.; Mizuguchi, E.; Isshiki, Y.; Aso, K.; Yoshinari, K.; Shirai, H.; Miwa, M.; Inagaki, Y.; et al. Design and synthesis of novel prodrugs of 2'-deoxy-2'-methylidenecytidine activated by membrane dipeptidase overexpressed in tumor tissues. Bioorg. Med. Chem. Lett. 2007, 17, 2241–2245. [Google Scholar]
- Major Jourden, J.L.; Cohen, S.M. Enzymatic activation of a matrix metalloproteinase inhibitor. Chem. Commun. (Camb) 2010, 46, 1241–1243. [Google Scholar] [CrossRef]
- Major Jourden, J.L.; Cohen, S.M. Hydrogen peroxide activated matrix metalloproteinase inhibitors: A prodrug approach. Angew. Chem. Int. Ed. Engl. 2010, 49, 6795–6797. [Google Scholar]
- Tabata, T.; Katoh, M.; Tokudome, S.; Nakajima, M.; Yokoi, T. Identification of the cytosolic carboxylesterase catalyzing the 5'-deoxy-5-fluorocytidine formation from capecitabine in human liver. Drug Metab. Dispos. 2004, 32, 1103–1110. [Google Scholar]
- Alevizos, I.; Mahadevappa, M.; Zhang, X.; Ohyama, H.; Kohno, Y.; Posner, M.; Gallagher, G.T.; Varvares, M.; Cohen, D.; Kim, D.; et al. Oral cancer in vivo gene expression profiling assisted by laser capture microdissection and microarray analysis. Oncogene 2001, 20, 6196–6204. [Google Scholar]
- Elie, B.T.; Gocheva, V.; Shree, T.; Dalrymple, S.A.; Holsinger, L.J.; Joyce, J.A. Identification and pre-clinical testing of a reversible cathepsin protease inhibitor reveals anti-tumor efficacy in a pancreatic cancer model. Biochimie 2010, 92, 1618–1624. [Google Scholar]
- Nagler, D.K.; Kruger, S.; Kellner, A.; Ziomek, E.; Menard, R.; Buhtz, P.; Krams, M.; Roessner, A.; Kellner, U. Up-regulation of cathepsin X in prostate cancer and prostatic intraepithelial neoplasia. Prostate 2004, 60, 109–119. [Google Scholar]
- Turk, V.; Turk, B.; Guncar, G.; Turk, D.; Kos, J. Lysosomal cathepsins: structure, role in antigen processing and presentation, and cancer. Adv. Enzyme Regul. 2002, 42, 285–303. [Google Scholar]
- Devetzi, M.; Scorilas, A.; Tsiambas, E.; Sameni, M.; Fotiou, S.; Sloane, B.F.; Talieri, M. Cathepsin B protein levels in endometrial cancer: Potential value as a tumour biomarker. Gynecol. Oncol. 2009, 112, 531–536. [Google Scholar]
- Lah, T.T.; Nanni, I.; Trinkaus, M.; Metellus, P.; Dussert, C.; De Ridder, L.; Rajcevic, U.; Blejec, A.; Martin, P.M. Toward understanding recurrent meningioma: the potential role of lysosomal cysteine proteases and their inhibitors. J. Neurosurg. 2010, 112, 940–950. [Google Scholar]
- Szumilo, J.; Burdan, F.; Zinkiewicz, K.; Dudka, J.; Klepacz, R.; Dabrowski, A.; Korobowicz, E. Expression of syndecan-1 and cathepsins D and K in advanced esophageal squamous cell carcinoma. Folia Histochem. Cytobiol. 2009, 47, 571–578. [Google Scholar]
- Linebaugh, B.E.; Sameni, M.; Day, N.A.; Sloane, B.F.; Keppler, D. Exocytosis of active cathepsin B enzyme activity at pH 7.0, inhibition and molecular mass. Eur. J. Biochem. 1999, 264, 100–109. [Google Scholar] [CrossRef]
- Pohl, J.; Davinic, S.; Blaha, I.; Strop, P.; Kostka, V. Chromophoric and fluorophoric peptide substrates cleaved through the dipeptidyl carboxypeptidase activity of cathepsin B. Anal. Biochem. 1987, 165, 96–101. [Google Scholar]
- Polgar, L.; Csoma, C. Dissociation of ionizing groups in the binding cleft inversely controls the endo- and exopeptidase activities of cathepsin B. J. Biol. Chem. 1987, 262, 14448–14453. [Google Scholar]
- Ju, B.G.; Kim, W.S. Upregulation of cathepsin D expression in the dedifferentiating salamander limb regenerates and enhancement of its expression by retinoic acid. Wound Repair Regen. 1998, 6, 349–357. [Google Scholar]
- Offermann, M.K.; Chlebowski, J.F.; Bond, J.S. Action of cathepsin D on fructose-1,6-bisphosphate aldolase. Biochem. J. 1983, 211, 529–534. [Google Scholar]
- Hulkower, K.I.; Butler, C.C.; Linebaugh, B.E.; Klaus, J.L.; Keppler, D.; Giranda, V.L.; Sloane, B.F. Fluorescent microplate assay for cancer cell-associated cathepsin B. Eur. J. Biochem. 2000, 267, 4165–4170. [Google Scholar]
- Carvelli, L.F.; Bannoud, N.; Aguilera, C.A.; Morales, C.R.; Sosa, M.A. Castration induces changes in the cation-dependent mannose-6-phosphate receptor in rat epididymis: Possible implications in secretion of lysosomal enzymes. J. Cell. Biochem. 2010, 110, 1101–1110. [Google Scholar]
- Landowski, C.P.; Lorenzi, P.L.; Song, X.; Amidon, G.L. Nucleoside ester prodrug substrate specificity of liver carboxylesterase. J. Pharmacol. Exp. Ther. 2006, 316, 572–580. [Google Scholar]
- Murray, H.; Turner, A.J.; Kenny, A.J. The aminopeptidase activity in the human T-cell lymphoma line (Jurkat) is not at the cell surface and is not aminopeptidase N (CD-13). Biochem. J. 1994, 298, 353–360. [Google Scholar]
- Landowski, C.P.; Anderle, P.; Sun, D.; Sadee, W.; Amidon, G.L. Transporter and ion channel gene expression after Caco-2 cell differentiation using 2 different microarray technologies. AAPS J. 2004, 6, e21. [Google Scholar]
- Sun, D.; Lennernas, H.; Welage, L.S.; Barnett, J.L.; Landowski, C.P.; Foster, D.; Fleisher, D.; Lee, K.D.; Amidon, G.L. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm. Res. 2002, 19, 1400–1416. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsume, Y.; Amidon, G.L. The Feasibility of Enzyme Targeted Activation for Amino Acid/Dipeptide Monoester Prodrugs of Floxuridine; Cathepsin D as a Potential Targeted Enzyme. Molecules 2012, 17, 3672-3689. https://doi.org/10.3390/molecules17043672
Tsume Y, Amidon GL. The Feasibility of Enzyme Targeted Activation for Amino Acid/Dipeptide Monoester Prodrugs of Floxuridine; Cathepsin D as a Potential Targeted Enzyme. Molecules. 2012; 17(4):3672-3689. https://doi.org/10.3390/molecules17043672
Chicago/Turabian StyleTsume, Yasuhiro, and Gordon L. Amidon. 2012. "The Feasibility of Enzyme Targeted Activation for Amino Acid/Dipeptide Monoester Prodrugs of Floxuridine; Cathepsin D as a Potential Targeted Enzyme" Molecules 17, no. 4: 3672-3689. https://doi.org/10.3390/molecules17043672