Spatial Control of DNA Reaction Networks by DNA Sequence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
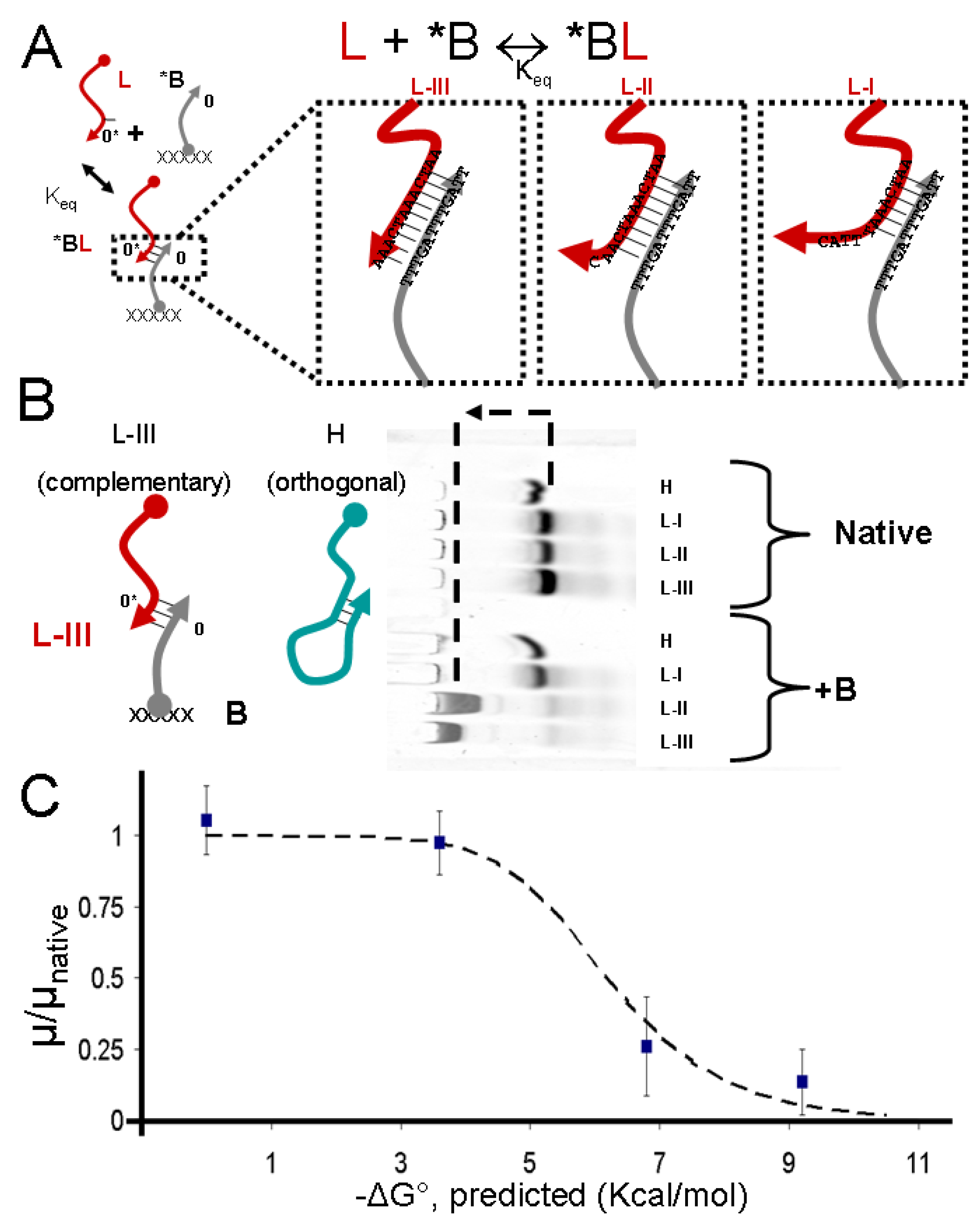
2.1. Control of DNA Electrophoretic Mobility in a DNA-Functionalized Polyacrylamide Gel
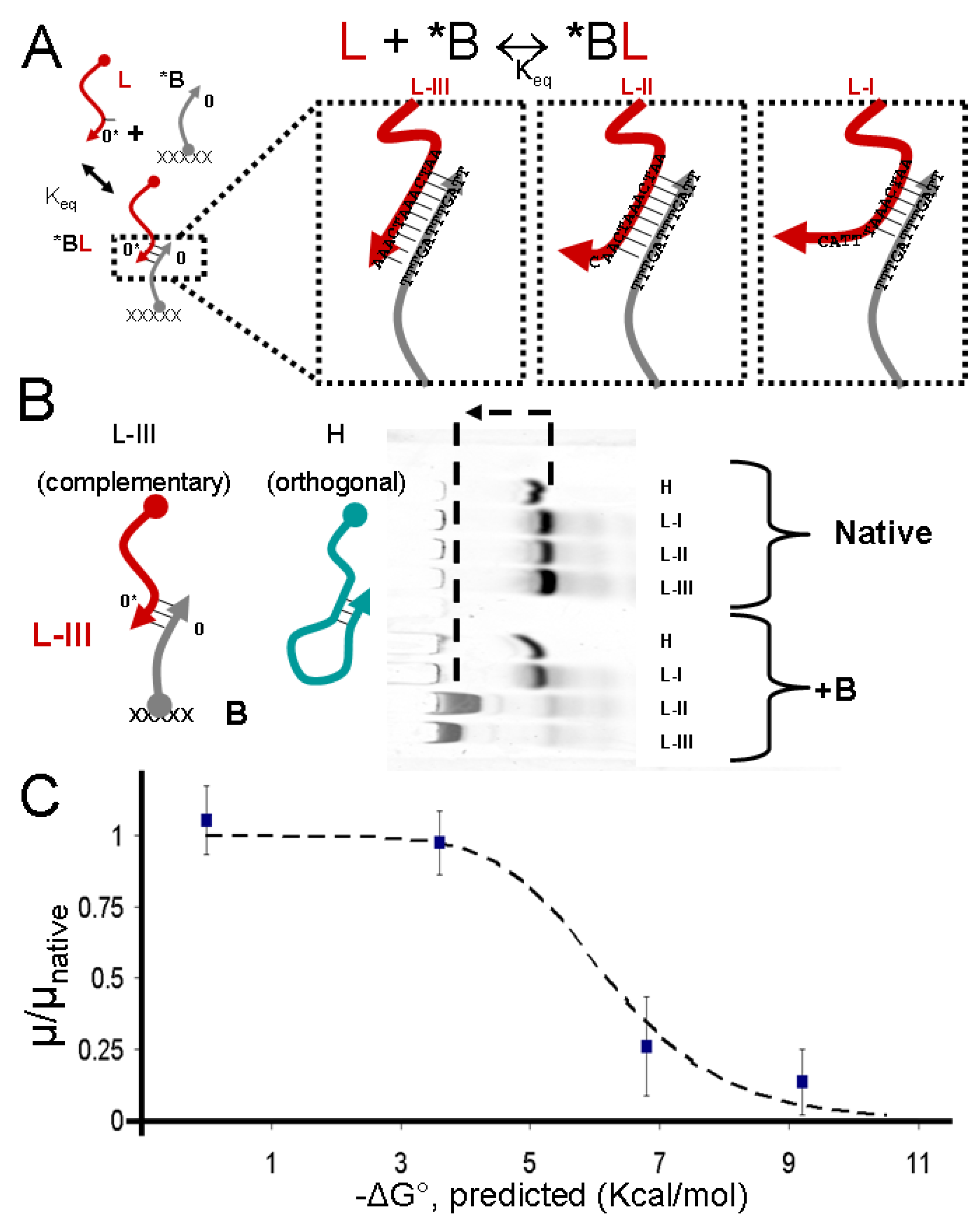
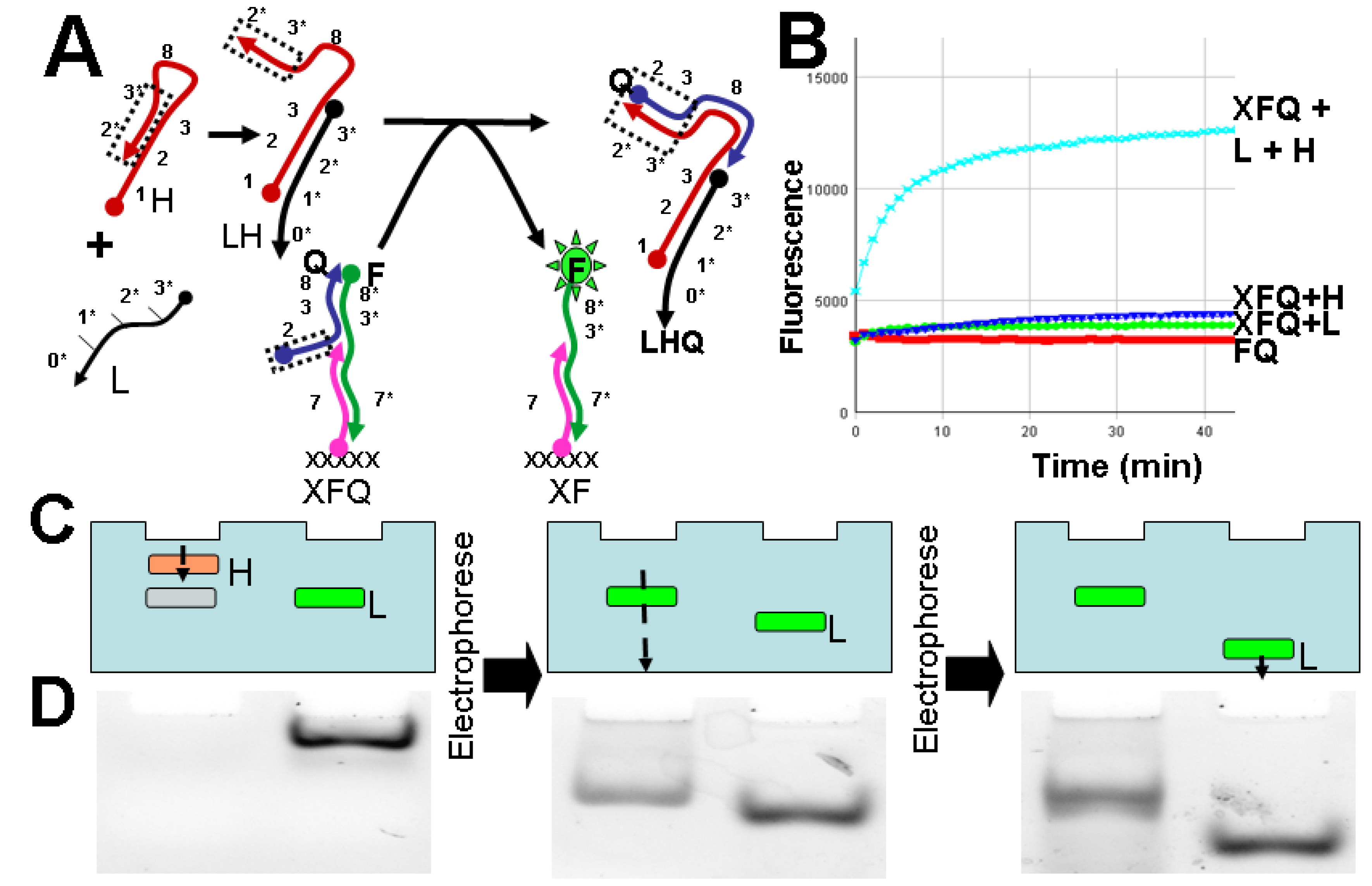
2.2. Design and Assay of DNA Circuits
2.3. Execution of Circuits within an Electrophoretic Gel

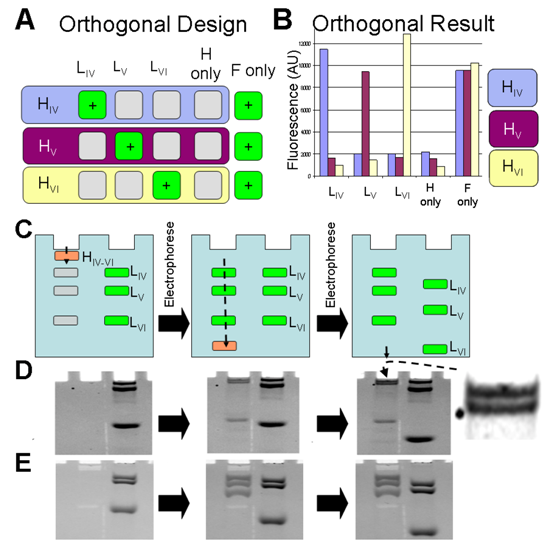
2.4. Generation of Three Lines from Three, Orthogonal Reactions
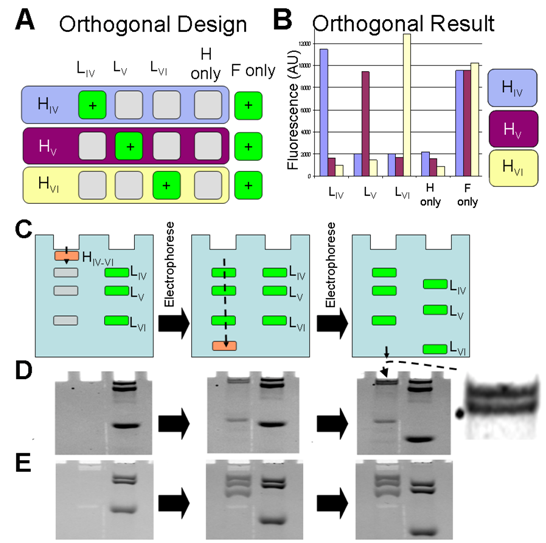
