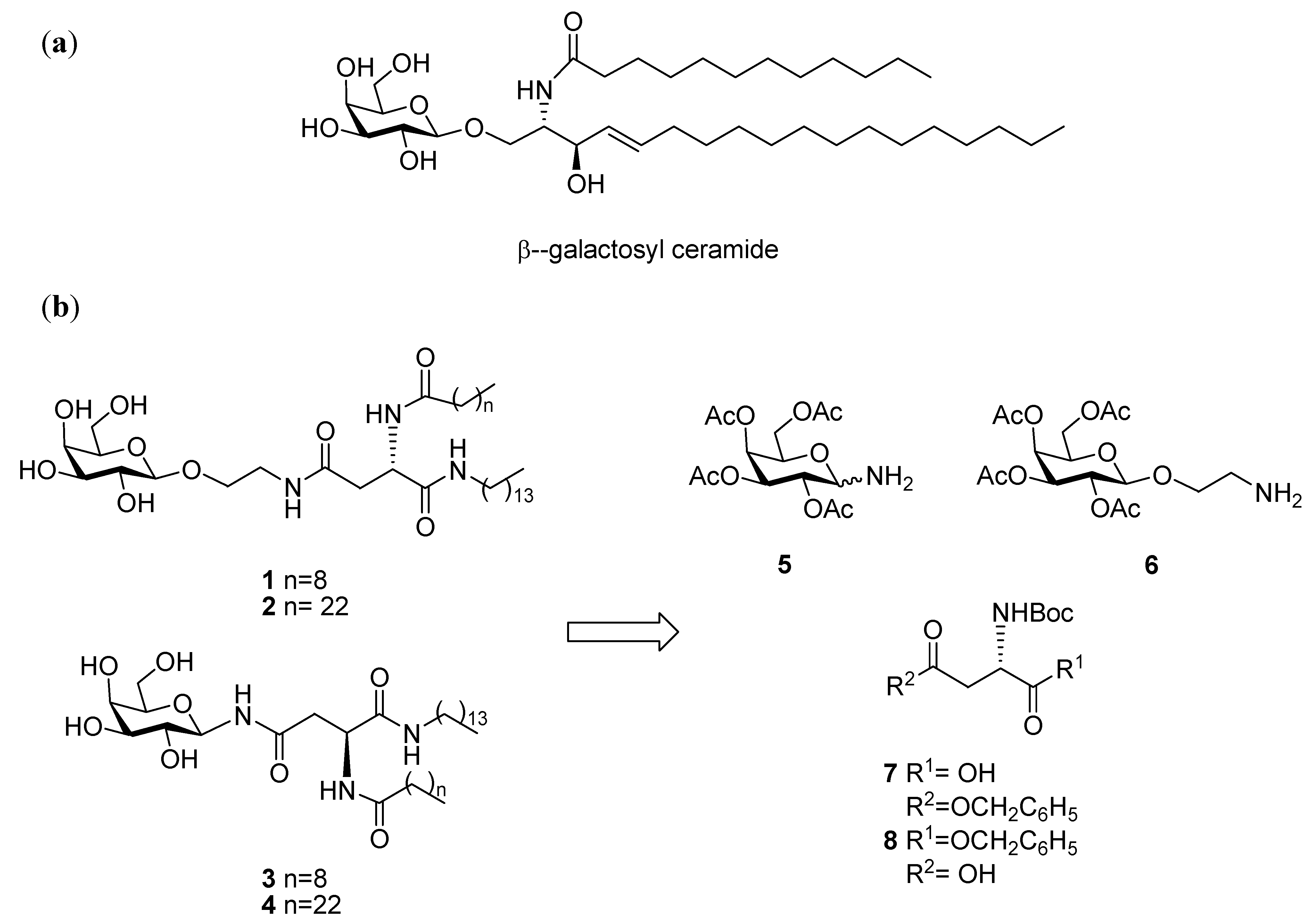
General Methods
All chemicals purchased were reagent grade and used without further purification unless stated otherwise. Dichloromethane was freshly distilled over CaH2 prior use. Anhydrous dimethylformamide (DMF) was purchased from Sigma Aldrich. Molecular sieves (MS) used for glycosylation and coupling reactions were 8–12 mesh and were flame dried prior to use. Reactions were monitored with thin layer chromatography (TLC) on Merck Silica Gel F254 plates, using mixtures of hexane/ethyl acetate unless otherwise stated. Detection was effected either by visualisation in UV light and/or charring in a mixture of 5% sulphuric acid-EtOH or phosphomolybdic acid-EtOH. NMR spectra were obtained on a Bruker Avance 300 spectrometer. Proton and carbon signals were assigned with the aid of 2D-NMR experiments and DEPT experiments for novel compounds. The 2D-NMR experiments included COSY and HCCOSW, which is an HSQC type of experiment. Better resolution of the signals was observed when using the HCCOSW experiments than with conventional HSQC experiments. Chemicals shifts for 1H-NMR are reported in ppm relative to residual solvent proton. Flash chromatography was performed with Merck Silica Gel 60, using adjusted mixtures of hexane/ethyl acetate unless otherwise stated. Optical rotations were obtained using an AA-100 polarimeter. [α]25 values are given in 10−1 cm2·g−1. The melting points were obtained using a Stuart Scientific SMP1 melting point apparatus and are uncorrected. High resolution mass spectrometry (HRMS) were performed on an Agilent-LC 1200 Series coupled to a 6210 Agilent Time-Of-Flight (TOF) mass spectrometer equipped with an electrospray source both positive and negative (ESI+/−) or in a MALDI-QTOF Premier MS SYSTEM, using an α-cyano-4-hydroxy cinnamic acid matrix. Infrared spectra were obtained as a film on NaCl plates in the region 4000–400 cm−1 on a Nicolet Impact 400D spectrophotometer.
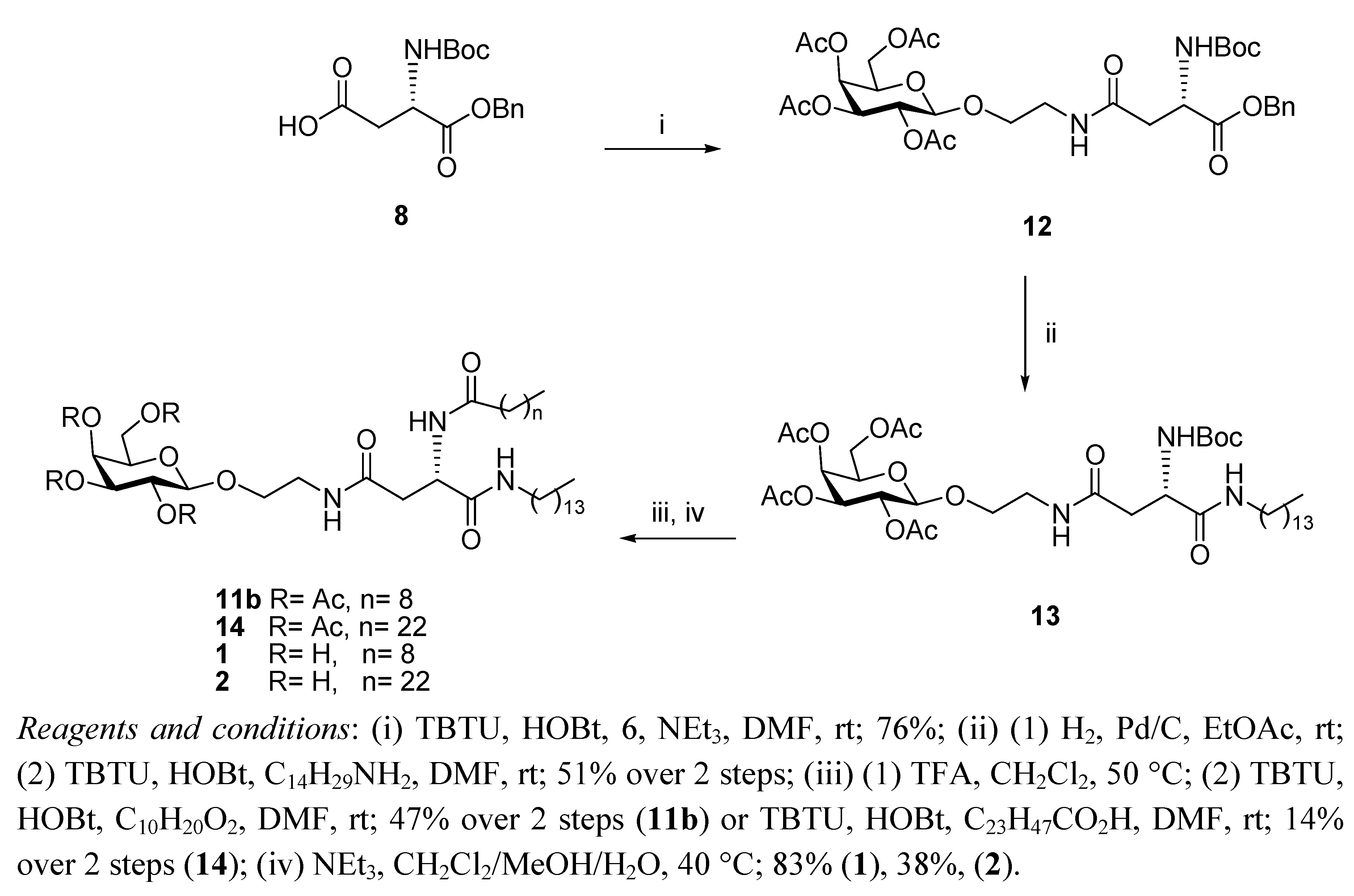
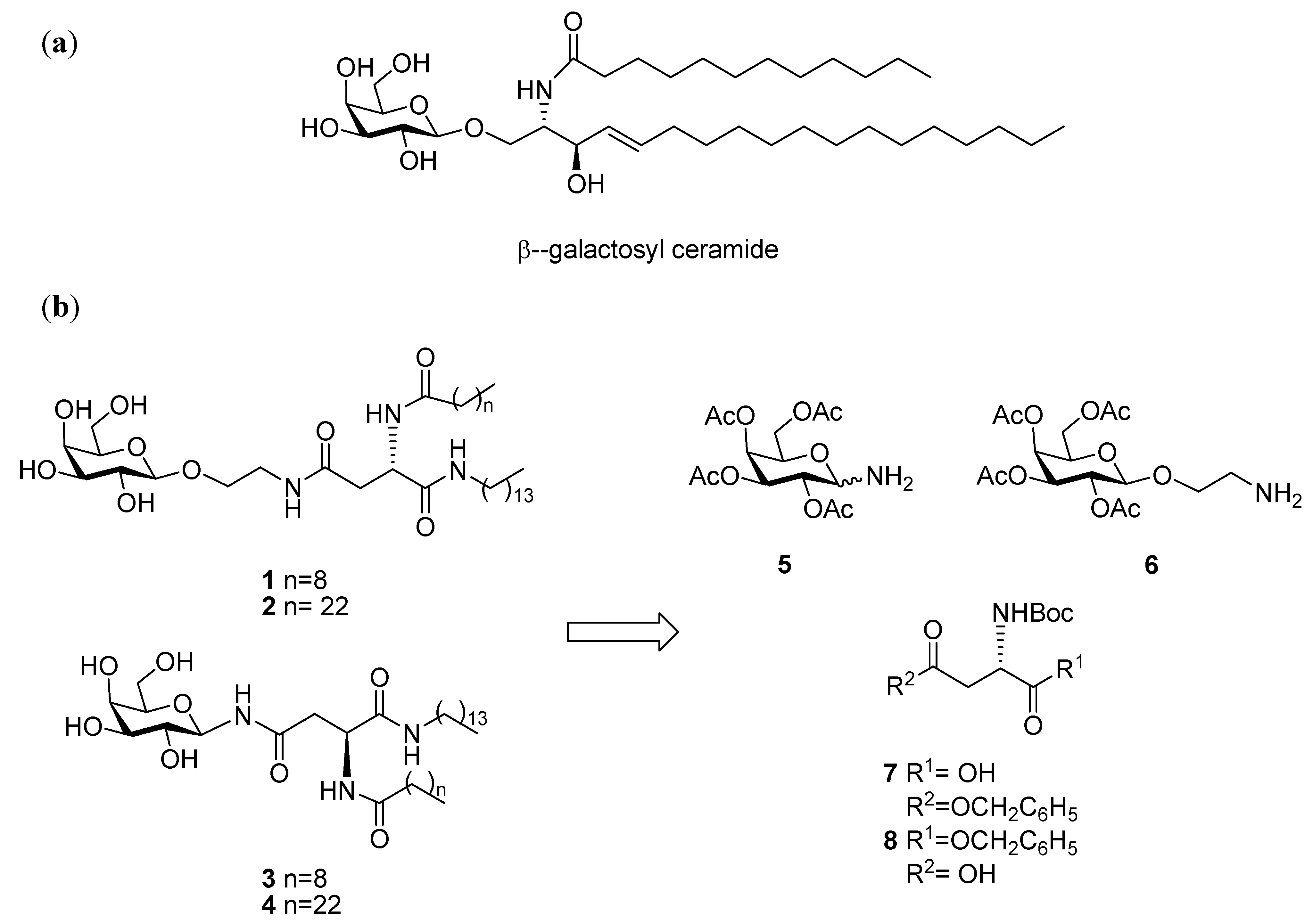
