Novel Steroidal Components from the Underground Parts of Ruscus aculeatus L.




Abstract
:1. Introduction
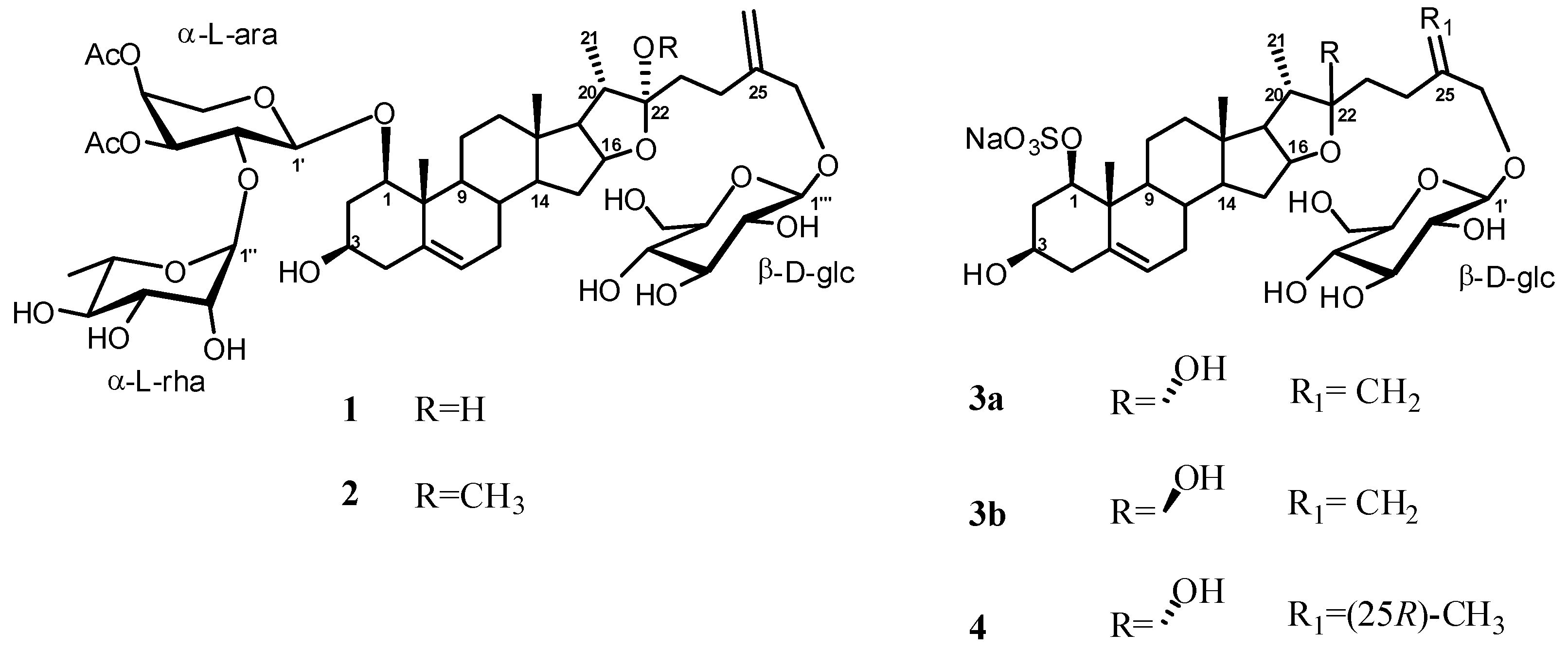
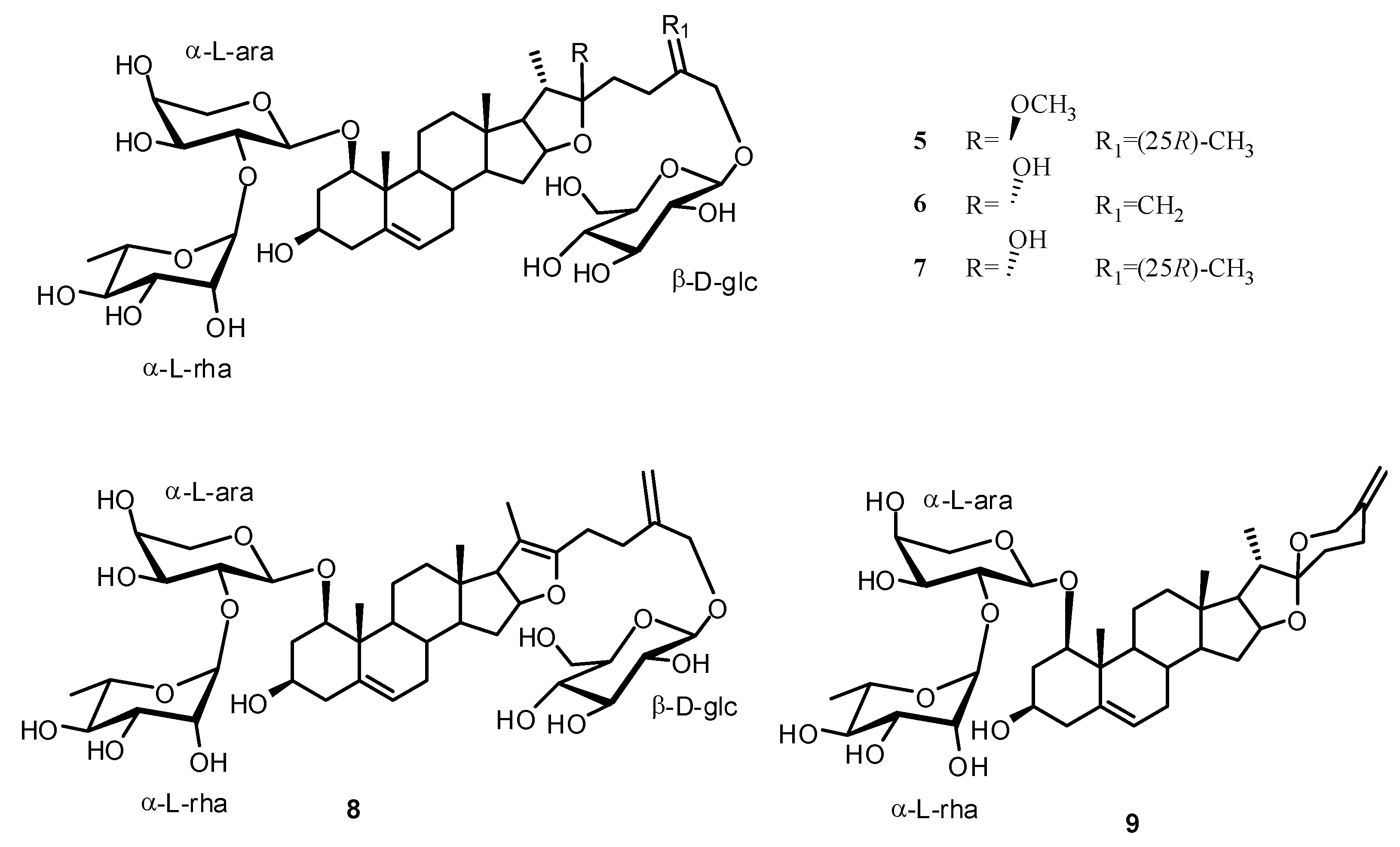
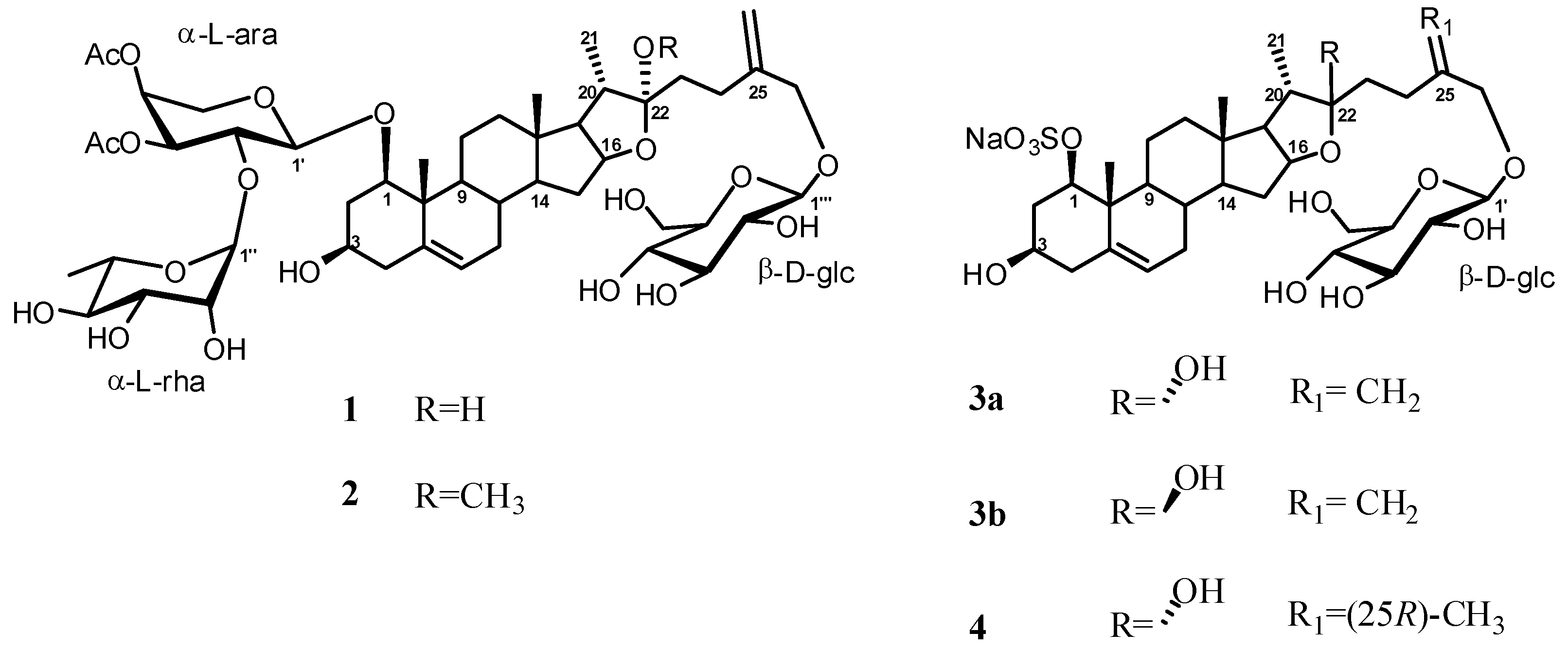
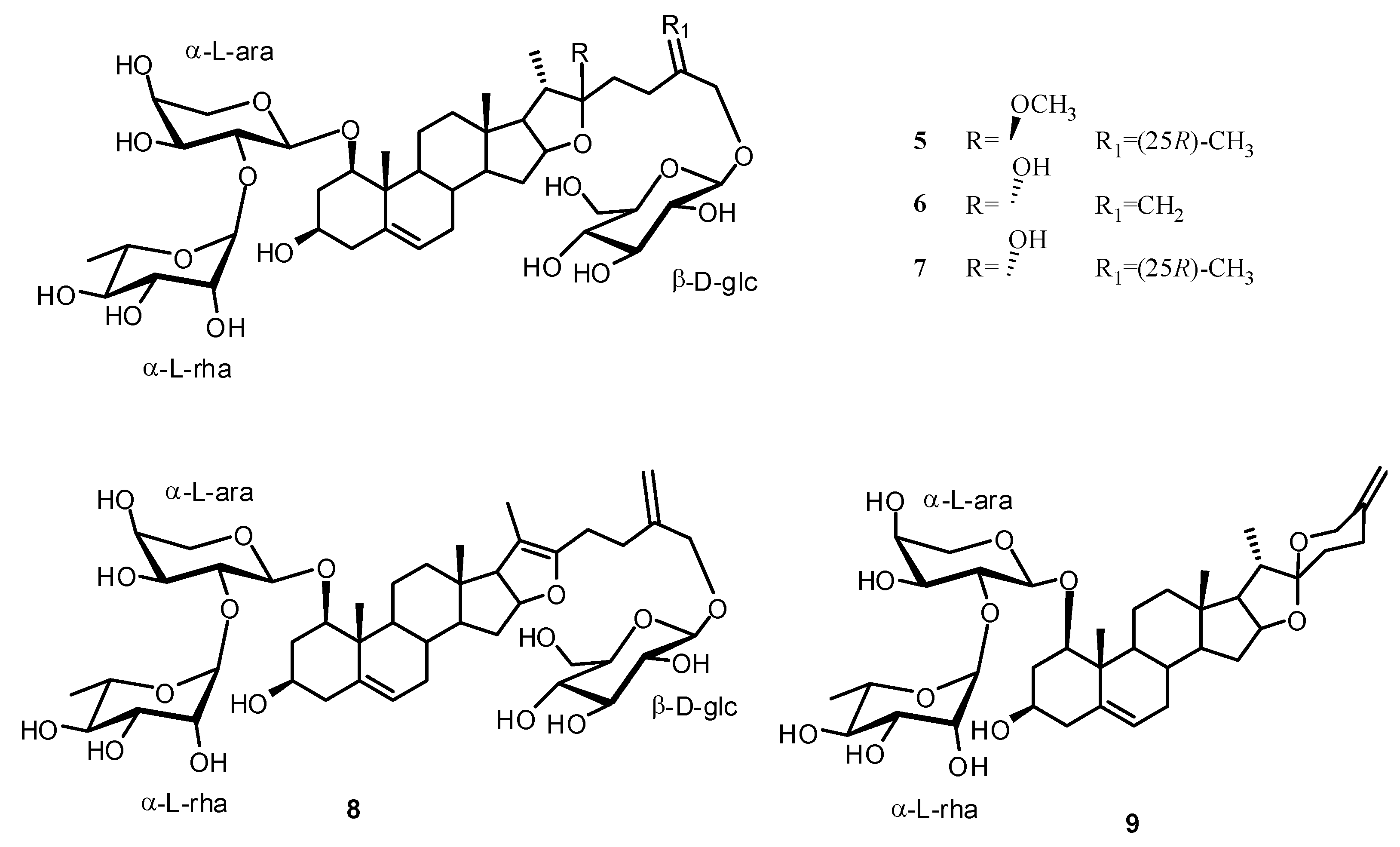
2. Results and Discussion
Structure Analysis and Characterization of Compounds 1–4


{kind=link}
{kind=link}
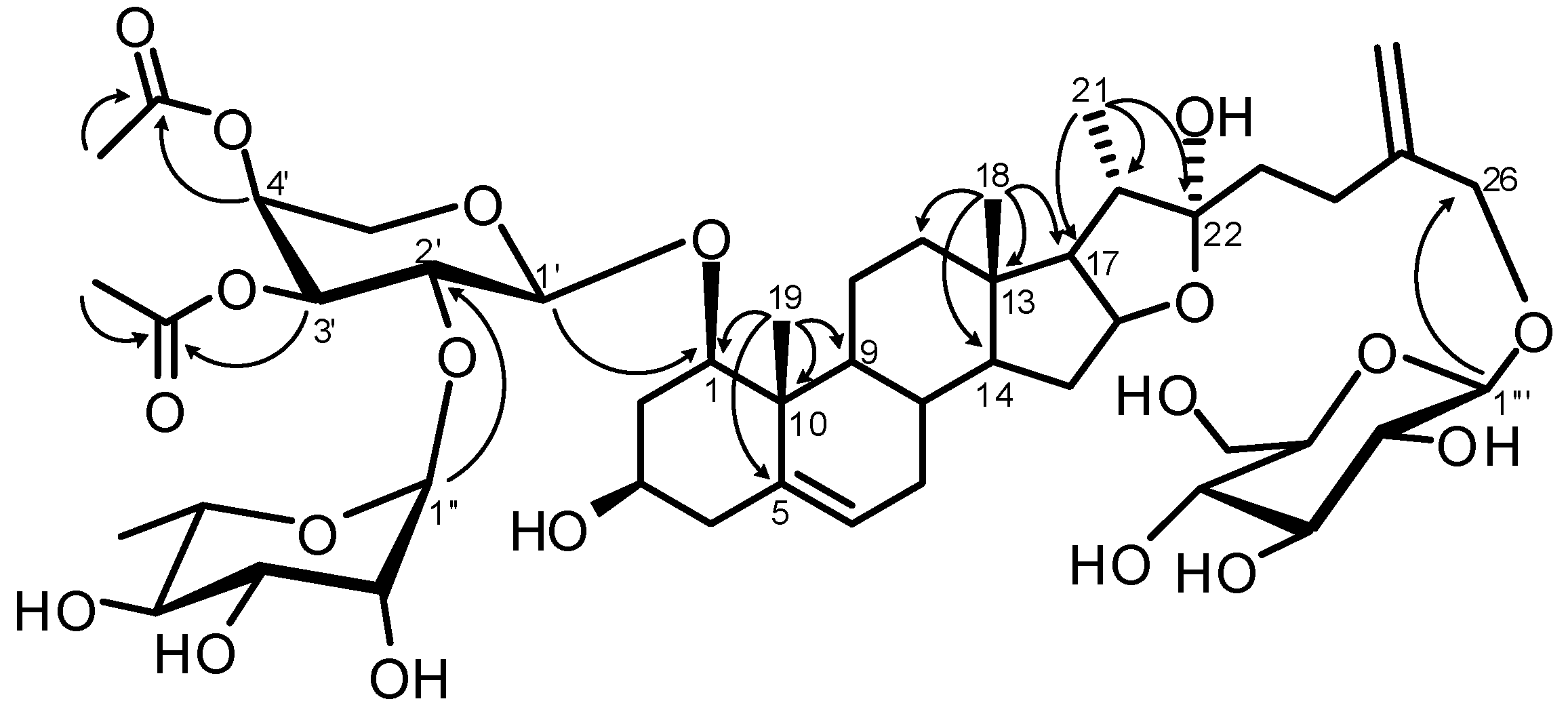{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δHa | δC | δHa | δC | |
| 1 | 3.38 m | 83.9 | 3.44 dd (11.8, 3.7) | 83.7 |
| 2a | 2.10 m | 36.8 | 2.10 m | 37.0 |
| 2b | 1.69 m | 1.69 m | ||
| 3 | 3.33 m | 68.6 | 3.35 m | 69.0 |
| 4 | 2.23 ovl | 43.0 | 2.21 ovl | 43.2 |
| 5 | - | 139.2 | - | 139.2 |
| 6 | 5.55 br d (5.6) | 125.4 | 5.56 br d (5.4) | 125.6 |
| 7a | 1.97 ovl | 32.3 | 1.96 ovl | 32.6 |
| 7b | 1.53 ovl | 1.53 ovl | ||
