A Facile Synthesis of 5'-Fluoro-5'-deoxyacadesine (5'-F-AICAR): A Novel Non-phosphorylable AICAR Analogue

 ,
,
Abstract
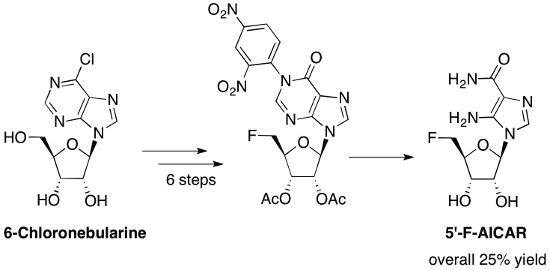
:
{kind=link}
{kind=link}
{kind=link}
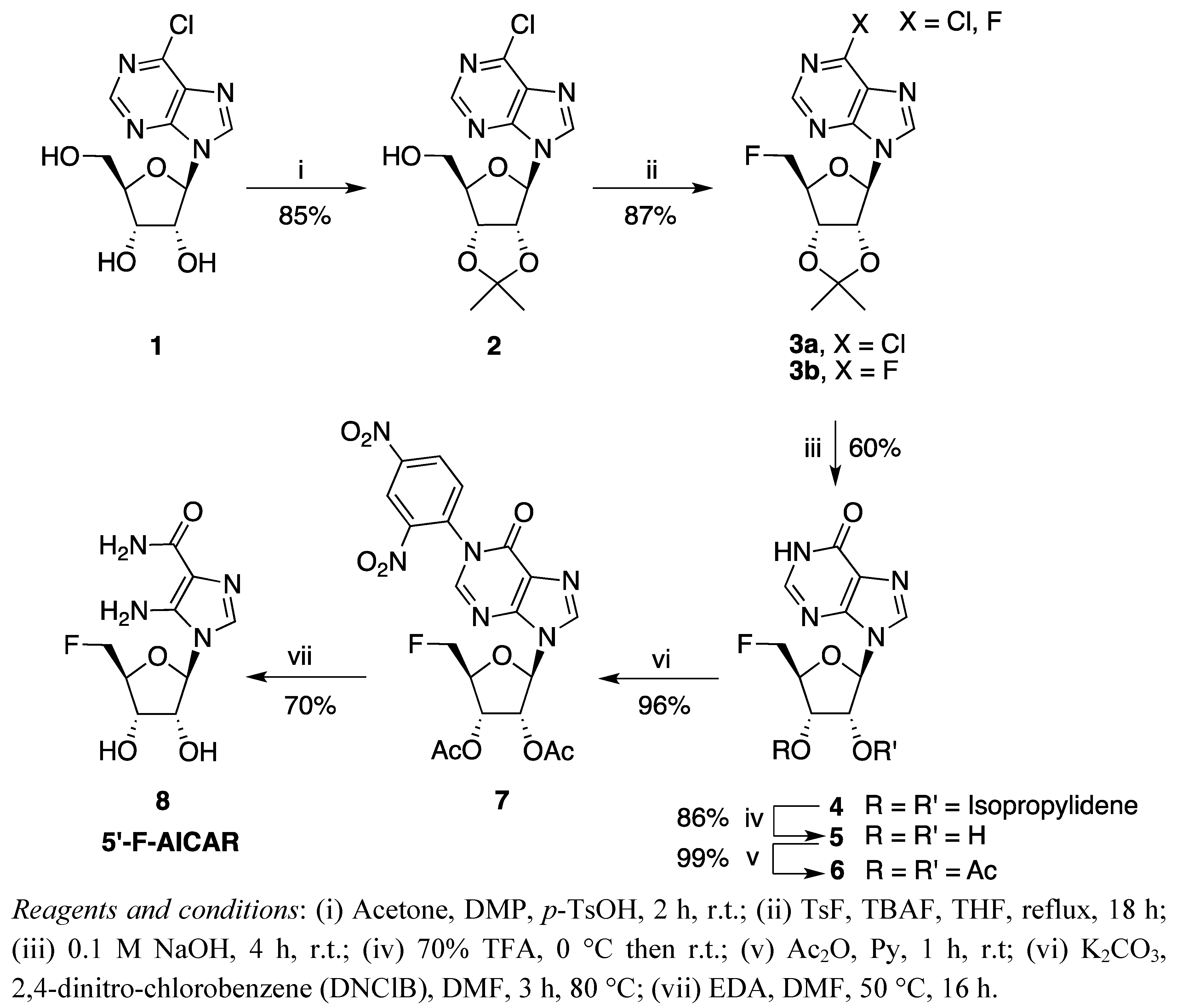{kind=link}
1. Introduction

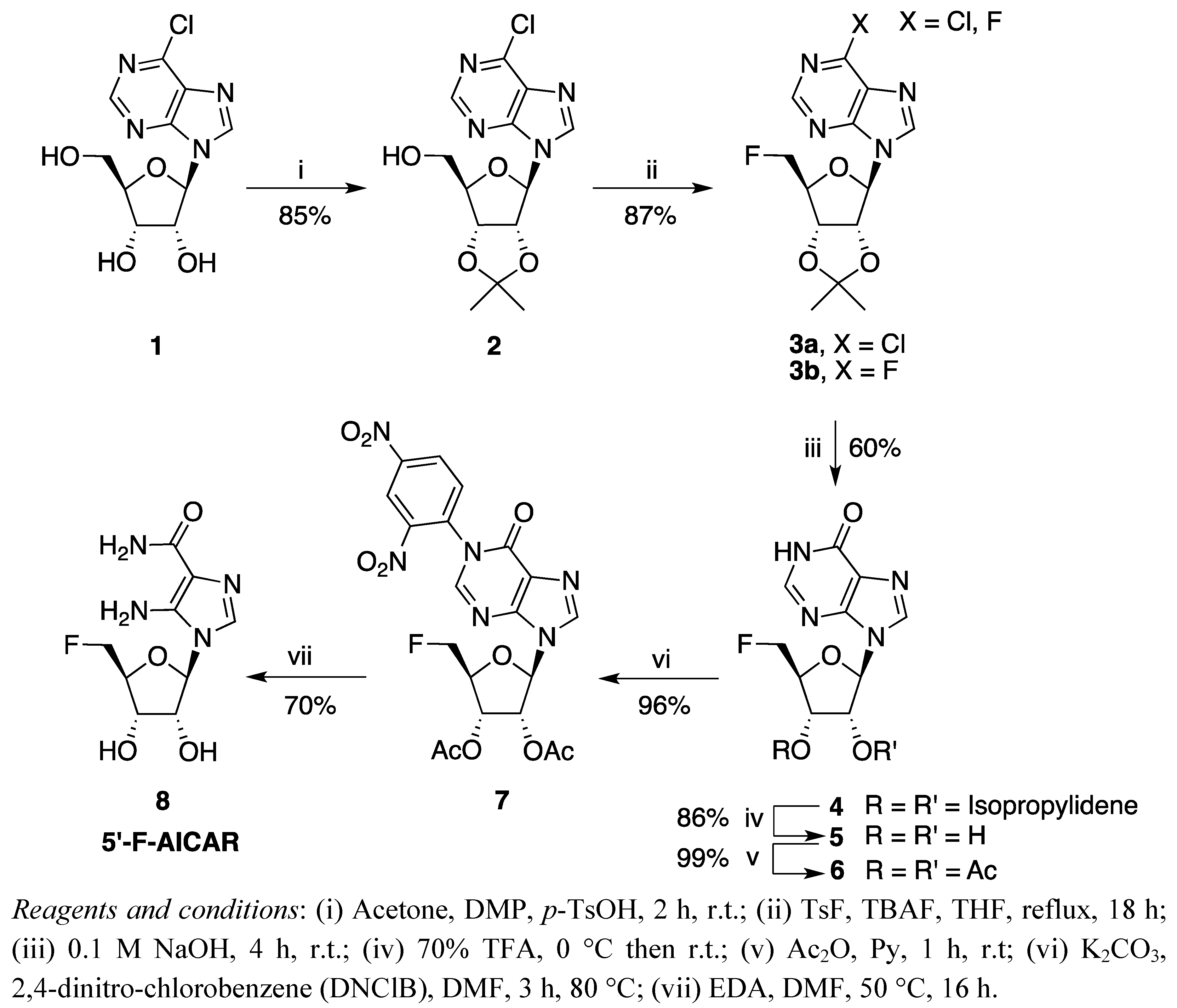
2. Results and Discussion
3. Experimental
General
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds 6–8 are available from the authors.