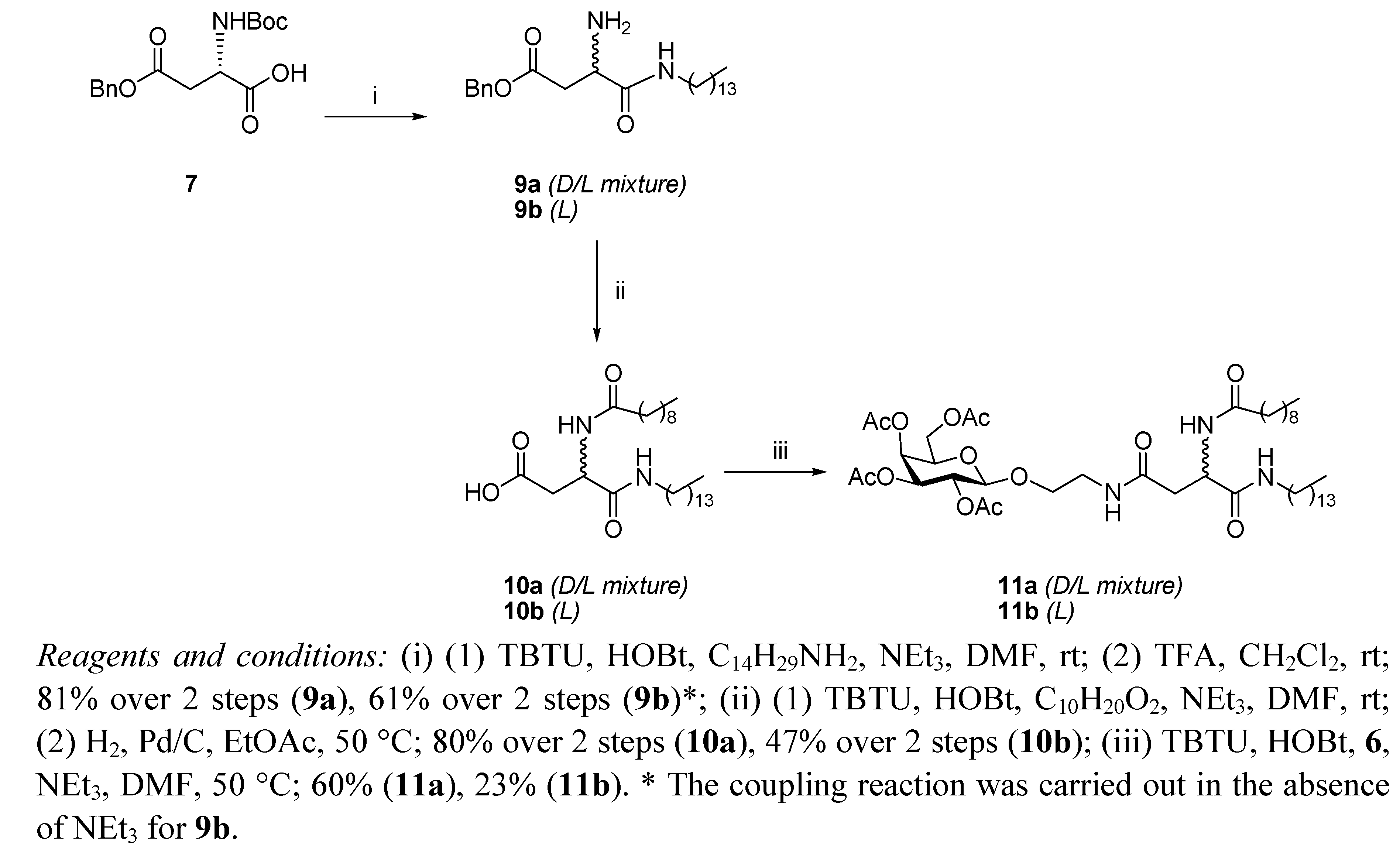
N4-[2-O-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-ethyl]-N2-tert-butoxycarbonyl-L-asparagine benzyl ester (12). HOBt (0.09 g, 0.68 mmol), followed by NEt3 (0.18 mL, 1.23 mmol), were added to a stirring solution of N-Boc-L-Asp-OBn 8 (0.2 g, 0.61 mmol) and TBTU (0.22 g, 0.6 mmol) dissolved in anhydrous DMF (10 mL), under N2 at rt. It was stirred for 30 min and 6 (0.29 g, 0.74 mmol) dissolved in anhydrous DMF (1.2 mL) was added dropwise. It was stirred for 18 h. The reaction mixture was concentrated under reduced pressure, diluted with ethyl acetate and washed succesively with HCl 0.1 N, aqueous sat. NaHCO3 solution, and brine. Flash chromotagraphy (hexane:ethyl acetate, 1:1) afforded 12 as a white solid (0.33 g, 76%). [α]22D +6.9 (c 1.35, CH2Cl2,); IR (NaCl film): 3374.7, 2978.0, 1750.7, 1665.8, 1499.3, 1368.8, 1224.3, 1167.9, 1124.3,1057.2 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.34 (bs, 5 H, H-Ph), 6.01 (t,J = 5.1 Hz, 1 H, CH2CH2NHCO), 5.76 (d, J = 8.1 Hz, 1 H, NHCOC(CH3)3), 5.39 (dd, J =0.6 Hz, J = 3.3 Hz, 1 H, H-4), 5.20–5.16 (m, 3 H, overlap of H-2, CH2Ph), 5.02 (dd, J = 3.3 Hz, J = 10.2 Hz, 1 H, H-3), 4.57–4.54 (m, 1 H, H-α), 4.44 (d, J = 7.8 Hz, 1 H, H-1), 4.18–4.13 (m, 2 H, overlap of H-6, H-6'), 3.93–3.89 (m, 1 H, H-5), 3.86–3.80 (m, 1 H, 1 H of OCH2CH2NH), 3.66–3.59 (m, 1 H, 1 H of OCH2CH2NH), 3.46–3.38 (m, 2 H, OCH2CH2NH), 2.91 (dd, J = 5.7 Hz, J = 17.4 Hz, 1 H, H-β), 2.71 (dd, J = 4.5 Hz, J = 15.9 Hz, 1 H, H-β'), 2.15 (s, 3 H, O(CO)CH3), 2.05 (s, 6 H, O(CO)CH3 × 2), 1.99 (s, 3 H, O(CO)CH3), 1.42 (s, 9 H, COC(CH3)3); 13C-NMR (75 Hz, CDCl3): δ 171.38, 170.37, 169.76 (each CO), 155.55 (C-Ph), 128.51, 128.09 (CH-Ph), 101.42 (C-1), 79.01 (COC(CH3)3), 70.89 (C-5), 70.68 (C-2), 68.91 (C-3), 67.25 (CH2Ph), 66.96 (C-4), 61.35 (C-6), 50.47 (C-α), 39.18, 37.72 (each OCH2CH2NH), 37.12 (C-β), 28.29 (COC(CH3)3), 20.82, 20.57 (overlap of O(CO)CH3); HRMS (MS-TOF): [M+H]+ calcd. for C32H44N2O15: 697.2181 found 697.2800.