| 8 | 1.54 ovl | 33.6 | 1.55 ovl | 33.8 |
| 9 | 1.25 ovl | 50.9 | 1.25 ovl | 51.1 |
| 10 | - | 43.5 | - | 43.4 |
| 11a | 2.55 m | 24.7 | 2.56 m | 25.0 |
| 11b | 1.49 m | 1.48 m | ||
| 12a | 1.68 ovl | 40.7 | 1.70 ovl | 41.1 |
| 12b | 1.21 m | 1.21 m | ||
| 13 | - | 41.6 | - | 41.4 |
| 14 | 1.13 m | 57.2 | 1.13 m | 57.5 |
| 15a | 1.98 ovl | 32.5 | 1.97 ovl | 32.6 |
| 15b | 1.29 m | 1.26 ovl | ||
| 16 | 4.56 m | 81.8 | 4.38 m | 82.4 |
| 17 | 1.77 m | 63.7 | 1.73 ovl | 65.0 |
| 18 | 0.86 s | 16.7 | 0.86 s | 17.1 |
| 19 | 1.09 s | 14.9 | 1.12 s | 15.1 |
| 20 | 2.14 m | 40.4 | 2.22 ovl | 41.0 |
| 21 | 1.02 d (6.8) | 15.4 | 1.04 d (6.8) | 15.9 |
| 22 | - | 111.7 | - | 113.3 |
| 23a | 1.84 m | 37.4 | 1.90 m | 32.1 |
| 23b | 1.84 m | |||
| 24 | 2.29 ovl | 28.3 | 2.18 ovl | 28.5 |
| 25 | - | 147.4 | - | 147.1 |
