Efficient Preparation of α-Ketoacetals

Abstract
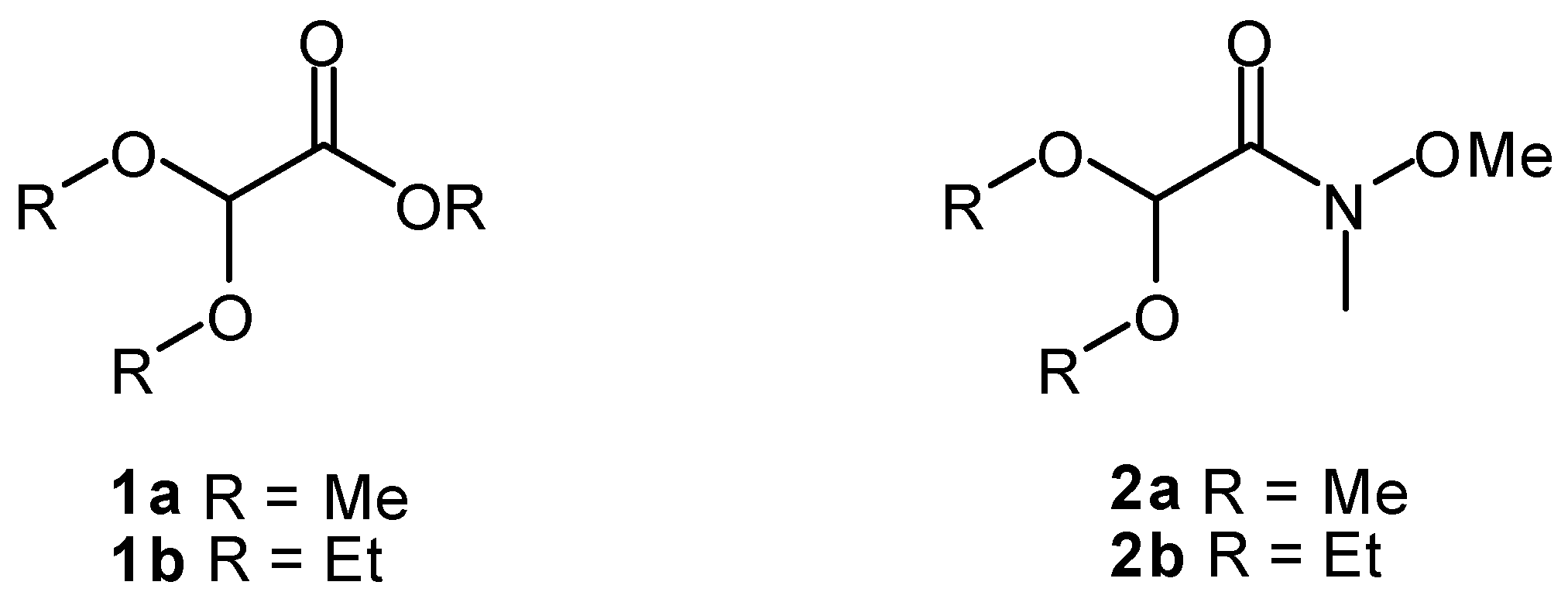
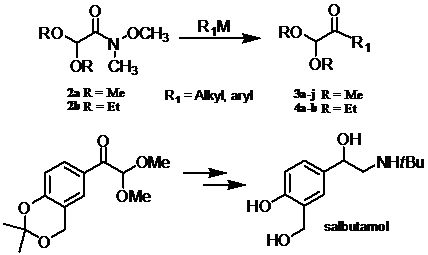
: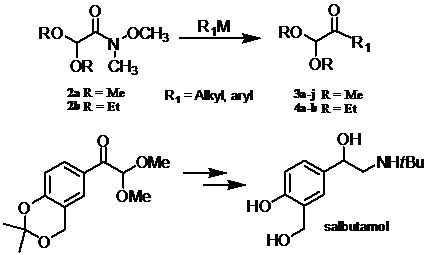
1. Introduction
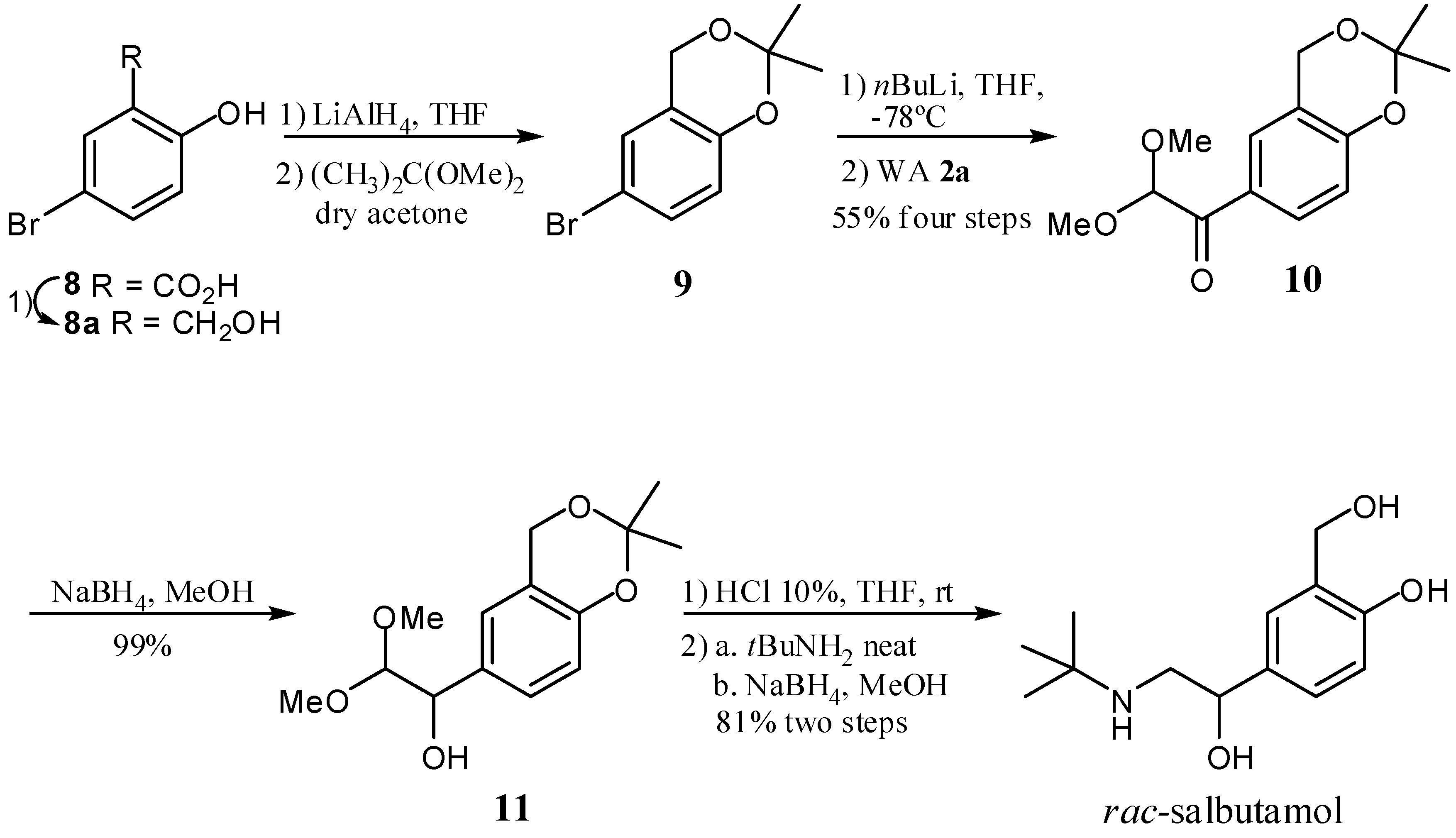
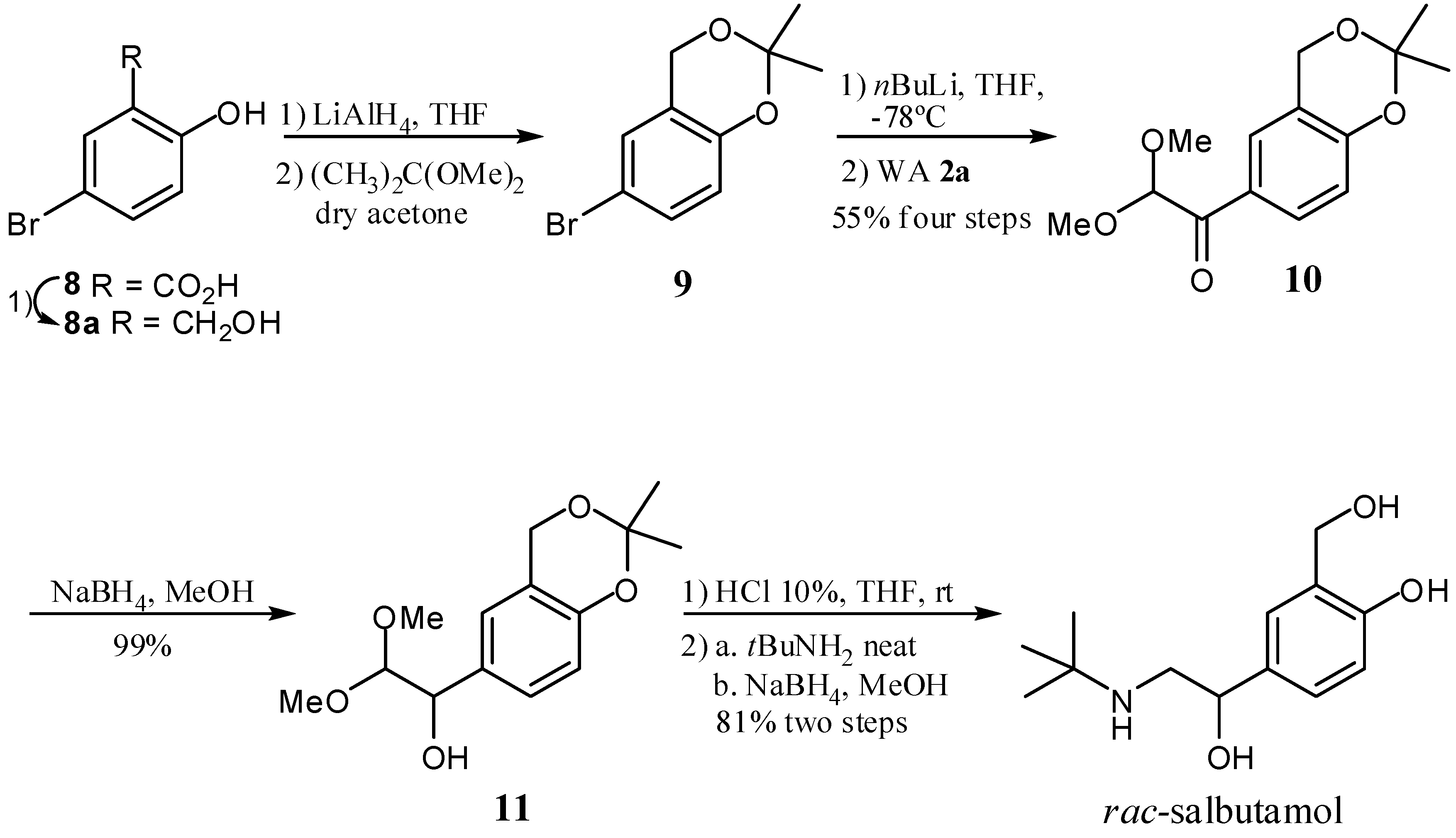
2. Results and Discussion
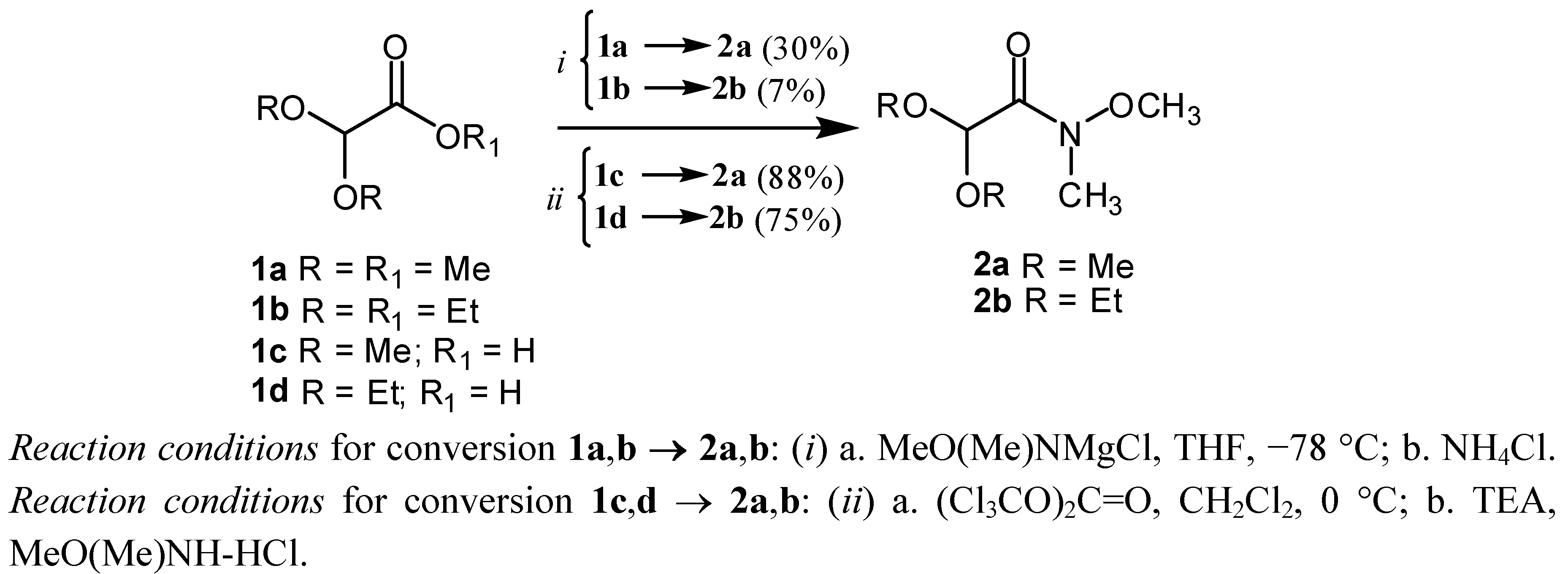