N4-[2-O-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-ethyl]-N2-tert-butoxycarbonyl-L-asparagine tetradecylamide (13). To a solution of 12 (0.120 g, 0.17 mmol) in ethyl acetate (6 mL), Pd/C 10% w/w (0.012 g, 10% w/w) was added. The resulting slurry was stirred under H2 gas for 4 h. The mixture was then filtered through a Celite cake and the filtrate was concentrated under vacuum to afford the corresponding carboxylic acid as an off-white solid, which was used without further purification (0.094 g, 90%). 1H-NMR (300 MHz, CDCl3): δ 6.53 (bs, 1 H, CH2CH2NHCO), 5.86 (d, J = 5.4 Hz, 1 H, NHCOC(CH3)3), 5.41 (d, J = 3.3 Hz, 1 H, H-4), 5.17 (dd, J = 7.8 Hz, J = 10.2 Hz, 1 H, H-2), 5.00 (dd, J = 3.3 Hz, J = 13.8 Hz, 1 H, H-3), 4.49 (d, J = 7.8 Hz, 1 H, H-1), 4.45–4.42 (m, 1 H, H-α), 4.20–4.11 (m, 2 H, overlap of H-6, H-6'), 3.96–3.92 (m, 1 H, H-5) 3.90–3.85 (m,1 H, 1 H of OCH2CH2NH), 3.75–3.68 (m, 1 H, 1 H of OCH2CH2NH), 3.54–3.39 (m, 2 H, OCH2CH2NH), 2.91 (d, J = 15.6 Hz, 1 H, H-β), 2.72 (dd, J = 8.49 Hz, J = 15.9 Hz, 1 H, H-β'), 2.17, 2.13, 2.05, 1.98 (each s, 3 H, O(CO)CH3), 1.42 (s, 9 H, COC(CH3)3); HRMS (MS-TOF): [M+K]+ calcd. for C25H38N2O15: 645.1904 found 645.1899. HOBt (0.034 g, 0.25 mmol) was added to a stirring solution of the carboxylic acid obtained as described above (0.140 g, 0.23 mmol), tetradecylamine (0.06 g, 0.28 mmol), and TBTU (0.081 g, 0.25 mmol) dissolved in anhydrous DMF, (12 mL) at rt. It was stirred for 18 h. The reaction mixture was concentrated in vacuo, diluted with ethyl acetate and washed with brine. Flash chromotagraphy (ethyl acetate) afforded 13 as a white solid (0.120 g, 56%). [α]22D = +8.8 (c 0.75, CH2Cl2,); IR (NaCl film): 3316.3, 3091.3 2919.9, 2851.3, 1748.0, 1687.1, 1646.1, 1548.9, 1524.1, 1467.4, 1434.6, 1369.2, 1368.8, 1230.2, 1171.0, 1055.3 cm−1; 1H-NMR (300 MHz, CDCl3): δ 6.87 (bs, 1 H, CONHC14CH29), 6.23 (t, J = 10.5 Hz, 1 H, CH2CH2NHCO), 6.14 (d, J = 8.1 Hz, 1 H, NHCOC(CH3)3), 5.38 (d, J = 2.7 Hz, 1 H, H-4), 5.15 (dd, J = 7.8 Hz, J = 10.5 Hz, 1 H, H-2), 5.01 (dd, J = 3.3 Hz, J = 10.5 Hz, 1 H, H-3), 4.49 (d, J = 7.8 Hz, 1 H, H-1), 4.40–4.39 (m, 1 H, H-α), 4.15–4.11 (m, 2 H, overlap of H-6, H-6'), 3.94–3.87 (m, 1 H, H-5), 3.86–3.81 (m, 1 H, 1 H of OCH2CH2NH), 3.67–3.60 (m, 1 H, 1 H of OCH2CH2NH), 3.49–3.34 (m, 2 H, OCH2CH2NH), 3.22–3.15 (m, 2 H, NHCH2C13H27), 2.71 (dd, J = 4.2 Hz, J = 15.6 Hz, 1 H, H-β), 2.51 (dd, J = 6.6 Hz, J = 15.6 Hz, 1 H, H-β'), 2.14, 2.07, 2.02, 1.96 (each s, 3 H, O(CO)CH3), 1.42 (m, 11 H, overlap of COC(CH3)3, NHCH2CH2C12H25), 1.23 (bs, 22 H, NHC2H4(CH2)11CH3), 0.87–0.83 (t, J = 6.6 Hz, 3 H, NHC13H26CH3); 13C-NMR (75 Hz, CDCl3): δ 171.17, 170.88, 170.35, 170.18, 170.05, 169.85, 155.73 (CO), 101.31 (C-1), 80.14 (COC(CH3)3), 70.81, 70.72 (C-5, C-3), 68.91. (C-2), 68.56 (NHCH2C13H27NH), 66.98 (C-4), 61.33 (C-6), 51.08 (C-α), 39.61, 39.22 (each OCH2CH2NH), 37.54 (C-β), 31.9, 29.67, 29.63, 29.59, 29.53, 29.41, 29.33, 29.22 (each CH2), 28.30 (COC(CH3)3), 26.83, 22.67 (each CH2), 20.82–20.57 (overlap of O(CO)CH3), 14.93 (NHC13H26CH3); HRMS (MS-TOF): [M+H]+ calcd. for C39 H67N3O14: 801.4623 found 801.4613.