| 26a | 4.33 d (12.5) | 72.4 | 4.34 d (12.6) | 72.4 |
| 26b | 4.13 d (12.5) | 4.13 d (12.6) | ||
| 27a | 5.09 br s | 112.1 | 5.08 br s | 112.2 |
| 27b | 4.92 br s | 4.93 br s | ||
| -OCH3 | 3.16 s | 47.5 | ||
| Position | 1 a | 6 b | ||
|---|---|---|---|---|
| δHc | δC | δHc | δC | |
| α-L-Ara | ||||
| 1′ | 4.50 d (7.4) | 100.1 | 4.26 d (7.1) | 100.7 |
| 2′ | 3.85 ovl | 73.5 | 3.70 ovl | 75.4 |
| 3′ | 5.05 dd (9.7, 3.1) | 75.7 | 3.64 dd (9.5, 3.2) | 75.7 |
| 4′ | 5.30 br s | 70.6 | 3.74 ovl | 70.5 |
| 5′ | 3.89 ovl3.73 ovl | 64.5 | 3.85 dd (12.1, 2.0)3.48 dd (12.1, 3.0) | 67.2 |
| 3′-COCH3 | 2.04 s | 20.8 | ||
| 3′-COCH3 | 170.9 | |||
| 4′-COCH3 | 2.03 s | 20.7 | ||
| 4′-COCH3 | 170.8 | |||
| α-L-Rha | ||||
| 1′′ | 4.95 br d (1.2) | 101.7 | 5.29 br d (1.2) | 101.3 |
| 2′′ | 3.72 ovl | 72.1 | 3.88 ovl | 72.0 |
| 3′′ | 3.62 dd (9.6, 3.3) | 71.8 | 3.69 ovl | 71.8 |