References
- Madela, K.; McGuigan, C. Progress in the development of anti-hepatitis C virus nucleoside and nucleotide prodrugs. Fut. Med. Chem. 2012, 4, 625–650. [Google Scholar] [CrossRef]
- Zhou, C.; Chattopadhyaya, J. The synthesis of therapeutic locked nucleos(t)ides. Curr. Opin. Drug Discov. Dev. 2009, 12, 876–898. [Google Scholar]
- Carell, T.; Brandmayr, C.; Hienzsch, A.; Müller, M.; Pearson, D.; Reiter, V.; Thoma, I.; Thumbs, P.; Wagner, M. Structure and function of noncanonical nucleobases. Angew. Chem. Int. Ed. 2012, 51, 7110–7131. [Google Scholar]
- D'Errico, S.; Oliviero, G.; Amato, J.; Borbone, N.; Cerullo, V.; Hemminki, A.; Piccialli, V.; Zaccaria, S.; Mayol, L.; Piccialli, G. Synthesis and biological evaluation of unprecedented ring-expanded nucleosides (RENs) containing the imidazo[4,5-d][1,2,6]oxadiazepine ring system. Chem. Commun. 2012, 48, 9310–9312. [Google Scholar]
- Romeo, G.; Chiacchio, U.; Corsaro, A.; Merino, P. Chemical synthesis of heterocyclic-Sugar nucleoside analogues. Chem. Rev. 2010, 110, 3337–3370. [Google Scholar] [CrossRef]
- Hosmane, R.S. Ring-expanded (“Fat”) purines and their nucleoside/nucleotide analogues as broad-spectrum therapeutics. In Progress in Heterocyclic Chemistry, 1st; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Oxford, UK, 2009; Volume 21, pp. 35–68. [Google Scholar]
- D’Alonzo, D.; Guaragna, A.; Van Aerschot, A.; Herdewijn, P.; Palumbo, G. De novo approach to L-Anhydrohexitol nucleosides as building blocks for the synthesis of l-Hexitol Nucleic Acids (L-HNA). Tetrahedron Lett. 2008, 49, 6068–6070. [Google Scholar] [CrossRef]
- D'Alonzo, D.; Van Aerschot, A.; Guaragna, A.; Palumbo, G.; Schepers, G.; Capone, S.; Rozenski, J.; Herdewijn, P. Synthesis and base pairing properties of 1′,5′-Anhydro-l-Hexitol Nucleic Acids (L-HNA). Chem. Eur. J. 2009, 15, 10121–10131. [Google Scholar] [CrossRef]
- Marquez, V.E. The properties of locked methanocarba nucleosides in biochemistry, biotechnology, and medicinal chemistry. In Modified Nucleosides: In Biochemistry, Biotechnology and Medicine, 1st; Herdewijn, P., Ed.; Wiley-VCH Verlag GmbH Co. KGaA: Weinheim, Germany, 2008; pp. 307–341. [Google Scholar]