N4-[2-O-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-ethyl]-N2-decanoyl-L-asparagine tetradecylamide (11b). A solution of 13 (0.11 g, 0.13mmol) in anhydrous CH2Cl2, (6 mL) was cooled in an ice bath and TFA (0.15 mL, 1.37 mmol) was added. The reaction mixture was heated to 50 °C for 1.5 h. The organic solvent was removed in vacuo and the residue obtained was diluted with ethyl acetate and washed with aqueous sat. NaHCO3 solution and brine, dried (MgSO4) and concentrated to yield the corresponding deprotected amine as a brown oil, which was used without further purification (0.071 g, 74%). 1H-NMR (300 MHz, CDCl3): δ 7.41 (bs, 1 H, CONHC14CH29), 6.42 (t, J = 5.1 Hz, 1 H, CH2CH2NHCO), 5.38 (d, J = 2.4 Hz, 1 H, H-4), 5.16 (dd, J = 7.8 Hz, J = 10.5 Hz, 1 H, H-2), 5.01 (dd, J = 3.3 Hz, J = 10.5 Hz, 1 H, H-3), 4.48 (d, J = 7.8 Hz, 1 H, H-1), 4.2-4.1 (m, 2 H, overlap of H-6, H-6'), 3.94-3.89 (m, 1 H, H-5) 3.87-3.82 (m,1 H, 1 H of OCH2CH2NH), 3.67-3.64 (m, 2 H, overlap of 1 H of OCH2CH2NH, H-α), 3.42-3.46 (bm, 2 H, OCH2CH2NH), 3.20-3.22 (m, 2 H, NHCH2C13H27),2.68 (dd, J = 3.9 Hz, J = 15 Hz, 1 H, H-β), 2.46 (dd, J = 7.8 Hz, J = 15 Hz, 1 H, H-β'), 2.15, 2.07, 2.03, 1.97 (each s, 3 H, O(CO)CH3), 1.50-1.43 (m, 2 H, NHCH2CH2C12H25), 1.23 (bs, 22 H, NHC2H4(CH2)11CH3), 0.87-0.83 (t, J = 6.6 Hz, 3 H, NHC13H26CH3); HRMS (MS-TOF): [M+H]+ calcd. for C34H59N3O12: 701.4099 found 701.4088. HOBt (0.041 g, 0.3 mmol) was added to a stirring solution of decanoic acid (0.048 g, 0.27 mmol) and TBTU (0.098 g, 0.3 mmol) dissolved in anhydrous DMF (6 mL), under N2 at rt. It was stirred for 10 min and the amine obtained from 13 as described above (0.06 g, 0.28 mmol) was dissolved in anhydrous DMF (8 mL) and added slowly. It was stirred for 18 h. The reaction mixture was concentrated in vacuo, diluted with ethyl acetate, washed with brine, dried (MgSO4) and concentrated . The residue obtained was purified by flash chromotagraphy (ethyl acetate) to afford 11b as a white solid (0.15 g, 63%). [α]22D = +5.8 (c 0.8, CH2Cl2,); IR (NaCl film): 3289.5, 3098.3, 2919.3, 2850.8, 1750.8, 168.1, 1646.5, 1542.4, 1467.4, 1370.4, 1225.5, 1174.9, 1058.5 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.43 (d, J = 6.9 Hz, 1 H, NHCOC9H19), 7.08 (t, J = 5.4 Hz, 1 H, CONHC14CH29), 6.26 (t, J = 5.4 Hz, 1 H, CH2CH2NHCO), 5.40 (d, J = 2.4 Hz, 1 H, H-4), 5.18 (dd, J = 7.8 Hz, J = 10.5 Hz, 1 H, H-2), 5.04 (dd, J = 3.3 Hz, J = 10.5 Hz, 1 H, H-3), 4.70–4.64 (m, 1 H, H-α), 4.54 (d, J = 7.8 Hz, 1 H, H-1), 4.22–4.11 (m, 2 H, overlap of H-6, H-6'), 3.97–3.94 (m, 1 H, H-5) 3.92–3.85 (m, 1 H, 1 H of OCH2CH2NH), 3.69–3.64 (m, 1 H, 1 H of OCH2CH2NH), 3.57–3.39 (m, 2 H, OCH2CH2NH), 3.22–3.16 (m, 2 H, NHCH2C13H27),2.81 (dd, J = 3.3 Hz, J = 15.3 Hz, 1 H, H-β), 2.46 (dd, J = 6.9, J = 15.6, 1 H, H-β'), 2.25–2.20 (t, J = 7.5 Hz, 2 H, COCH2C8H17), 2.16, 2.09, 2.05, 1.9 (each s, 3 H, O(CO)CH3), 1.62–1.60 (m, 2 H, COCH2CH2C7H15), 1.46–1.45 (m, 2 H, NHCH2CH2C12H25), 1.26 (bs, 34 H, overlap of COC2H4(CH2)6CH3, NHC2H4(CH2)11CH3), 0.90–0.85 (t, J = 6.6 Hz, 6 H, overlap of COC8H16CH3, NHC13H26CH3); 13C-NMR (75 Hz, CDCl3): δ 173.67, 171.68, 170.61, 170.38, 170.20, 170.06, 169.90 (each CO), 101.33 (C-1), 70.81, 70.73 (C-5, C-3), 68.97. (C-2), 68.51 (NHCH2C13H27), 66.98 (C-4), 61.30 (C-6), 49.72 (C-α), 39.67, 39.31 (each OCH2CH2NH), 36.98 (C-β), 36.62, 31.92, 29.70, 29.65, 29.56, 29.52, 29.36, 29.29, 28.31, 26.88, 25.63, 22.62 (each CH2), 20.87–20.85 (overlap of O(CO)CH3), 14.12 (overlap of COC8H16CH3, NHC13H26CH3); HRMS (MS-TOF): [M+H]+ calcd. for C44H77N3O13: 855.5456 found 855.5492.
N4-[2-O-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-ethyl]-N2-tetracosanoyl-L-asparagine tetradecylamide (14). HOBt (0.022 g, 0.16 mmol) was added to a stirring solution of tetracosanoic acid (0.056 g, 0.15 mmol) and TBTU (0.056 g, 0.16 mmol) dissolved in anhydrous DMF (6 mL), under N2 at rt. It was stirred for 10 min and the amine obtained from 13 as described above (0.128 g, 0.18 mmol) was dissolved in anhydrous DMF (8 mL) and added slowly. It was stirred for 3 h. The reaction mixture was concentrated in vacuo, diluted with ethyl acetate, washed with brine, dried (MgSO4) and concentrated. The residue obtained was purified by flash chromotagraphy (ethyl acetate) to afford 14 as a white solid, (0.030 g, 18%). [α]22D = +3.8 (c 0.83, CH2Cl2,); IR (NaCl film): 3423.0, 2918.4, 2850.3, 1749.6, 1644.4,1543.1, 1465.6, 1369.9, 1223.4, 1058.3 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.44 (d, J = 6.9 Hz, 1 H, NHCOC23H47), 7.09 (t, J = 4.8 Hz, 1 H, CONHC14CH29) 6.26 (bs, 1 H, CH2CH2NHCO), 5.40 (d, J = 3 Hz, 1 H, H-4), 5.17 (dd, J = 7.8 Hz , J = 10.5 Hz, 1 H, H-2), 5.04 (dd, J = 2.7 Hz, J = 10.2 Hz, 1 H, H-3), 4.70-4.64 (m, 1 H, H-α), 4.53 (d, J = 7.8 Hz, 1 H, H-1), 4.21-4.10 (m, 2 H, overlap of H-6, H-6'), 3.97–3.92 (tm, 1 H, H-5) 3.90–3.85 (m,1 H, 1 H of OCH2CH2NH), 3.71–3.64 (m, 1 H, 1 H of OCH2CH2NH), 3.54–3.44 (m, 2 H, OCH2CH2NH), 3.22–3.15 (m, 2 H, NHCH2C13H27),2.77 (dd, J = 3.3 Hz, J = 15.3 Hz, 1 H, H-β), 2.45 (dd, J = 6.9 Hz, J = 15.6 Hz, 1 H, H-β'), 2.25–2.20 (t, J = 7.5 Hz, 2 H, COCH2C22 H45), 2.16, 2.09, 2.04, 1.90 (each s, 3 H, O(CO)CH3), 1.62–1.59 (m, 2 H, COCH2CH2C21H43), 1.46–1.45 (m, 2 H, NHCH2CH2C12H25), 1.25 (bs, 62 H, overlap of COC2H4(CH2)20CH3, NHC2H4(CH2)11CH3), 0.89–0.85 (t, J = 6.3 Hz, 6 H, overlap of COC22H44CH3, NHC13H26CH3); 13C-NMR (75 Hz, CDCl3): δ 173.57, 171.72, 170.58, 170.38, 170.20, 170.07, 169.90 (each CO), 101.34 (C-1), 70.82, 70.73 (C-5, C-3), 68.98. (C-2), 68.54 (NHCH2C13H27), 66.98 (C-4), 61.31 (C-6), 49.72 (C-α), 39.66, 39.24 (each OCH2CH2NH), 36.95 (C-β), 36.64 (COCH2(CH2)20CH3), 31.92, 31.86, 29.66,29.56, 29.45, 29.36, 29.28, 26.89, 22.69, 22.66 (each CH2), 20.88 (overlap of O(CO)CH3), 14.12 (overlap of COC22H44CH3, NHC13H26CH3); HRMS (MS-TOF): [M+H]+ calcd. for C58H105N3O13: 1052.772 found 1052.775.