| 4′′ | 3.39 t (9.6) | 73.8 | 3.40 t (9.7) | 73.8 |
| 5′′ | 4.08 m | 69.8 | 4.08 m | 69.5 |
| 6′′ | 1.26 d (6.3) | 18.3 | 1.26 d (6.2) | 18.0 |
| β-D-Glc (C26) | ||||
| 1′′′ | 4.28 d (7.7) | 103.1 | 4.28 d (7.6) | 103.0 |
| 2′′′ | 3.22 t (8.3) | 74.9 | 3.21 t (8.4) | 74.9 |
| 3′′′ | 3.35 ovl | 77.9 | 3.35 ovl | 77.9 |
| 4′′′ | 3.27 ovl | 71.5 | 3.28 ovl | 71.4 |
| 5′′′ | 3.25 ovl | 77.7 | 3.26 ovl | 77.6 |
| 6′′′ | 3.87 ovl3.66 dd (12.1, 4.5) | 62.6 | 3.87 ovl3.67 dd (12.0, 4.5) | 62.6 |
| Position | 3a | 3b | 4 | |||
|---|---|---|---|---|---|---|
| δHa | δC | δHa | δC | δHa | δC | |
| 1 | 4.03 dd (11.9, 3.8) | 85.5 | 4.02 dd (11.9, 3.8) | 85.4 | 4.01 dd (11.8, 4.0) | 85.6 |
| 2a 2b | 2.55 m 1.69 m | 38.8 | 2.56 m 1.69 m | 38.7 | 2.55 m 1.67 m | 38.8 |