{kind=link}
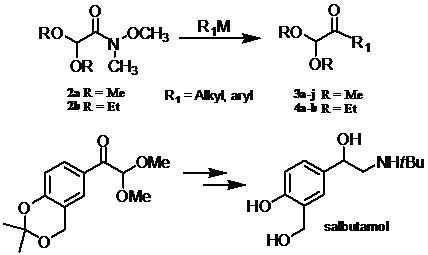{kind=link}
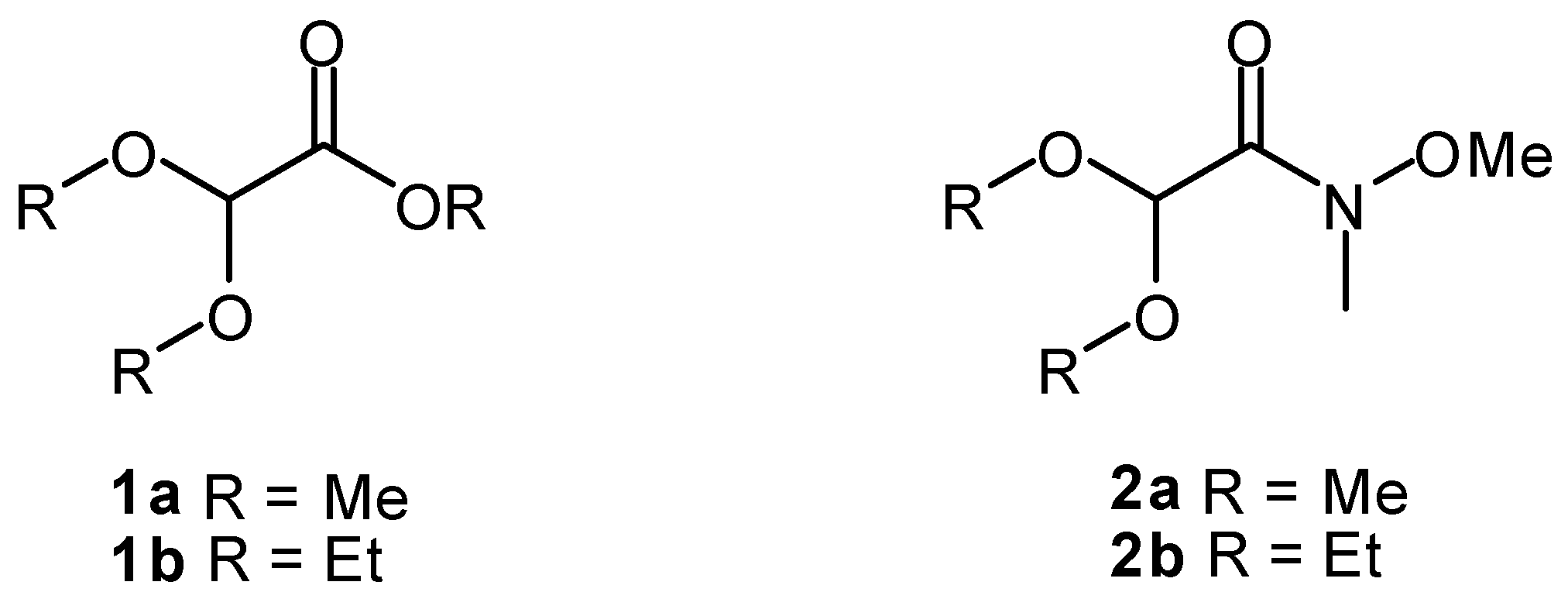{kind=link}
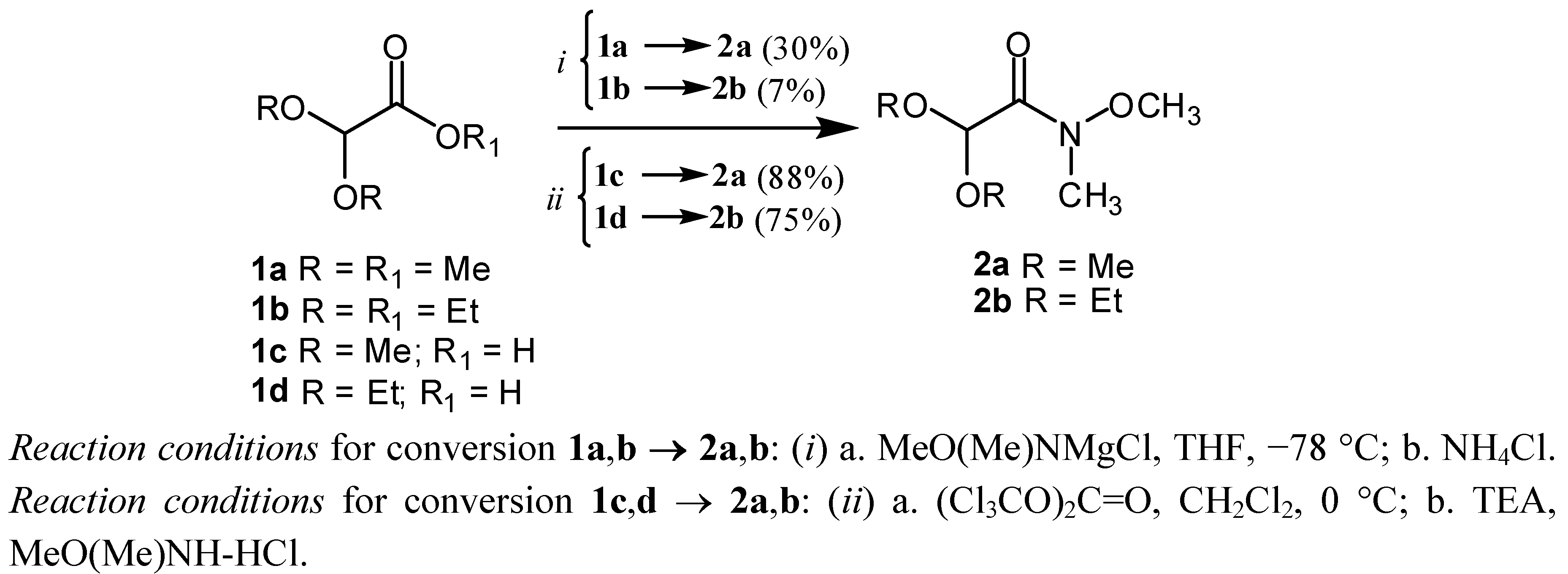{kind=link}
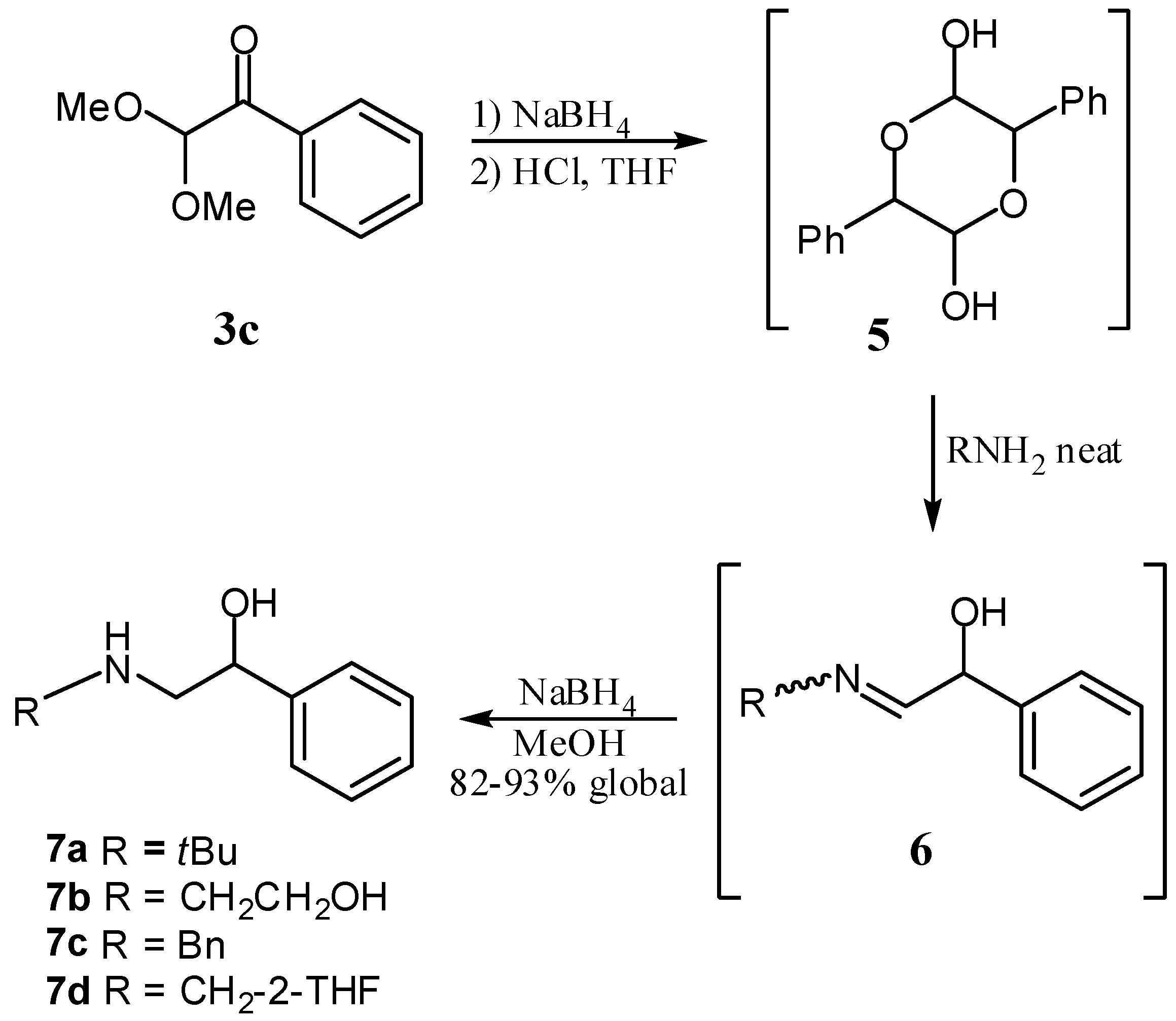{kind=link}
{kind=link}
| Entry | R1M | R1 | Product (% yield) |
|---|---|---|---|
| 1 | MeLi | -CH3 | 3a (95) |