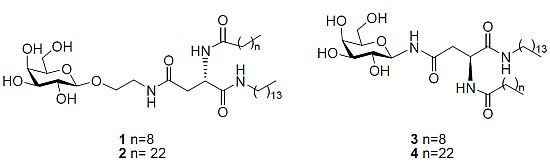
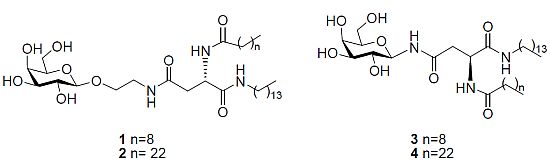
N4-[2-O-(β-D-Galactopyranosyl)-ethyl]-N2-decanoyl-L-asparagine tetradecylamide (1). Triethylamine (0.1 mL) was added to a stirring solution of 11b (0.120 g, 0.14 mmol) dissolved in CH2Cl2/MeOH/H2O (3 mL/6 mL/3 mL) at 40 °C. It was stirred for 18 h. The reaction mixture was concentrated under reduced pressure to afford 1 as a white solid (0.080 g, 83%). [α]22D= −6.0 (c 0.33, C5H5N); 1H-NMR (300 MHz, d5-Pyr): δ 8.95 (d, J = 8.1 Hz, 1 H, NHCOC9H19), 8.87 (t, J = 5.4 Hz, 1 H, CH2CH2NHCO), 8.55 (t, J = 5.7 Hz, 1 H, CONHC14CH29), 7.05, 6.79, 6.63, 6.39 (each bs, 1 H, OH), 5.55–5.53 (m, 1 H, H-α), 4.79 (d, J = 7.5 Hz, 1 H, H-1), 4.51–4.33 (m, 4 H, overlap of H-2, H-4, H-6, H-6'), 4.18–4.07 (m, 3 H, overlap of H-3, H-5, 1 H of OCH2CH2NH), 3.99–3.95 (m, 1 H, 1 H of OCH2CH2NH), 3.77–3.65 (m, 2 H, OCH2CH2NH), 3.22–3.16 (m, 2 H, NHCH2C13H27),3.18 (td, J = 6.6 Hz, J = 1.2 Hz, 2 H, overlap of H-β, H-β'), 2.39–2.34 (t, J = 7.5 Hz, 2 H, COCH2C8H17), 1.79–1.69 (m, 2 H, COCH2CH2C7H15), 1.60–1.50 (m, 2 H, NHCH2CH2C12H25), 1.20 (bs, 34 H, overlap of COC2H4(CH2)6CH3, NHC2H4(CH2)11CH3), 0.89–0.82 (m, 6 H, overlap of COC8H16CH3, NHC13H26CH3); 13C-NMR (75 Hz, d5-Pyr): δ 175.40, 173.98, 173.04 (each CO), 107.68 (C-1), 78.96, 77.22, 74.52, 72.25 (C-2, C-4, C-3, C-5), 71.68 (NHCH2C13H27), 64.53 (C-6), 53.27 (C-α), 42.50, 41.80 (each OCH2CH2NH), 40.71 (C-β), 38.49, 38.49, 34.09, 34.01,32.04, 31.95, 31.90, 31.68, 31.66, 31.63, 31.58, 31.49, 29.22, 28.09 (each CH2), 18.63 (overlap of COC8H16CH3, NHC13H26CH3); HRMS (MS-TOF): [M+H]+ calcd. for C36H69N3O9: 688.5107 found 688.5099.
N4-[2-O-(β-D-Galactopyranosyl)-ethyl]-N2-tetracosanoyl-L-asparagine tetradecylamide (2). Triethylamine (0.1 mL) was added to a stirring solution of 14 (0.016 g, 0.015mmol) dissolved in CH2Cl2/MeOH/H2O/THF (1 mL/2 mL/1 mL/2 mL) at 40 °C. The reaction mixture was stirred and its progress was followed by 1H-NMR spectra of aliquots. The reaction was deemed complete after 36 h. The reaction was concentrated under reduced pressure to afford 2 as a white solid (0.05 g, 38%). 1H-NMR (300 MHz, d5-Pyr): δ 9.02–8.96 (m, 1 H, NHCOC23H47), 8.89–8.79 (m, 1 H, CH2CH2NHCO), 8.54–8.52 (m, 1 H, CONHC14CH29), 5.54 (dd, J = 6.3, 12.9 Hz, 1 H, H-α), 4.80 (d, J = 7.8 Hz, 1 H, H-1), 4.52–4.36 (m, 4 H, overlap of H-2, H-4, H-6,H-6'), 4.18–4.10 (m, 3 H, overlap of H-3, H-5, 1 H of OCH2CH2NH), 3.99–3.95 (m, 1 H, 1 H of OCH2CH2NH), 3.77–3.65 (m, 2 H, OCH2CH2NH), 3.48–3.38 (m, 2 H, NHCH2C13H27), 3.18 (m, 2 H, H-β, H-β'), 2.40–2.35 (m, 2 H, COCH2C22H45), 1.79–1.69 (m, 2 H, COCH2CH2C21H43), 1.60–1.50 (m, 2 H, NHCH2CH2C12H25), 1.20 (bs, 62 H, overlap of COC2H4(CH2)20CH3, NHC2H4(CH2)11CH3), 0.89–0.82 (m, 6 H, overlap of COC22H44CH3, NHC13H26CH3); HRMS (MS-TOF): [M+H]+ calcd. for C50H97N3O9: 883.7225 found 883.7278.
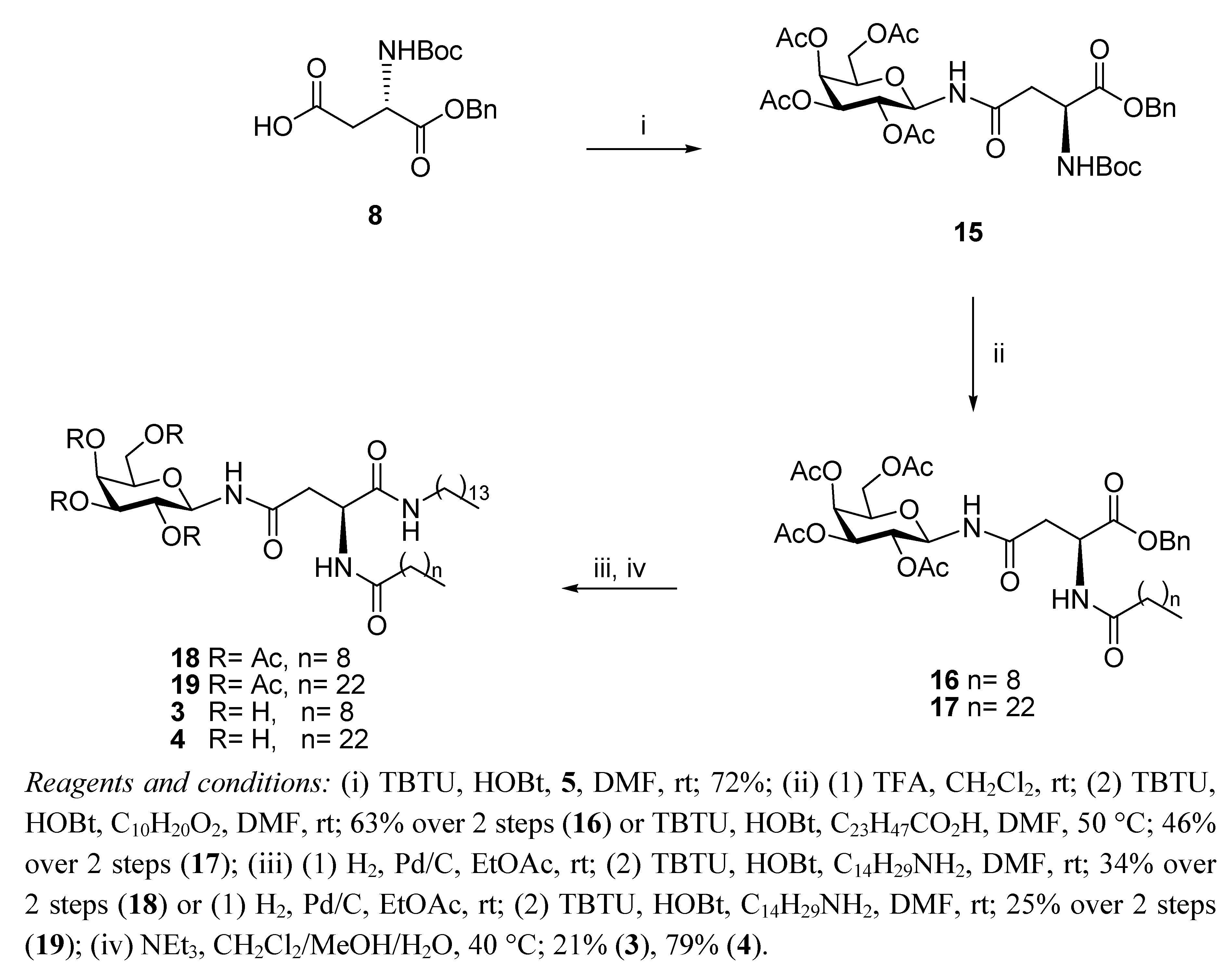
N4-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-N2-tert-butoxycarbonyl-L-asparagine benzyl ester (
15) [
30]. HOBt (1.30 g, 9.60 mmol) was added to a stirring solution of
N-Boc-L-Asp-OBn
8 (1.55 g, 4.80 mmol) and TBTU (3.08 g, 0.720 mmol) in anhydrous DMF (25 mL) under N
2 at rt. It was stirred for 30 min and galactosyl amine
5 (2 g, 5.76 mmol) dissolved in anhydrous DMF (10 mL) was added dropwise to the solution. It was stirred for 18 h. The reaction mixture was concentrated
in vacuo, diluted with ethyl acetate, washed with water, HCl 0.1 N and aqueous sat. NaHCO
3solution, dried (MgSO
4) and concentrated. Flash chromatography (hexane/ethyl acetate 1:1) afforded
15 as a white solid (1.70 g, 72%). This was used without further purification. A small sample of
15 was recrystallised in CHCl
3/hexane to give white crystals used for characterisation. [α]
22D = +30 (c 1.2, CHCl
3); m.p. = 148–150 °C; IR (NaCl film): 3348.7, 2965.2, 1749.6, 1499.7, 1369.0, 1221.7, 1054.5 771.3 cm
−1;
1H-NMR (300 MHz, CDCl
3): δ 7.34–7.33 (m, 5 H, Ph-
H), 6.38 (d,
J = 9 Hz, N
HCOC(CH
3)
3), 5.70 (d,
J = 9 Hz,1 H, N
HCOCH
2), 5.43 (d,
J = 3 Hz, 1 H,
H-4), 5.21–5.04 (m, 5 H, overlap of CH
2Ph,
H-1,
H-2,
H-3), 4.58 (t,
J = 6 Hz, 1 H,
H-α), 4.15–3.97 (m, 3 H, overlap of
H-5,
H-6,
H-6'), 2.95–2.84 (m, 1 H,
H-β'), 2.71 (dd,
J = 3 Hz,
J = 15 Hz, 1
H, H-β), 2.13 , 2.03,1.99, 1.98 (each s, 3 H, O(CO)C
H3), 1.41 (9 H, O(CO)C
H3)
3);
13C-NMR (75 MHz, CDCl
3): δ171.53, 171.12, 170.54, 170.33, 169.95, 169.74 (each
CO), 135.33 (
C-Ph), 128.50, 128.26, 127.88 (
CH-Ph), 80.10 (
C(CH
3)
3), 78.44 (
C-1), 72.40 (
C-5), 70.67 (
C-3), 68.17 (
C-2), 67.29 (CH
2Ph), 67.06 (
C-4), 61.08 (
C-6), 50.09 (
C-α), 37.85 (
C-β), 28.25 (C(
CH
3)
3), 20.66, 20.59, 20.57, 20.52 (each O(CO)
CH
3); HRMS (MS-TOF): [M+H]
+ calcd. for C
30H
41N
2O
14: 653.2552 found 653.2541.