- D’Errico, S.; Oliviero, G.; Piccialli, V.; Amato, J.; Borbone, N.; D’Atri, V.; D’Alessio, F.; Di Noto, R.; Russo, F.; Salvatore, F.; Piccialli, G. Solid-phase synthesis and pharmacological evaluation of novel nucleoside-tethered dinuclear platinum (II) complexes. Bioorg. Med. Chem. Lett. 2011, 21, 5835–5838. [Google Scholar]
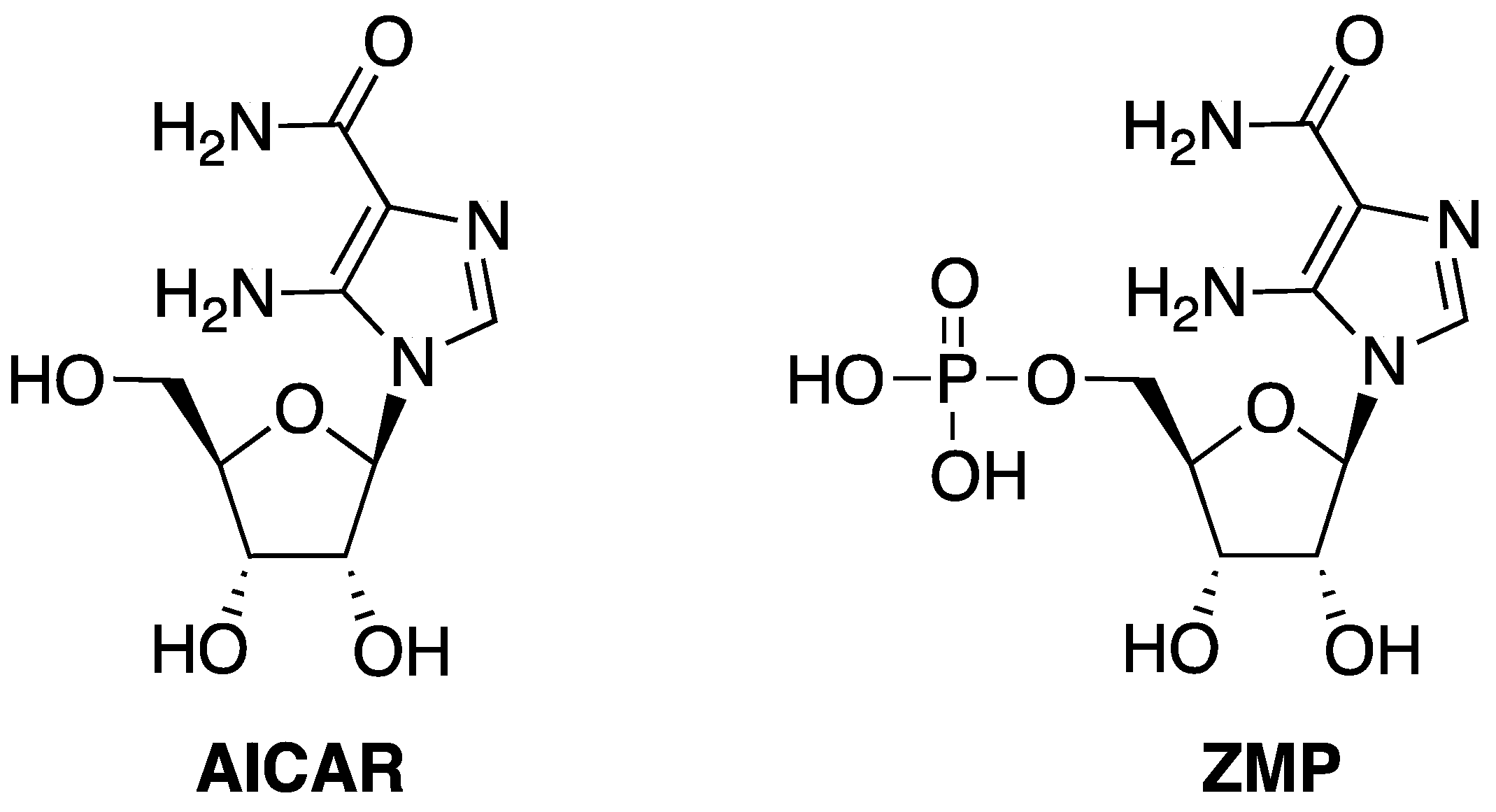
- Daignan-Fornier, B.; Pinson, B. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranosyl 5′-Monophosphate (AICAR), a highly conserved purine intermediate with multiple effects. Metabolites 2012, 2, 292–302. [Google Scholar] [CrossRef]
- Gruzman, A.; Babai, G.; Sasson, S. Adenosine monophosphate-activated protein kinase (AMPK) as a new target for antidiabetic drugs: A review on metabolic, pharmacological and chemical considerations. Rev. Diabet. Stud. 2009, 6, 13–36. [Google Scholar] [CrossRef]
- Merrill, G.F.; Kurth, E.J.; Hardie, D.G.; Winder, W.W. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. Endocrinol. Metab. 1997, 273, E1107–E1112. [Google Scholar]
- Sepe, V.; D’Orsi, R.; Borbone, N.; D’Auria, M.V.; Bifulco, G.; Monti, M.C.; Catania, A.; Zampella, A. Callipeltins F-I: New antifungal peptides from the marine sponge Latrunculia sp. Tetrahedron 2006, 62, 833–840. [Google Scholar] [CrossRef]
- Ewart, M.A.; Kennedy, S. Diabetic cardiovascular disease AMP-activated protein kinase (AMPK) as a therapeutic target. Cardiovasc. Hematol. Agents Med. Chem. 2012, 10, 190–211. [Google Scholar] [CrossRef]
- Sengupta, T.K.; Leclerc, G.M.; Hsieh-Kinser, T.; Leclerc, G.J.; Singh, I.; Barredo, J.C. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: Implication for targeted therapy. Mol. Cancer 2007. [Google Scholar] [CrossRef]
- Xiang, X.; Saha, A.K.; Wen, R.; Ruderman, N.B.; Luo, Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem. Biophys. Res. Commun. 2004, 321, 161–167. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Dagnino-Acosta, A.; Ainbinder, A.; Cheng, Q.; Joshi, A.D.; Chen, Z.; Yarotskyy, V.; Oakes, J.M.; Lee, C.S.; et al. AICAR prevents heat-induced sudden death in RyR1 mutant mice independent of AMPK activation. Nat. Med. 2012, 18, 244–251. [Google Scholar]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Mayol, L. Synthesis of N-1 and ribose modified inosine analogues on solid support. Tetrahedron Lett. 2007, 48, 397–400. [Google Scholar]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Bucci, E.; Piccialli, V.; Mayol, L. Synthesis of 4-N-alkyl and ribose-modified AICAR analogues on solid support. Tetrahedron 2008, 64, 6475–6481. [Google Scholar] [CrossRef]
- Oliviero, G.; D Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Piccialli, G.; Mayol, L. A solid-phase approach to the synthesis of N-1-alkyl analogues of cyclic inosine-diphosphate-ribose (cIDPR). Tetrahedron 2010, 66, 1931–1936. [Google Scholar] [CrossRef]
- Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Piccialli, G.; Mayol, L. Facile solid-phase synthesis of AICAR 5′-Monophosphate (ZMP) and its 4-N-alkyl derivatives. Eur. J. Org. Chem. 2010. [Google Scholar] [CrossRef]
- D’Errico, S.; Piccialli, V.; Oliviero, G.; Borbone, N.; Amato, J.; D’Atri, V.; Piccialli, G. Probing the reactivity of nebularine N1-oxide. A novel approach to C-6 C-substituted purine nucleosides. Tetrahedron 2011, 67, 6138–6144. [Google Scholar]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Mayol, L.; Piccialli, G. Solid-phase synthesis of a New Diphosphate 5-Aminoimidazole-4-carboxamide Riboside (AICAR) derivative and studies toward cyclic AICAR diphosphate ribose. Molecules 2011, 16, 8110–8118. [Google Scholar] [CrossRef]
- Kirk, K.L. Fluorine in medicinal chemistry: Recent therapeutic applications of fluorinated small molecules. J. Fluorine Chem. 2006, 127, 1013–1029. [Google Scholar] [CrossRef]
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluorine Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Pankiexicz, K.W. Fluorinated nucleosides. Carbohydr. Res. 2000, 327, 87–105. [Google Scholar] [CrossRef]
- Peng, L. Fluorinated nucleosides: Synthesis and biological implication. J. Fluorine Chem. 2008, 129, 743–766. [Google Scholar] [CrossRef]
- Maruyama, T.; Ikejiri, M.; Izawa, K.; Onishi, T. Synthesis and biological activity of fluorinated nucleosides. In Medicinal Chemistry and Chemical Biology, 1st; Ojima, I., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2009; pp. 165–198. [Google Scholar]