| 3 | 3.43 dddd (12.0, 12.0, 6.4, 6.4) | 68.6 | 3.41 dddd (12.0, 12.0, 6.4, 6.4) | 68.6 | 3.45 dddd (12.1, 12.1, 6.5, 6.5) | 68.7 |
| 4 | 2.21 ovl | 43.2 | 2.23 ovl | 43.2 | 2.21 m (2H) | 43.0 |
| 5 | - | 138.7 | - | 139.0 | - | 138.8 |
| 6 | 5.61 br d (5.2) | 126.5 | 5.60 br d (5.2) | 126.4 | 5.60 br d (5.4) | 126.5 |
| 7a 7b | 1.98 ovl 1.55 ovl | 32.8 | 1.99 ovl 1.57 ovl | 33.0 | 1.96 m 1.54 ovl | 32.6 |
| 8 | 1.55 ovl | 33.8 | 1.56 ovl | 33.8 | 1.54 ovl | 33.8 |
| 9 | 1.37 ovl | 50.6 | 1.37 ovl | 50.7 | 1.35 ovl | 50.7 |
| 10 | - | 43.7 | - | 43.6 | - | 43.7 |
| 11a 11b | 2.36 br d (12.6) 1.52 ovl | 24.0 | 2.35 br d (12.6) 1.52 ovl | 24.0 | 2.36 br d (12.4) 1.48 ovl | 24.0 |
| 12a 12b | 1.71 ovl 1.26 m | 40.8 | 1.70 ovl 1.28 m | 40.9 | 1.70 ovl 1.24 ovl | 40.9 |
| 13 | - | 41.2 | - | 41.4 | - | 41.4 |
| 14 | 1.17 m | 57.4 | 1.17 m | 57.5 | 1.18 m | 57.5 |