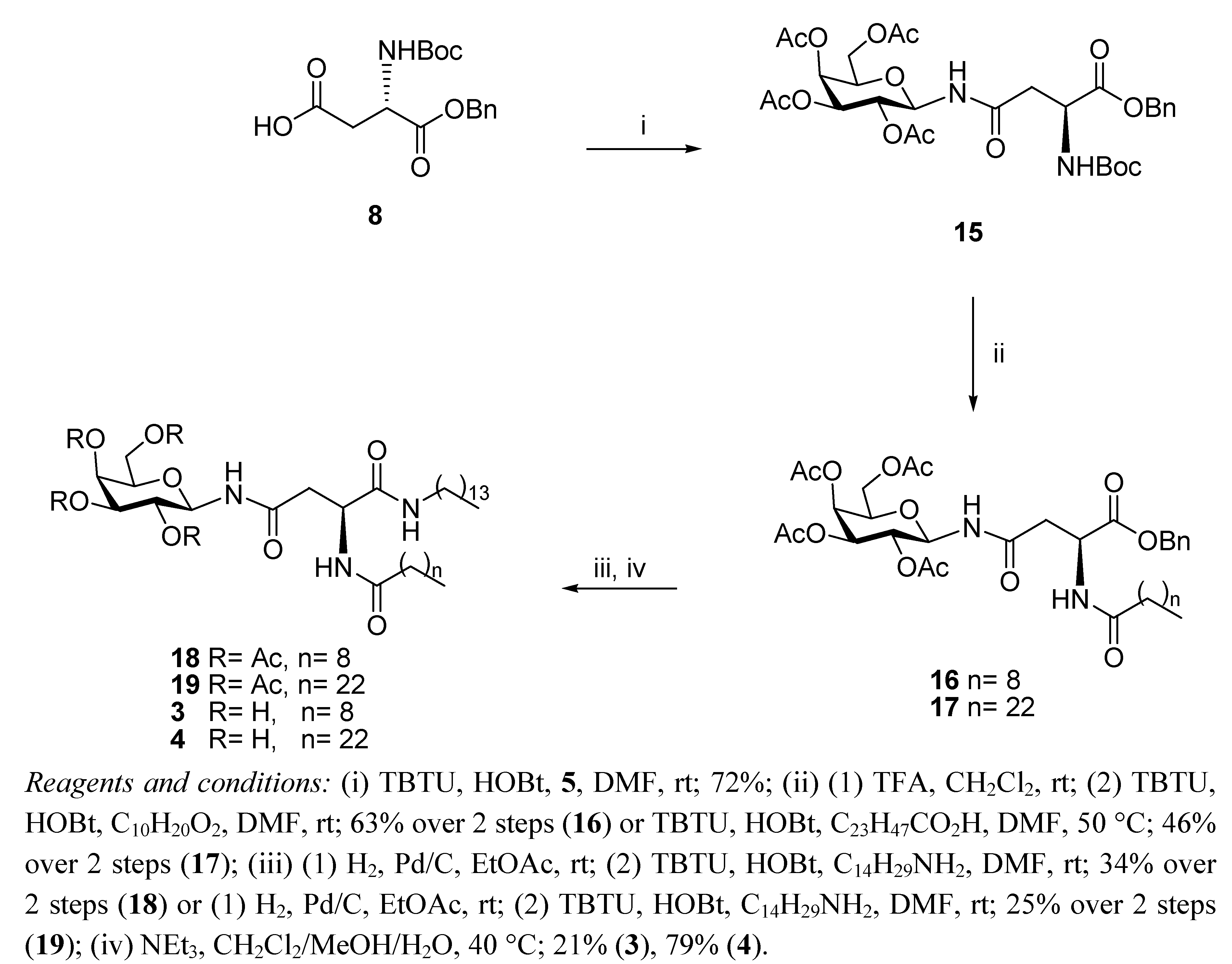
N4-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-N2-decanoyl-L-asparagine benzyl ester (16). TFA (1.34 mL, 17.93 mmol) in anhydrous CH2Cl2 (1.34 mL) was added dropwise to a solution of 15 (1.17 g, 1.79 mmol) in anhydrous CH2Cl2 (10 mL). It was stirred for 6 h. The reaction mixture was concentrated in the rotary evaporator and the residue was dissolved in CH2Cl2 and washed with aqueous sat. NaHCO3 solution, brine and water. The organic phase was dried (MgSO4) and concentrated in vacuo to yield the corresponding amine as a white foam (0.73 g, 74%). The compound was used without further purification. 1H-NMR (300 MHz, CDCl3): δ 8.10 (d, J = 9.3 Hz, 1 H, NHCOCH2), 7.51–7.31 (m, 5 H, Ph-H), 5.40 (d, J = 1.4 Hz, 1 H, H-4), 5.22 (t, J = 9.3 Hz, 1 H, H-1), 5.12–5.07 (m, 4 H, overlap of CH2Ph, H-2, H-3), 4.11–3.97 (m, 3 H, overlap of H-5, H-6, H-6'), 3.67 (bs, 1 H, H-α), 2.67–2.63 (m, 1 H, H-β), 2.39 (dd, J = 9.6 Hz, J = 5.3 Hz, 1 H, H-β'), 2.10, 2.00, 1.99, 1.95 (each s, 3 H, O(CO)CH3); HRMS (MS-TOF): [M+Na]+ calcd. for C25H32O12N2Na: 553.2028 found 553.2024. TBTU (56 mg, 0.18 mmol) and HOBt (24 mg, 0.18 mmol) were added to a solution of decanoic acid (27 mg, 0.16 mmol) in anhydrous DMF (2 mL) under N2 at rt. It was stirred for 20 min and the free amine obtained from 15 as described above (88 mg, 0.16 mmol) in anhydrous DMF (1 mL) was added dropwise to the solution. It was stirred for 18 h. The reaction mixture was concentrated in vacuo, diluted with ethyl acetate, washed with water and brine, dried (MgSO4) and concentrated. The residue obtained was purified by flash chromotagraphy (hexane/ethyl acetate 1:1) to yield 16 as a colourless oil (94 mg, 85%). [α]22D = +27.2 (c 1.76, CHCl3); IR (NaCl film): 3330.9, 2926.4, 1751.0, 1674.3, 1530.8, 1370.1, 1222.5, 1179.4, 1052.8, 698.5 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.33–7.31 (m, 5 H, Ph-H), 6.73 (d, J = 8.3 Hz, 1 H, NHCOC9H19), 6.50 (d, J = 8.7 Hz, 1 H, NHCOCH2), 5.43 (d, J = 2.0 Hz, 1 H, H-4), 5.20–5.04 (m, 5 H, overlap of H-1, H-2, H-3, CH2Ph), 4.93–4.87 (m, 1 H, H-α), 4.15–3.97 (m, 3 H, overlap of H-5, H-6, H-6'), 2.90 (dd, J = 4.1 Hz, J = 16.5 Hz, 1 H, H-β), 2.70 (dd, J = 4.4 Hz, J = 16.4 Hz, 1 H, H-β'), 2.22–2.17 (t, J = 7.3 Hz, 2 H, COCH2C8H17), 2.13, 2.03, 1.99, 1.98 (each s, 3 H, O(CO)CH3), 1.61–1.56 (m, 2 H, COCH2CH2C7H15), 1.24 (bs, 12 H, COC2H4(CH2)6CH3), 0.88–0.84 (t, J = 7.0 Hz, 3 H, COC8H16CH3); 13C-NMR (75 MHz, CDCl3): δ 173.06, 171.49, 170.90, 170.82, 170.28, 169.95, 169.76 (each CO), 135.23, (Ph-C), 128.53, 128.33, 127.92 (Ph-CH), 78.44 (C-1), 72.37 (C-5), 70.66 (C-3), 68.11 (C-2), 67.36 (CH2Ph), 67.00 (C-4), 61.00 (C-6), 48.42 (C-α), 37.43 (C-β), 36.49, 31.81, 29.36, 29.27, 29.22, 29.18, 25.49, 22.62 (each CH2), 20.62, 20.55, 20.53, 20.49 (each O(CO)CH3), 14.06 (COC8H16CH3); HRMS (MS-TOF,): [M+Na]+ calcd. for C35H50O13N2Na: 707.3386 found 707.3376.