- Ashton, T.D.; Scammels, P.J. An improved synthesis of 5′-fluoro-5′-deoxyadenosines. Bioorg. Med. Chem. Lett. 2005, 15, 3361–3363. [Google Scholar] [CrossRef]
- Li, W.; Yin, X.; Schneller, S.W. 5′-Fluoro-5′-deoxyaristeromycin. Bioorg. Med. Chem. Lett. 2008, 18, 220–222. [Google Scholar] [CrossRef]
- Kappler, F.; Hampton, A. Approaches to isozyme-specific inhibitors. 17. Attachment of a selectivity-inducing substituent to a multisubstrate adduct. Implications for facilitated design of potent, isozyme-selective inhibitors. J. Med. Chem. 1990, 33, 2545–2551. [Google Scholar] [CrossRef]
- Spitale, R.C. Efficient Syntheses of 5“-Deoxy-5-”fluoroguanosine and-inosine. J. Org. Chem. 2007, 72, 8551–8554. [Google Scholar] [CrossRef]
- De Napoli, L.; Messere, A.; Montesarchio, D.; Piccialli, G.; Varra, M. 1-Substituted 2′-deoxyinosine analogues. J. Chem. Soc. Perkin Trans. I 1997. [Google Scholar] [CrossRef]
- Galeone, A.; Mayol, L.; Oliviero, G.; Piccialli, G.; Varra, M. Synthesis of a novel N-1 carbocyclic, N-9 butyl analogue of cyclic ADP ribose (cADPR). Tetrahedron 2002, 58, 363–368. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D'Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; D'Alonzo, D.; Piccialli, V.; Mayol, L.; Piccialli, G. A Facile Synthesis of 5'-Fluoro-5'-deoxyacadesine (5'-F-AICAR): A Novel Non-phosphorylable AICAR Analogue. Molecules 2012, 17, 13036-13044. https://doi.org/10.3390/molecules171113036
D'Errico S, Oliviero G, Borbone N, Amato J, D'Alonzo D, Piccialli V, Mayol L, Piccialli G. A Facile Synthesis of 5'-Fluoro-5'-deoxyacadesine (5'-F-AICAR): A Novel Non-phosphorylable AICAR Analogue. Molecules. 2012; 17(11):13036-13044. https://doi.org/10.3390/molecules171113036
Chicago/Turabian StyleD'Errico, Stefano, Giorgia Oliviero, Nicola Borbone, Jussara Amato, Daniele D'Alonzo, Vincenzo Piccialli, Luciano Mayol, and Gennaro Piccialli. 2012. "A Facile Synthesis of 5'-Fluoro-5'-deoxyacadesine (5'-F-AICAR): A Novel Non-phosphorylable AICAR Analogue" Molecules 17, no. 11: 13036-13044. https://doi.org/10.3390/molecules171113036