| 2 | MeLi | -CH3 | 4a (90) * |
| 3 | EtLi | -CH2CH3 | 3b (99) |
| 4 | EtLi | -CH2CH3 | 4b (89) * |
| 5 | PhLi | -C6H5 | 3c (92) |
| 6 | MeMgBr | -CH3 | 3a (97) |
| 7 | MeMgBr | -CH3 | 4a (93)* |
| 8 | EtMgBr | -CH2CH3 | 4b (91)* |
| 9 | nPrMgBr | -(CH2)2CH3 | 3d (97) |
| 10 | CH3CCMgBr | -C≡C-CH3 | 3e (78) |
| 11 | PhCCMgBr | -C≡C-C6 H5 | 3f (83) |
| 12 | 4-MeC6H5MgBr | - pC6H4-CH3 | 3g (79) |
| 13 | 4-FC6H5MgBr | - pC6H4-F | 3h (92) |
| 14 | 3-MeOC6H5MgBr | - mC6H4-OCH3 | 3i (77) |
| 15 | BnMgBr | -CH2C6H5 | 3j (81) |
3. Experimental
3.1. General Procedures
N-Trimethoxy-N-methyl-acetamide (2a):
Method A
Method B
3.2. General Procedure for the Preparation of α-Ketoacetals
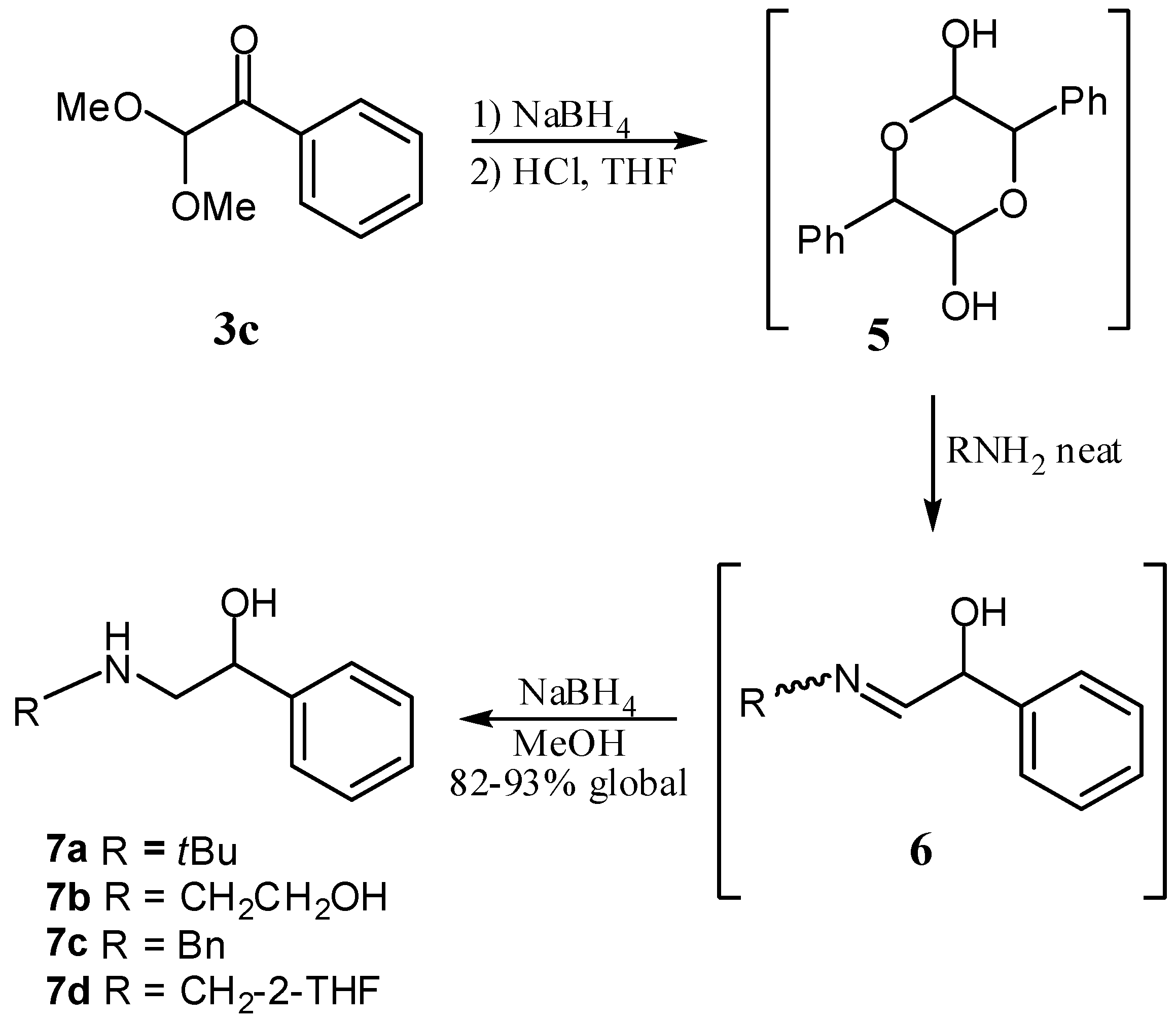
3.3. General Procedure for the Synthesis of β-Aminoalcohols
4. Conclusions
Supplementary Materials
Acknowledgments
- Sample Availability: Samples of the compounds 2a, 2b, 3a–j, 4a, 4b, 7a–d, 9, 10, 11 and rac-salbutamol are available from the authors.