N4-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-N2-tetracosanoyl-L-asparagine benzyl ester (17). TBTU (59 mg, 0.183 mmol) and HOBt (25 mg, 0.183 mmol) were added to tetracosanoic acid (62 mg, 0.167 mmol) in anhydrous DMF (3 mL) containing 4 Å MS under N2 at rt. It was stirred for 30 min and the free amine obtained from 15 as described above (88 mg, 0.16 mmol) in anhydrous DMF (2 mL) was added dropwise to the solution. It was stirred for 2 h at 50 °C. The reaction mixture was concentrated in vacuo, diluted with ethyl acetate, washed with water and brine, dried (MgSO4) and concentrated under reduced pressure. The residue obtained was purified by flash chromotagraphy (hexane/ethyl acetate 1:1) to yield 17 as a white solid (93 mg, 62%). [α]25D = +18.9 (c 0.95, ethyl acetate); IR (NaCl film): 2918.7, 2850.5, 1750.5, 1371.1, 1231.8, 1054.9, 913.2, 743.7 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.33–7.31 (m, 5 H, H-Ph), 6.73 (d, J = 8.3 Hz, 1 H, NHCOC23H47), 6.48 (d, J = 8.6 Hz, 1 H, NHCOCH2), 5.43 (d, J = 2.0 Hz, 1 H, H-4), 5.20–5.07 (m, 5 H, overlap of H-1, H-2, H-3, CH2Ph), 4.93–4.88 (m, 1 H, H-α), 4.15–3.97 (m, 3 H, overlap of H-5, H-6, H-6'), 2.91 (dd, J = 3.9 Hz, J = 16.4 Hz, 1 H, H-β'), 2.71 (dd, J = 4.4 Hz, J = 16.4 Hz, 1 H, H-β'), 2.22–2.17 (t, J = 7.5 Hz, 2 H, COCH2C22H45), 2.13, 2.04, 2.03, 1.99 (each s, 3 H, O(CO)CH3), 1.61–1.56 (m, 2 H, COCH2CH2C21H43), 1.25 (bs, 40 H, COC2H4(CH2)20CH3, 0.89–0.85 (t, J = 7.0 Hz, 3 H, COC22H44CH3); 13C-NMR (75 Hz, CDCl3): δ 173.09, 171.53, 170.91, 170.82, 170.29, 169.97, 169.77 (each CO), 135.23, 128.54, 128.35 (CH-Ph), 127.93 (C-Ph), 78.46 (C-1), 72.38 (C-5), 70.66 (C-3), 68.12 (C-2), 67.39 (CH2Ph), 67.00 (C-4), 61.00 (C-6), 48.43 (C-α), 37.45 (C-β), 36.52, 31.89, 29.67, 29.62, 29.45, 29.33, 29.31, 29.21, 25.51, 22.66 (each CH2), 20.64, 20.57, 20.55, 20.51 (each O(CO)CH3), 14.09 (COC22H44CH3); HRMS (MS-TOF): [M+H]+ calcd. for C49 H79O13N2: 904.5610 found 904.5632.
N4-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-N2-decanoyl-L-asparagine tetradecylamide (18). H2 gas was bubbled through a suspension of 16 (56 mg, 0.079 mmol) in ethyl acetate (10 mL) and Pd/C 10% w/w (6 mg, 10% w/w) was added. It was left to stir for 3 h and then the reaction mixture was filtered through Celite washing with ethyl acetate and concentrated in vacuo, to yield the corresponding carboxylic acid as a colourless oil, which was used without further purification (30 mg, 61%). 1H-NMR (300 MHz, CDCl3): δ 7.24 (d, J = 6.8 Hz, 1 H, NHCOC9H19), 6.57 (d, J = 9.4 Hz, 1 H, NHCOCH2), 5.53 (d, J = 1.7 Hz, 1 H, H-4), 5.35 (m, 1 H, H-1), 5.16–5.10 (m, 2 H, overlap of H-2, H-3), 4.74–4.71 (m, 1 H, H-α), 4.25–4.00 (m, 3 H, overlap of H-5, H-6, H-6'), 2.87 (dd, J = 3.6 Hz, J = 16.4 Hz, 1 H, H-β), 2.77 (dd, J = 4.8 Hz, J = 16.5 Hz, 1 H, H-β'), 2.32–2.27 (t, J = 8.0 Hz, 2 H, COCH2C8H17), 2.15 (s, 3 H, O(CO)CH3), 2.05 (s, 6 H, O(CO)CH3 × 2), 2.00 (s, 3 H, O(CO)CH3), 1.65–1.60 (m, 2 H, COCH2CH2C7H15), 1.25 (bs, 12 H, COC2H4(CH2)6CH3), 0.89–0.85 (t, J = 6.9 Hz, 3 H, COC8H16CH3); HRMS (MS-TOF): [M+H]+ calcd. for C28H45O13N2: 617.2916 found 617.2900. TBTU (14 mg, 0.045 mmol) and HOBt (6 mg, 0.045 mmol) were added to a solution of the carboxylic acid obtained from 16 as described above (25 mg, 0.041 mmol) in anhydrous DMF (3 mL) under N2 at rt. It was stirred for 30 min and tetradecylamine (9 mg, 0.041 mmol) was added to the solution and it was stirred for 3 h. The reaction mixture was concentrated in vacuo, diluted with ethyl acetate, washed with brine and water, dried (MgSO4) and concentrated. The residue obtained was purified by flash chromotagraphy (hexane/ethyl acetate 1:1) to yield 18 as a white solid (18 mg, 55%). [α]25D = +20.0 (c 0.75, CH2Cl2); IR (NaCl film): 3286.1, 2924.1, 2853.8, 1751.2, 1642.6, 1546.1, 1466.7, 1371.0, 1227.6, 1054.9 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.58 (d, J = 7.6 Hz, 1 H, NHCOC9H19), 6.79–6.74 (m, 2 H, overlap of NHCOCH2, CONHC14CH29), 5.44 (d, J = 1.6 Hz, 1 H, H-4), 5.23–5.10 (m, 3 H, overlap of H-1, H-2, H-3), 4.71–4.67 (m, 1 H, H-α), 4.16–3.99 (m, 3 H, overlap of H-5, H-6, H-6'), 3.18–3.11 (m, 2 H, NHCH2C13H27), 2.69 (dd, J = 3.4 Hz, J = 15.6 Hz, 1 H, H-β), 2.44 (dd, J = 5.6 Hz, J = 15.5 Hz, 1 H, H-β'), 2.25–2.21 (m, 2 H, COCH2C8H17), 2.17, 2.14, 2.04, 2.00 (each s, 3 H, O(CO)CH3), 1.65 (bs, 2 H, COCH2CH2C7H15), 1.46–1.42 (m, 2 H, NHCH2CH2C12H25), 1.25 (bs, 34 H, overlap of COC2H4(CH2)6CH3, NHC2H4(CH2)11CH3), 0.90–0.85 (t, J = 6.9 Hz, 6 H, overlap of COC8H16CH3, NHC13H26CH3); 13C-NMR (75 MHz, CDCl3): δ 173.77, 173.03, 172.25, 170.35, 170.30, 170.01, 169.79 (each CO), 78.49 (C-1), 72.31 (C-5), 70.73 (C-3), 67.84 (C-2), 67.05 (C-4), 61.06 (C-6), 49.75 (C-α), 39.59 (NHCH2C13H27), 36.60 (C-β), 36.21, 31.91, 31.84, 29.68, 29.65, 29.61, 29.55, 29.44, 29.35, 29.31, 29.26, 26.89, 25.63, 22.68, 22.65 (each CH2), 20.89, 20.67, 20.59, 20.54, (each O(CO)CH3), 14.11 (overlap of COC8H16CH3, NHC13H26CH3); HRMS (MS-TOF): [M+Na]+ calcd. for C42 H73O12N3Na: 834.5086 found 834.5079.