| 15a 15b | 1.99 ovl 1.32 ovl | 32.7 | 2.00 ovl 1.33 ovl | 32.6 | 1.95 ovl, 1.26 ovl | 32.7 |
| 16 | 4.57 m | 81.8 | 4.38 m | 82.2 | 4.57 q (5.6) | 82.0 |
| 17 | 1.78 ovl | 64.0 | 1.74 ovl | 64.1 | 1.71 ovl | 64.1 |
| 18 | 0.86 s | 16.8 | 0.87 s | 16.9 | 0.83 s | 16.9 |
| 19 | 1.10 s | 14.5 | 1.10 s | 14.5 | 1.10 s | 14.6 |
| 20 | 2.18 ovl | 40.8 | 2.19 ovl | 40.9 | 2.16 ovl | 41.1 |
| 21 | 1.04 d (6.7) | 15.8 | 1.01 d (6.7) | 16.0 | 0.99 d (7.1) | 16.1 |
| 22 | - | 111.1 | - | 113.7 | - | 111.3 |
| 23a 23b | 1.90 ovl 1.86 ovl | 37.5 | 1.91 ovl 1.85 ovl | 37.3 | 1.61 ovl | 31.4 |
| 24a 24b | 2.22 ovl 2.15 ovl | 28.5 | 2.21 ovl 2.15 ovl | 28.4 | 1.57 ovl 1.12 m | 28.6 |
| 25 | - | 147.0 | - | 147.3 | 1.72 m | 34.8 |
| 26a 26b | 4.34 d (12.5) 4.11 d (12.5) | 72.6 | 4.33 d (12.4) 4.12 d (12.4) | 72.4 | 3.74 dd (9.4, 6.5) 3.39 ovl | 75.6 |