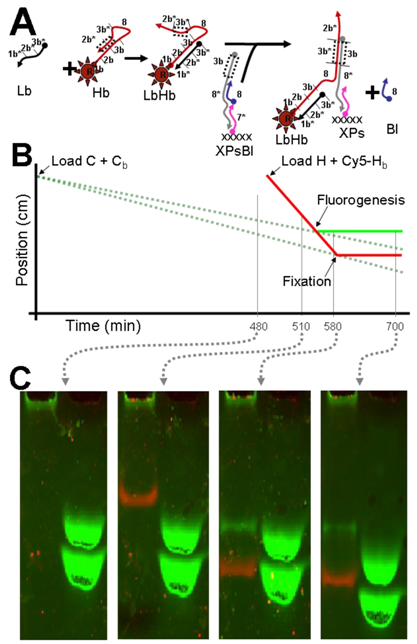
2.5. Modular Manipulation of DNA Circuits; Fixing a Mobile, Fluorescent Species

3. Experimental
3.1. Control of DNA Electrophoretic Mobility in a DNA-Functionalized Polyacrylamide Gel

3.2. Solution Assay of Fluorogenic DNA Circuits
3.3. Execution of Circuits within an Electrophoretic Gel
3.4. Generation of Three Lines from Three, Orthogonal Reactions
3.5. Modular Manipulation of DNA Circuits; Fixing a Mobile, Fluorescent Species
3.6. DNA Sequences
- B 5'-acrydite-TTTGATTTGATT
- LI 5'-fluorescein-CGACATCTAACCTAGCAAATACGATGAATCAAATTTAC