References and Notes
- Qin, B.; Liu, X.; Shi, J.; Zheng, K.; Zhao, H.; Feng, X. Enantioselective Cyanosilylation of α,α-Dialkoxy Ketones Catalyzed by Proline-Derived in-Situ-Prepared N-Oxide as Bifunctional Organocatalyst. J. Org. Chem. 2007, 72, 2374–2378. [Google Scholar]
- Graef, E.; Troschuetz, R. Synthesis of 6-Phenyl Substituted 2-Formylnicotinates. Synthesis 1999, 7, 1216–1222. [Google Scholar]
- Garcia Ruano, J.L.; Maestro, M.C.; Sanchez-Sancho, F. Enantiomerically pure 3-p-tolylsulfinyl acrolein and crotonaldehyde dimethylacetals. Stereoselective reduction of β-keto-γ,γ-dialkoxysulfoxides. Tetrahedron: Asymmetry 1995, 6, 2299–2312. [Google Scholar] [CrossRef]
- Török, B.; Balázsik, K.; Bartók, M.; Felföldi, K.; Bartók, M. New synthesis of a useful C3 chiral building block by a heterogeneous method: enantioselective hydrogenation of pyruvaldehyde dimethyl acetal over cinchona modified Pt/Al2O3 catalysts. Chem. Commun. 1999, 17, 1725–1726. [Google Scholar]
- Studer, M.; Burkhardt, S.; Blaser, H.-U. Enantioselective hydrogenation of alfa-keto acetals with cinchona modified Pt catalyst. Chem. Commun. 1999, 17, 1727–1728. [Google Scholar]
- Szőllősi, G.; Makra, Z.; Fülöp, F.; Bartók, M. The First Case of Competitive Heterogeneously Catalyzed Hydrogenation using Continuous-Flow Fixed-Bed Reactor System: Hydrogenation of Binary Mixtures of Activated Ketones on Pt-Alumina and on Pt-Alumina-Cinchonidine Catalysts. Catal. Lett. 2011, 141, 1616–1620. [Google Scholar] [CrossRef]
- Tamura, Y.; Kondo, H.; Annoura, H.; Takeuchi, R.; Fujioka, F. Diastereoselective nucleophilic addition to chiral open-chain α-ketoacetals: Synthesis of (R)- and (S)-mevalolactone. Tetrahedron Lett. 1986, 27, 2117–2120. [Google Scholar] [CrossRef]
- Becerra-Martínez, E.; Velázquez-Ponce, P.; Sánchez-Aguilar, M.A.; Rodríguez-Hosteguín, A.; Joseph-Nathan, P.; Tamariz, J.; Zepeda, L.G. New 2-acyl-1,3-dioxane derivatives from (1R)-(−)-myrtenal: Stereochemical effect on their relative ability as chiral auxiliaries. Tetrahedron: Asymmetry 2007, 18, 2727–2737. [Google Scholar] [CrossRef]
- Vargas-Díaz, M.E.; Joseph-Nathan, P.; Tamariz, J.; Zepeda, L.G. Synthesis of a New (1R)-(−)-Myrtenal-Derived Dioxadithiadodecacycle and Its Use as an Efficient Chiral Auxiliary. Org. Lett. 2007, 9, 13–16. [Google Scholar] [CrossRef]
- Vargas-Díaz, M.E.; Lagunas-Rivera, S.; Joseph-Nathan, P.; Tamariz, J.; Zepeda, L.G. New S,O-acetals from (1R)-(−)-myrtenal as chiral auxiliaries in nucleophilic additions. Tetrahedron Lett. 2005, 46, 3297–3300. [Google Scholar] [CrossRef]
- Verhe, R.; Courtheyn, D.; de Kimpe, N.; de Buyck, L.; Schamp, N. Preparation of 1,1-Dialkoxy-2-alkanones. Synthesis 1982, 8, 667–670. [Google Scholar]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Chianelli, D.; Bartoli, D. Selenium-mediated conversion of alkynes into α-dicarbonyl compounds. J. Org. Chem. 1991, 56, 4529–4534. [Google Scholar]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Bartoli, D. Selenium-catalyzed conversion of methyl ketones intoα-keto acetals. J. Org. Chem. 1990, 55, 4523–4528. [Google Scholar] [CrossRef]
- Nair, V.; Nair, L.G.; Panicker, S.B.; Sheeba, V.; Augustine, A. Novel Cerium(IV) Ammonium Nitrate Mediated Transformation of Styrenes to α-Methoxy Acetophenones. Chem. Lett. 2000, 6, 584–585. [Google Scholar]
- Wegner, G.; Karbach, S.; Smuda, H.; Hickmann, E.; Kober, R.; Seele, R.; Zierke, T. Process for the preparation of a,a-dialkoxy ketones by treatment of aldehydes or ketones with alkyl nitrite. Eur. Pat. Appl. EP 472118 A1. 26 February 1992. [Google Scholar]
- Tang, H.; Chen, S.; Zhang, P. (1.3)-Sigmatropic rearrangements of 1,3-dialkyloxy-acetylactones and-acetones. Huaxue Xuebao 1985, 43, 72–78. [Google Scholar]
- Yu, Y.; Chen, G.; Zhu, J.; Zhang, X.; Chen, S.; Tang, H.; Zhang, P. A study of rearrangement of some 1,3-dimethoxyalkan-2-ones. J. Chem. Soc. Perkin Trans. 1 1990, 8, 2239–2243. [Google Scholar]
- Devos, A.; Remion, J.; Frisque-Hesbain, A.-M.; Colens, A.; Ghosez, L. Synthesis of acyl halides under very mild conditions. J. Chem. Soc. Chem. Commun. 1979, 24, 1180–1181. [Google Scholar]
- Adamczyk, M.; Johnson, D.D.; Mattingly, P.G.; Pan, Y.; Reddy, R.E. A convenient method for the preparation of α-ketoacetals. Synth. Comm. 2002, 32, 3199–3205. [Google Scholar] [CrossRef]
- These α-ketoacetals can be purchased from Sigma-Aldrich, Co., St. Louis, MO, USA.
