Cyclization vs. Cyclization/Dimerization in o-Amidostilbene Radical Cation Cascade Reactions: The Amide Question

Abstract
:
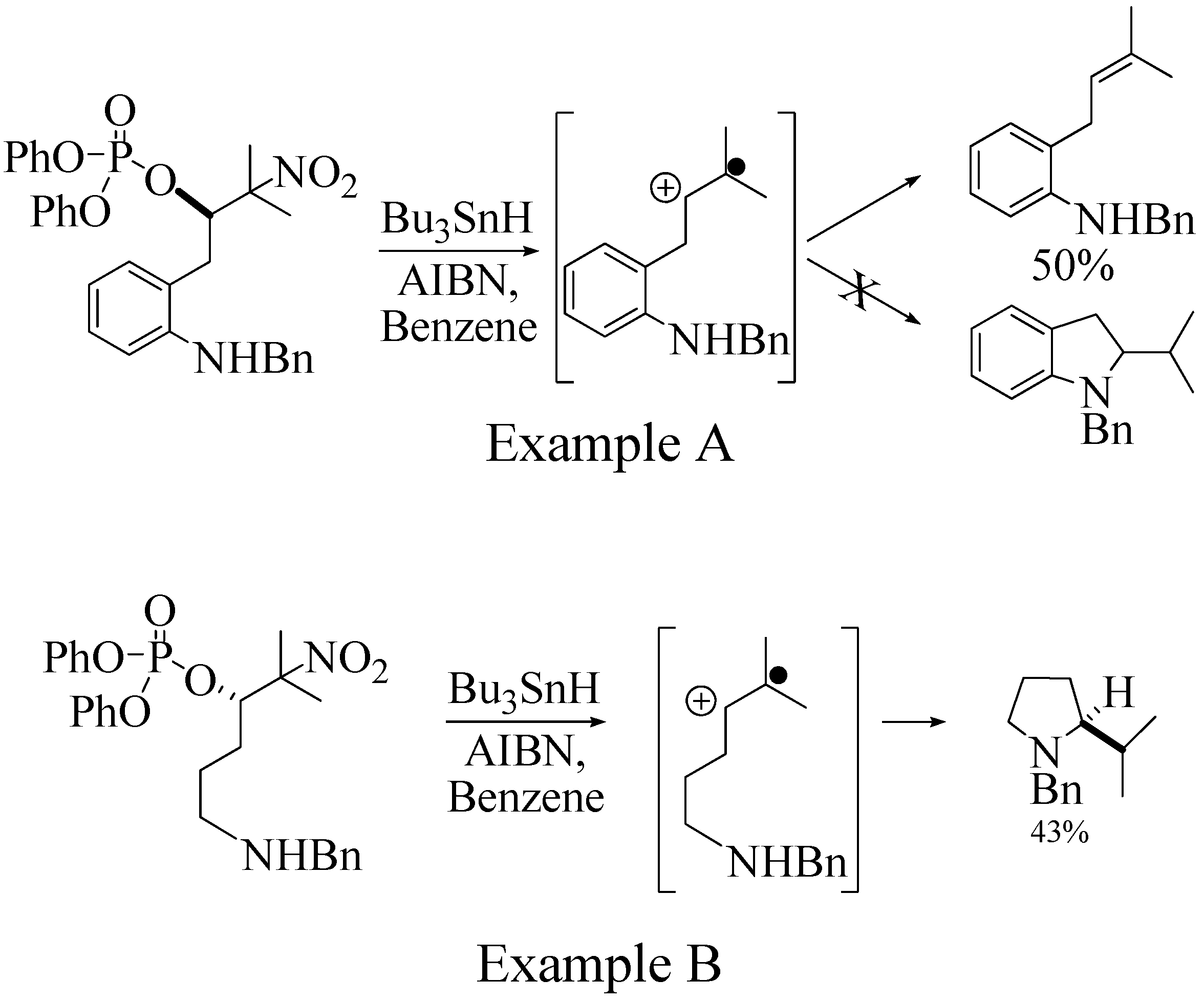
1. Introduction



| Entry | Stilbene | C(7)-H (J, Hz) | C(8)-H (J, Hz) | N-H (J, Hz) |
|---|---|---|---|---|
| 1 |  | 6.85 | 6.95 | 7.56 |