- LII 5'-fluorescein-CGACATCTAACCTAGCCTCCTCCTCCAATCAAATCAAC
- LIII 5'-fluorescein-CGACATCTAACCTAGCCTCCTCCTCCAATCAAATCAAA
- H 5'-GTCAGTGATGCTAGGTTAGATGTCGCCATGTGTAGACGACATCTAACCTAGCCCTTGTCATAGA GCAC
- L 5'-CGACATCTAACCTAGCATCACTGACAATCAAATCTTC
- F 5'-GAATTGTCAGACTTCTCCTTGTCATAGAGCAC-3’-fluorescein
- X 5'-acrydite-AGAAGTCTGACAATTC
- Q 5'-Black Hole Quencher-GTGCTCTATGACAAGGGCTAGGTT
- Ps 5'-GTTTACCG CTCCCTCCTTTTAGTTGGAATTGTCAGACTTCT
- Bl 5'-CAACTAAAAGGAGGGAG
- Hb 5'-Cy5-TCGTATTTGGTTTTTGGTTTACCGCAACTAAAAGGAGGGAGCGGTAAACCAAAAACC
- Lb 5'-CGGTAAACCAAAAACCAAATACGAAATCAAATCAAA
- HVI 5'-GGAGGAGGAGGCTAGGTTAGATGTCGCCATGTGTAGACGACATCTAACCTAGCCCTTGTCATA GAGCAC
- LVI 5'-fluorescein-CGACATCTAACCTAGCCTCCTCCTCCAATCAAATCAAC
- HV 5'-CATCGTATTTGCTAGGTTAGATGTCGCCATGTGTAGACGACATCTAACCTAGCCCTTGTCATAG AGCAC
- LVa 5'-fluorescein-CGACATCTAACCTAGCAAATACGATGAATCAAATCATG
- LVb 5'-fluorescein-CGACATCTAACCTAGCAAATACGATGTCCCAAATCAAA
- HIV 5'-CACCACCCACGCTAGGTTAGATGTCGCCATGTGTAGACGACATCTAACCTAGCCCTTGTCATA GAGCAC
- LIV 5'-fluorescein-CGACATCTAACCTAGCGTGGGTGGTGAATCAAATCTTC
4. Conclusions
Acknowledgments
- Sample Availability: Not available.