- A very complete review concerning synthesis and use of Weinreb amides is highly recommendable: Balasubramaniam, S.; Aidhen, I.S. The growing synthetic utility of the Weinreb amide. Synthesis 2008, 23, 3707–3738. [Google Scholar]
- Nahm, S.; Weinreb, S.M. N-methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar] [CrossRef]
- Williams, J.M.; Jobson, R.B.; Yasuda, N.; Marchesini, G.; Dolling, U.H.; Grabowski, E.J.J. A new general method for preparation of N-Methoxy-N-methylamides. Application in direct conversion of an ester to a ketone. Tetrahedron Lett. 1995, 36, 5461–5464. [Google Scholar]
- Toda, N.; Ori, M.; Takami, K.; Tago, K.; Kogen, H. Total Synthesis of (+)-Benzastatin E via Diastereoselective Grignard Addition to 2-Acylindoline. Org. Lett. 2003, 5, 269–271. [Google Scholar] [CrossRef]
- Ki-Jong, H.; Misoo, K. Direct Synthesis of Weinreb Amides from Carboxylic Acids Using Triphosgene. Lett. Org. Chem. 2007, 4, 20–22. [Google Scholar]
- Rare Chemicals Catalogue 20461-86-3 and 3559-62. Supplier Name: Rare Chemicals GmbH, Catalog Publication Date: 6 June 2012.
- Graham, S.L.; Scholz, T.H. A new mode of reactivity of N-methoxy-N-methylamides with strongly basic reagents. Tetrahedron Lett. 1990, 31, 6269–6272. [Google Scholar]
- Meester, W.J.N.; Van Dijk, R.; Van Maarseveen, J.H.; Rutjes, F.P.J.T.; Hermkens, P.H.H.; Hiemstra, H. Highly diastereoselective synthesis of β-amino alcohols. J. Chem. Soc. Perkin Trans. 1 2001, 22, 2909–2911. [Google Scholar]
- Azizi, N.; Saidi, M.R. Highly Chemoselective Addition of Amines to Epoxides in Water. Org. Lett. 2005, 17, 3649–3651. [Google Scholar] [CrossRef]
- Goswami, S.; Maity, A.C.; Fun, H.-K.; Chantrapromma, S. The smallest vicinal tricarbonyl compound as a monohydrate and tetracarbonyl compound as a thiane derivative - first effective synthesis, characterization and chemistry. Eur. J. Org. Chem. 2009, 9, 1417–1426. [Google Scholar]
- Groening, Carsten; Ebel, Klaus; Kaibel, Gerd; Therre, Joerg; Koopmann, Juergen; Menig, Helmuth; Fritz, Gerard; Dietz, Rainer. Preparation of methylglyoxal dimethyl acetal. Eur. Pat. Appl. EP 704422 A1, 3 April 1996.
- Guseinov, F.I.; Tagiev, S.Sh.; Moskva, V.V. Reaction of α-Chloro- and α,α-Dichloro-β-oxo-substituted Aldehydes with Anionic Nucleophiles. Zh. Org. Khimii 1995, 31, 96–99. [Google Scholar]
- Khamliche, L.; Bakkas, S.; Robert, A. Selective protection of the functionalities of α-hydroxy unsaturated aldehydes. Synthesis 1994, 11, 1129–1131. [Google Scholar]
- Devulapally, R.; Hon, Y.-S. The first total synthesis of (±)-zenkequinone. Tetrahedron Lett. 2011, 25, 3183–3185. [Google Scholar]
- Wu, C.; Kawasaki, K.; Ohgiya, S.; Ohmiya, Y. Syntheses and evaluation of the bioluminescent activity of (S)-Cypridina luciferin and its analogs. Tetrahedron Lett. 2006, 47, 753–756. [Google Scholar] [CrossRef]
- Keiko, N.A.; Funtikova, E.A.; Stepanova, L.G.; Chuvashev, Y.A.; Larina, L.I. Reactions of 2-Alkoxypropenals with Thiols in Neutral and Acid Media. Russ. J. Org. Chem. 2002, 38, 970–976. [Google Scholar] [CrossRef]
- Noack, Rainer; Palm, Clemens; Groening, Carsten; Lipowsky, Gunter. Preparation of hydroxymethyl-1,2-diphenyloxiranes. PCT. Int. Appl. WO 2010089353 A1 12 August 2010.