| 27a 27b | 5.09 br s 4.93 br s | 111.9 | 5.10 br s 4.94 br s | 111.9 | 0.95 d (6.6) | 17.1 |
| Glucose | ||||||
| 1′ | 4.28 d (7.8) | 103.0 | 4.27 d (7.8) | 103.1 | 4.24 d (7.7) | 104.2 |
| 2′ | 3.22 t (8.5) | 74.9 | 3.20 t (8.5) | 75.0 | 3.19 t (8.4) | 74.8 |
| 3′ | 3.36 ovl | 77.8 | 3.34 ovl | 77.8 | 3.35 ovl | 77.9 |
| 4′ | 3.28 ovl | 71.5 | 3.28 ovl | 71.4 | 3.27 ovl | 71.5 |
| 5′ | 3.27 ovl | 77.7 | 3.26 ovl | 77.8 | 3.26 ovl | 77.7 |
| 6′ | 3.86 d (11.7) 3.66 dd (11.7, 5.2) | 62.6 | 3.88 d (11.7) 3.65 dd (11.7, 5.2) | 62.6 | 3.87 d (11.9) 3.68 dd (11.9, 5.1) | 62.7 |
3. Experimental
3.1. General
3.2. Plant Material
3.3. Compound Isolation
3.4. Solvolysis of Compound 4 Giving 4a
3.5. Methanolysis of 1–2: Sugar Analysis
4. Conclutions
Acknowledgments
Conflict of Interest
- Sample Availability: Samples of the pure compounds are available from the authors.