References
- Kondo, S.; Miura, T. Reaction-Diffusion Model as a Framework for Understanding Biological Pattern Formation. Science 2010, 329, 1616–1620. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef]
- Macfarlane, R.J.; Lee, B.; Jones, M.R.; Harris, N.; Schatz, G.C.; Mirkin, C.A. Nanoparticle Superlattice Engineering with DNA. Science 2011, 334, 204–208. [Google Scholar] [CrossRef]
- Yin, P.; Choi, H.M.T.; Calvert, C.R.; Pierce, N.A. Programming biomolecular self-assembly pathways. Nature 2008, 451, 318–322. [Google Scholar] [CrossRef]
- Phillips, A.; Cardelli, L. A programming language for composable DNA circuits. J. Roy. Chem. Soc. 2009, 6 (Suppl. 4), S419–S436. [Google Scholar]
- Ouchterlony, O. Diffusion-in-gel methods for immunological analysis. Prog. Allergy 1958, 5, 1–78. [Google Scholar]
- Rehman, F.N.; Audeh, M.; Abrams, E.S.; Hammond, P.W.; Kenney, M.; Boles, T.C. Immobilization of acrylamide-modified oligonucleotides by co-polymerization. Nucleic Acids Res. 1999, 27, 649–655. [Google Scholar] [CrossRef]
- Chan, A.; Krull, U.J. Capillary electrophoresis for capture and concentrating of target nucleic acids by affinity gels modified to contain single-stranded nucleic acid probes. Anal. Chim. Acta 2006, 578, 31–42. [Google Scholar] [CrossRef]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef]
- SantaLucia, J., Jr. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Turberfield, A.J.; Yurke, B.; Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science 2007, 318, 1121–1125. [Google Scholar] [CrossRef]
- Turing, A.M. The Chemical Basis of Morphogenesis. Philos. T. Roy. Soc. B 1952, 237, 37–72. [Google Scholar] [CrossRef]
- Zhu, Z.; Wu, C.; Liu, H.; Zou, Y.; Zhang, X.; Kang, H.; Yang, C.J.; Tan, W. An Aptamer Cross-Linked Hydrogel as a Colorimetric Platform for Visual Detection. Angew. Chem. Int. Ed. Engl. 2010, 49, 1052–1056. [Google Scholar] [CrossRef]
- Hoque, M.E.; Chuan, Y.L.; Pashby, I. Extrusion based rapid prototyping technique: An advanced platform for tissue engineering scaffold fabrication. Biopolymers 2011, 97, 83–93. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Allen, P.B.; Chen, X.; Ellington, A.D. Spatial Control of DNA Reaction Networks by DNA Sequence. Molecules 2012, 17, 13390-13402. https://doi.org/10.3390/molecules171113390
Allen PB, Chen X, Ellington AD. Spatial Control of DNA Reaction Networks by DNA Sequence. Molecules. 2012; 17(11):13390-13402. https://doi.org/10.3390/molecules171113390
Chicago/Turabian StyleAllen, Peter B., Xi Chen, and Andrew D. Ellington. 2012. "Spatial Control of DNA Reaction Networks by DNA Sequence" Molecules 17, no. 11: 13390-13402. https://doi.org/10.3390/molecules171113390
APA StyleAllen, P. B., Chen, X., & Ellington, A. D. (2012). Spatial Control of DNA Reaction Networks by DNA Sequence. Molecules, 17(11), 13390-13402. https://doi.org/10.3390/molecules171113390