- Studer, M. Production of optically active α-hydroxy acetals. PCT Int. Appl. WO 2001000545 A1, 4 January 2001. [Google Scholar]
- Durham, T.B.; Hahn, P.J.; Kohn, T.J.; McCarthy, J.R.; Broughton, H.B.; Dally, R.D.; Gonzalez-Garcia, M.R.; Henry, K.J., Jr.; Shepherd, T.A.; Erickson, J.A. Preparation of acylated 2-amino-1-(morpholin-3-yl)ethanols and derivatives as BACE inhibitors for treating Alzheimer’s. PCT Int. Appl. WO 2006034093 A2, 30 March 2006. [Google Scholar]
- Bringmann, G.; Geisler, J.P. A Simple, Chiral-Pool-Independent Synthesis of Enantiomerically Pure Alanine-Derived α-Amino Aldehyde Acetals. Synthesis 1989, 8, 608–610. [Google Scholar] [CrossRef]
- Panunzi, B.; Rotiroti, L.; Tingoli, M. Solvent directed electrophilic iodination and phenylselenenylation of activated alkyl aryl ketones. Tetrahedron Lett. 2003, 44, 8753–8756. [Google Scholar] [CrossRef]
- Nishio, T.; Omote, Y. The substitution reaction of 2-aralkylthio-1-alkenyl and 2-alkylsulfinyl-1-alkenyl ketones with alkoxides: preparation of 2-alkoxy-1-alkenyl ketones. Synthesis 1980, 12, 1013–1015. [Google Scholar] [CrossRef]
- Bedore, M.W.; Zaborenko, N.; Jensen, K.F.; Jamison, T.F. Aminolysis of Epoxides in a Microreactor System: A Continuous Flow Approach to β-Amino Alcohols. Org. Process Res. Dev. 2010, 14, 432–440. [Google Scholar] [CrossRef]
- Chung, J.Y. L.; Cvetovich, R.; Amato, J.; McWilliams, J.; Reamer, R.; DiMichele, L. Enantioselective Nitrile Anion Cyclization to Substituted Pyrrolidines. A Highly Efficient Synthesis of (3S,4R)-N-tert-Butyl-4-Arylpyrrolidine-3-Carboxylic Acid. J. Org. Chem. 2005, 70, 3592–3601. [Google Scholar] [CrossRef]
- Alcaide, B.; Escobar, G.; Perez-Ossorio, R.; Plumet, J.; Sanz, D. The reaction of phenylglyoxal with primary aliphatic and aromatic amines. Synthesis of phenylglyoxal monoimines and some derivatives. J. Chem. Res. Synop. 1984, 5, 144–145. [Google Scholar]
- Huerta, G.; Contreras‐Ordoñez, G.; Alvarez‐Toledano, C.; Santes, V.; Gómez, E.; Toscano, R.A. Facile Synthesis of Aminoalcohols by Ring Opening of Epoxides Under Solvent Free Conditions. Synth. Commun. 2004, 34, 2393–2406. [Google Scholar] [CrossRef]
- Maeda, H.; Furuyoshi, S.; Nakatsuji, Y.; Okahara, M. Synthesis of monoaza crown ethers from N,N-bis[oligo(oxyalkylene)]amines and oligoethylene glycol bis(p-toluenesulfonates) or corresponding dichlorides. Bull. Chem. Soc. Jap. 1983, 56, 212–18. [Google Scholar] [CrossRef]
- Negrón-Silva, G.; Hernández-Reyes, C.X.; Ángeles-Beltrán, D.; Lomas-Romero, L.; González-Zamora, E.; Méndez-Vivar, J. Comparative Study of Regioselective Synthesis of β-Aminoalcohols under Solventless Conditions Catalyzed by Sulfated Zirconia and SZ/MCM-41. Molecules 2007, 12, 2515–2532. [Google Scholar] [CrossRef]
- Desai, H.; D’Souza, B.R.; Foether, D.; Johnson, B.F.; Lindsay, H.A. Regioselectivity in a highly efficient, microwave-assisted epoxide aminolysis. Synthesis 2007, 6, 902–910. [Google Scholar]
- Bonollo, S.; Fringuelli, F.; Pizzo, F.; Vaccaro, L. A green route to β-amino alcohols via the uncatalyzed aminolysis of 1,2-epoxides by alkyl- and arylamines. Green Chem. 2006, 8, 960–964. [Google Scholar] [CrossRef]
- Placzek, A.T.; Donelson, J.L.; Trivedi, R.; Gibbs, R.A.; De, S.K. Scandium triflate as an efficient and useful catalyst for the synthesis of β-amino alcohols by regioselective ring opening of epoxides with amines under solvent-free conditions. Tetrahedron Lett. 2005, 46, 9029–9034. [Google Scholar]
- Munson, H.R., Jr.; Tankersley, R.W., Jr. Substituted dialkanolamines,sulfur analogs and condensed 1,4-oxazine derivatives thereof in viral disease treatment. U.S. Patent 4,803,200, 7 February 1989. [Google Scholar]
- Cox, P.J. 4-Bromo-2-(hydroxymethyl) phenol: helical hydrogen bonding, R22(12) rings and C-H···π interactions. Acta Cryst. Sect. C: Cryst. Struct. Commun. 2003, C59, o512–o513. [Google Scholar] [CrossRef]
- Bajwa, N.; Jennings, M. Efficient and Selective Reduction Protocols of the 2,2-Dimethyl-1,3-benzodioxan-4-one Functional Group to Readily Provide Both Substituted Salicylaldehydes and 2-Hydroxybenzyl Alcohols. J. Org. Chem. 2006, 71, 3646–3649. [Google Scholar] [CrossRef]
- Gisch, N.; Balzarini, J.; Meier, C. 5-Diacetoxymethyl-cycloSal-d4TMP-A prototype of enzymatically activated cycloSal-pronucleotides. Nucleos. Nucleot. Nucl. Acids 2007, 26, 861–864. [Google Scholar] [CrossRef]
- Ding, Y.S.; Shiue, C.Y.; Fowler, J.S.; Wolf, A.P.; Plenevaux, A. No-carrier-added (NCA) aryl [18F]fluorides via the nucleophilic aromatic substitution of electron-rich aromatic rings. J. Fluorine Chem. 1990, 48, 189–206. [Google Scholar]
- Aggarwal, K.; Esquivel-Zamora, B. Application of the Chiral Acyl Anion Equivalent, trans-1,3-Dithiane 1,3-Dioxide to an Asymmetric Synthesis of (R)-Salbutamol. J. Org. Chem. 2002, 67, 8618–8621. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ayala-Mata, F.; Barrera-Mendoza, C.; Jiménez-Vázquez, H.A.; Vargas-Díaz, E.; Zepeda, L.G. Efficient Preparation of α-Ketoacetals. Molecules 2012, 17, 13864-13878. https://doi.org/10.3390/molecules171213864
Ayala-Mata F, Barrera-Mendoza C, Jiménez-Vázquez HA, Vargas-Díaz E, Zepeda LG. Efficient Preparation of α-Ketoacetals. Molecules. 2012; 17(12):13864-13878. https://doi.org/10.3390/molecules171213864
Chicago/Turabian StyleAyala-Mata, Francisco, Citlalli Barrera-Mendoza, Hugo A. Jiménez-Vázquez, Elena Vargas-Díaz, and L. Gerardo Zepeda. 2012. "Efficient Preparation of α-Ketoacetals" Molecules 17, no. 12: 13864-13878. https://doi.org/10.3390/molecules171213864
APA StyleAyala-Mata, F., Barrera-Mendoza, C., Jiménez-Vázquez, H. A., Vargas-Díaz, E., & Zepeda, L. G. (2012). Efficient Preparation of α-Ketoacetals. Molecules, 17(12), 13864-13878. https://doi.org/10.3390/molecules171213864