N4-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-N2-tetracosanoyl-L-asparagine tetradecylamide (19). H2 gas was bubbled through a suspension of 17 (67 mg, 0.074 mmol) in ethyl acetate (5 mL) and Pd/C 10% w/w (7 mg, 10% w/w) was added. It was left to stir for 18 h and then the reaction mixture was filtered through Celite, washed with ethyl acetate and concentrated in vacuo to yield the corresponding carboxylic acid as a white solid, which was used without further purification (60 mg, 55%). 1H-NMR (300 MHz, CDCl3): δ 7.20 (d, J = 7.0 Hz, 1 H, NHCOC23H47), 6.62 (d, J = 9.2 Hz, 1 H, NHCOCH2), 5.51 (d, J = 1.2 Hz, 1 H, H-4), 5.34–5.28 (m, 1 H, H-1), 5.12–5.10 (m, 2 H, overlap of H-2, H-3), 4.76–4.71 (m, 1 H, H-α), 4.22–4.02 (m, 3 H, overlap of H-5, H-6, H-6'), 2.96–2.84 (m, 1 H, H-β), 2.75 (dd, J = 5.0 Hz, J = 16.5 Hz, 1 H, H-β'), 2.37–2.26 (m, 2 H, COCH2C22H45), 2.15, 2.06, 2.05, 2.00 (each s, 3 H, O(CO)CH3), 1.67–1.58 (m, 2 H, COCH2CH2C21H43), 1.25 (bs, 40 H, COC2H4(CH2)20CH3), 0.90–0.85 (t, J = 6.9 Hz, 3 H, COC22H44CH3); HRMS (MS-TOF): [M+H]+ calcd. for C42H73O13N2: 813.5107 found 813.5106. TBTU (12 mg, 0.037 mmol) and HOBt (5 mg, 0.037 mmol) were added to a solution of the carboxylic acid obtained from 17 as described above (27 mg, 0.033 mmol) in anhydrous DMF (3 mL), containing 4 Å MS, under N2 and at rt. It was stirred for 20 min and tetradecylamine (7 mg, 0.033 mmol) was added to the solution. It was stirred for 18 h. The reaction mixture was concentrated in vacuo, diluted with ethyl acetate, washed with brine and water, dried (MgSO4) and concentrated under reduced pressure. Flash chromatography (hexane/ethyl acetate 1:1) afforded 19 as a white solid (15 mg, 45%). [α]25D = +09.2 (c 0.65, CH2Cl2); IR (NaCl film): 3426.0, 2918.5, 2850.5, 1750.7, 1641.8, 1228.5 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.58 (d, J = 7.8 Hz, 1 H, NHCOC23H47), 6.78–6.73 (m, 2 H, overlap of NHCOCH2, CONHC14CH29), 5.45 (d, J = 1.8 Hz, 1 H, H-4), 5.26–5.10 (m, 3 H, overlap of H-1, H-2, H-3), 4.71–4.66 (m, 1 H, H-α), 4.17–3.98 (m, 3 H, overlap of H-5, H-6, H-6'), 3.26–3.11 (m, 2 H, NHCH2C13H27), 2.69 (dd, J = 3.5 Hz, J = 15.7 Hz, 1 H, H-β), 2.44 (dd, J = 5.6 Hz, J = 15.7 Hz, 1 H, H-β'), 2.24–2.21 (m, 2 H, COCH2C22H45), 2.17, 2.14, 2.04, 2.00 (each s, 3 H, O(CO)CH3), 1.66 (bs, 2 H, COCH2CH2C21H43), 1.46–1.42 (m, 2 H, NHCH2CH2C12H25), 1.25 (bs, 62 H, overlap of COC2H4(CH2)20CH3, NHC2H4(CH2)11CH3), 0.90–0.85 (t, J = 6.9 Hz, 6 H, overlap of COC22H44CH3, NHC13H26CH3); 13C-NMR (75 MHz, CDCl3): δ 173.77, 173.03, 172.24, 170.35, 170.29, 170.00, 169.78 (each CO), 78.47 (C-1), 72.30 (C-5), 70.72 (C-3), 67.83 (C-2), 67.04 (C-4), 61.04 (C-6), 49.73 (C-α), 39.57 (NHCH2C13H27), 36.59 (C-β), 36.18,31.90, 29.68, 29.33, 26.87, 25.62, 22.66, (each CH2), 20.87, 20.64, 20.57, 20.51, (each O(CO)CH3), 14.09 (overlap of COC22H44CH3, NHC13H26CH3); HRMS (MS-TOF): [M+H]+ calcd. for C56H102O12N3: 1008.7458 found 1008.7429.
N4-β-D-Galactopyranosyl-N2-decanoyl-L-asparagine tetradecylamide (3). Triethylamine (0.05 mL) was added to a stirring solution of 18 (0.043 g, 0.053 mmol) dissolved in CH2Cl2/MeOH/H2O (1 mL/2 mL/1 mL) at 40 °C. It was stirred for 18 h. The precipitate formed was filtered through a vacuum to afford 3 as white crystals (7 mg, 21%). [α]22D = −6.7 (c 0.6, C5H5N); 1H-NMR (300 MHz, d5-Pyr): δ 10.14 (d, J = 9.1 Hz, 1 H, NHCOCH2), 8.94 (d, J = 7.9 Hz, 1 H, NHCOC9H19), 8.53–8.48 (t, J = 5.7 Hz, 1 H, CONHC14CH29), 5.91–5.78 (t, J = 9.2 Hz, 1 H, H-1), 5.45 (dd, J = 6.8, 14.3 Hz, 1 H, H-α), 4.58 (d, J = 2.9 Hz, 1 H, H-4), 4.56–4.49 (t, J = 9.2 Hz, 1 H, H-2), 4.38 (dd, J = 2.1 Hz,J = 6.1 Hz, 2 H, H-6, H-6'), 4.17 (dd, J = 3.0 Hz, J = 9.0 Hz, 1 H, H-3), 4.13–4.09 (t, J = 5.8 Hz, 1 H, H-5), 3.45–3.34 (m, 2 H, NHCH2C13H27), 3.27 (d, J = 6.5 Hz, 2 H, H-β, H-β'), 2.36–2.31 (t, J = 7.5 Hz, 2 H, COCH2C8H17), 1.75–1.70 (m, 2 H, COCH2CH2C7H15), 1.58–1.50 (m, 2 H, NHCH2CH2C12H25), 1.24 (bs, 34 H, overlap of COC2H4(CH2)6CH3, NHC2H4(CH2)11CH3), 0.88–0.81 (m, 6 H, overlap of COC8H16CH3, NHC13H26CH3); 13C NMR (d5-Pyr, 75 Hz): δ 173.41, 171.98, 171.83, (each CO), 81.74 (C-1), 78.35 (C-5), 76.19 (C-3), 71.90 (C-2), 70.42 (C-4), 62.37 (C-6), 51.13 (C-α), 39.89 (NHCH2C13H27), 39.05 (C-β), 36.46, 32.08, 32.00, 29.94, 29.88, 29.67. 29.64, 29.61, 29.57, 29.48, 27.25, 26.02, 22.89, 22.85 (each CH2), 14.22 (overlap of COC8H16CH3, NHC13H26CH3); HRMS (MS-TOF): [M+Na]+ calcd for C34H65O8N3Na: calcd: 666.4664 found 666.4689.
N4-β-D-Galactopyranosyl-N2-tetracosanoyl-L-asparagine tetradecylamide (4). Triethylamine (0.05 mL) was added to a stirring solution of 19 (0.035 g, 0.035mmol) dissolved in CH2Cl2/MeOH/H2O (1 mL/2 mL/1 mL) at 40 °C. It was stirred for 18 h. The precipitate formed was filtered through a vacuum to afford 4 as a white solid (23 mg, 79%). 1H-NMR (300 MHz, d5-Pyr): δ 10.20–10.14 (m, 1 H, NHCOCH2), 9.02–8.90 (m, 1 H, NHCOC23H47), 8.50 (t, J = 5.5 Hz, 1 H, CONHC14CH29), 5.90 (dd, J = 8.9 Hz, J = 17.9 Hz, 1 H, H-1), 5.65–5.61 (m, 1 H, H-α), 4.73 (d, J = 6.1 Hz, 1 H, H-4), 4.60 (d, J = 2.9 Hz, 1 H, H-2), 4.56–4.36 (m, 3 H, overlap of H-6, H-6', H-3), 4.20–4.13 (m, 1 H, H-5), 3.49–3.38 (m, 2 H, NHCH2C13H27), 3.29–3.21 (m, 2 H, H-β, H-β'), 2.35 (t, J = 7.5 Hz, 2 H, COCH2C22H45), 1.77–1.73 (m, 2 H, COCH2CH2C21H43), 1.59–1.54 (m, 2 H, NHCH2CH2C12H25), 1.26 (bs,62 H, overlap of COC2H4(CH2)20CH3, NHC2H4(CH2)11CH3), 0.89–0.85 (m, 6 H, overlap of COC8H16CH3, NHC13H26CH3); HRMS (MALDI MS-QTOF): [M+Na]+ calcd for C48H93O8N3Na: calcd: 862.6860 found 862.6841.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}