References
- Hostettmann, K.; Marston, A. Saponins; Cambridge University Press: London, UK, 1995; pp. 302–303. [Google Scholar]
- Mimaki, Y.; Kuroda, M.; Kameyama, A.; Yokosuka, A.; Sashida, Y. New steroidal constituents of the underground parts of Ruscus aculeatus and their cytostatic activity on HL-60 cells. Chem. Pharm. Bull. 1998, 46, 298–303. [Google Scholar]
- Mari, A.; Napolitano, A.; Perrone, A.; Pizza, C.; Piacente, S. An analytical approach to profile steroidal saponins in food supplements: The case of Ruscus aculeatus. Food Chem. 2012, 134, 461–468. [Google Scholar]
- de Combarieu, E.; Falzoni, M.; Fuzzati, N.; Gattesco, F.; Giori, A.; Lovati, M.; Pace, R. Identification of Ruscus steroidal saponins by HPLC-MS analysis. Fitoterapia 2002, 73, 583–596. [Google Scholar]
- Perrone, A.; Muzashvili, T.; Napolitano, A.; Skhirtladze, A.; Kemertelidze, E.; Pizza, C.; Piacente, S. Steroidal glycosides from the leaves of Ruscus colchicus: isolation and structural elucidation based on a preliminary liquid chromatography-electrospray ionization tandem mass spectrometry profiling. Phytochemistry 2009, 70, 2078–2088. [Google Scholar]
- Napolitano, A.; Muzashvili, T.; Perrone, A.; Pizza, C.; Kemertelidze, E.; Piacente, S. Steroidal glycosides from Ruscus ponticus. Phytochemistry 2011, 72, 651–661. [Google Scholar]
- Dunouau, C.; Belle, R.; Oulad-Ali, A.; Anton, R.; David, B. Triterpenes and sterols from Ruscus aculeatus. Planta Med. 1996, 62, 189–190. [Google Scholar]
- Oulad-Ali, A.; Guillaume, R.B.; David, B.; Anton, R. Sulphated steroidal derivatives from Ruscus aculeatus. Phytochemistry 1996, 42, 895–897. [Google Scholar] [CrossRef]
- Corea, G.; Fattorusso, E.; Lanzotti, V.; Capasso, R.; Izzo, A.A. Antispasmodic saponins from bulbs of red onion, Allium cepa L. var. Tropea. J. Agric. Food. Chem. 2005, 53, 935–940. [Google Scholar]
- Hara, S.; Okabe, H.; Mihashi, K. Gas-liquid chromatographic separation of aldose enantiomers as trimethylsilyl ethers of methyl 2-(polyhydroxyalkyl)thiazolidine-4(R)-carboxylates. Chem. Pharm. Bull. 1987, 35, 501–506. [Google Scholar]
- Corea, G.; Fattorusso, E.; Lanzotti, V. Saponins and flavonoids of Allium triquetrum. J. Nat. Prod. 2003, 66, 1405–1411. [Google Scholar]
- Challinor, V.L.; Piacente, S.; De Voss, J.J. NMR assignment of the absolute configuration of C-25 in furostanol steroidal saponins. Steroids 2012, 77, 602–608. [Google Scholar]
- Yuan, L.; Ji, T.F.; Wang, A.G.; Yang, J.B.; Su, Y.L. Two new furostanol saponins from the seeds of Allium cepa L. Chin. Chem. Lett. 2008, 19, 461–464. [Google Scholar]
- Bombardelli, E.; Bonati, A.; Gabetta, B.; Mustich, G. Glycosides from rhizomes of Ruscus aculeatus. Fitoterapia 1972, 43, 3–10. [Google Scholar]
- Mimaki, Y.; Takaashi, Y.; Kuroda, M.; Sashida, Y.; Nikaido, T. Steroidal saponins from Nolina recurvata stems and their inhibitory activity on cyclic AMP phosphodiesterase. Phytochemistry 1996, 42, 1609–1615. [Google Scholar]
- Kupchan, S.M.; Britton, R.W.; Ziegler, M.F.; Sigel, C.W. Bruceantin, a new potent antileukemic simaroubolide from Brucea antidysenterica. J. Org. Chem. 1973, 38, 178–179. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Marino, S.D.; Festa, C.; Zollo, F.; Iorizzi, M. Novel Steroidal Components from the Underground Parts of Ruscus aculeatus L. Molecules 2012, 17, 14002-14014. https://doi.org/10.3390/molecules171214002
Marino SD, Festa C, Zollo F, Iorizzi M. Novel Steroidal Components from the Underground Parts of Ruscus aculeatus L. Molecules. 2012; 17(12):14002-14014. https://doi.org/10.3390/molecules171214002
Chicago/Turabian StyleMarino, Simona De, Carmen Festa, Franco Zollo, and Maria Iorizzi. 2012. "Novel Steroidal Components from the Underground Parts of Ruscus aculeatus L." Molecules 17, no. 12: 14002-14014. https://doi.org/10.3390/molecules171214002
APA StyleMarino, S. D., Festa, C., Zollo, F., & Iorizzi, M. (2012). Novel Steroidal Components from the Underground Parts of Ruscus aculeatus L. Molecules, 17(12), 14002-14014. https://doi.org/10.3390/molecules171214002