| 2 |  | 6.90 | 6.97 | 7.31 |
| 3 |  | 6.93 | 7.04 | 8.03 |
| 4 |  | 6.91 | 6.97 | 7.21 |
| 5 |  | 7.40 | 7.29 | 8.25 |
| 6 |  | 7.02 | 7.22 | 8.17 |
| 7 |  | 6.96 | 7.07 | 8.19 |
| 8 |  | 7.00 | 7.09 | 8.17 |
2. Results and Discussion
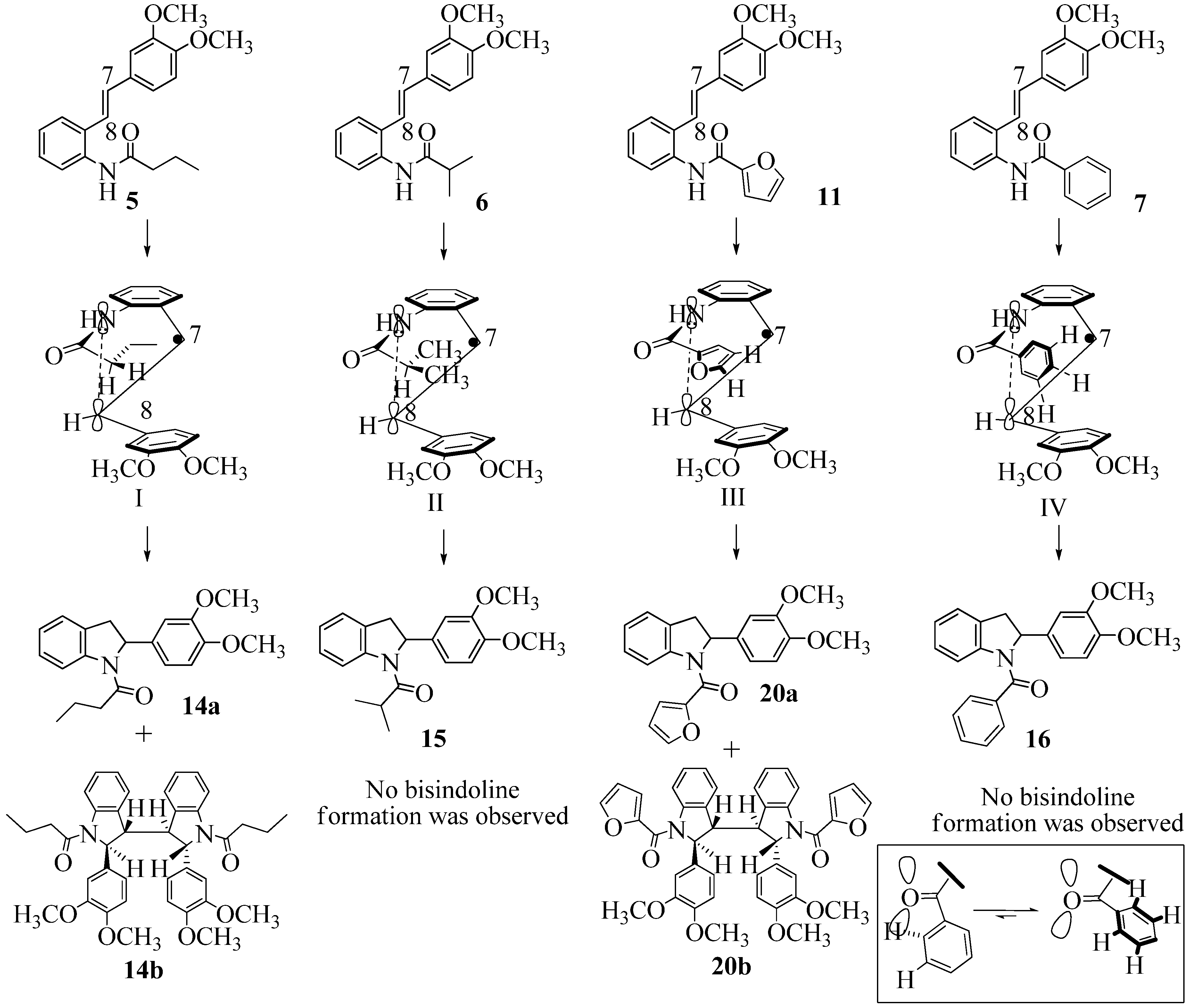
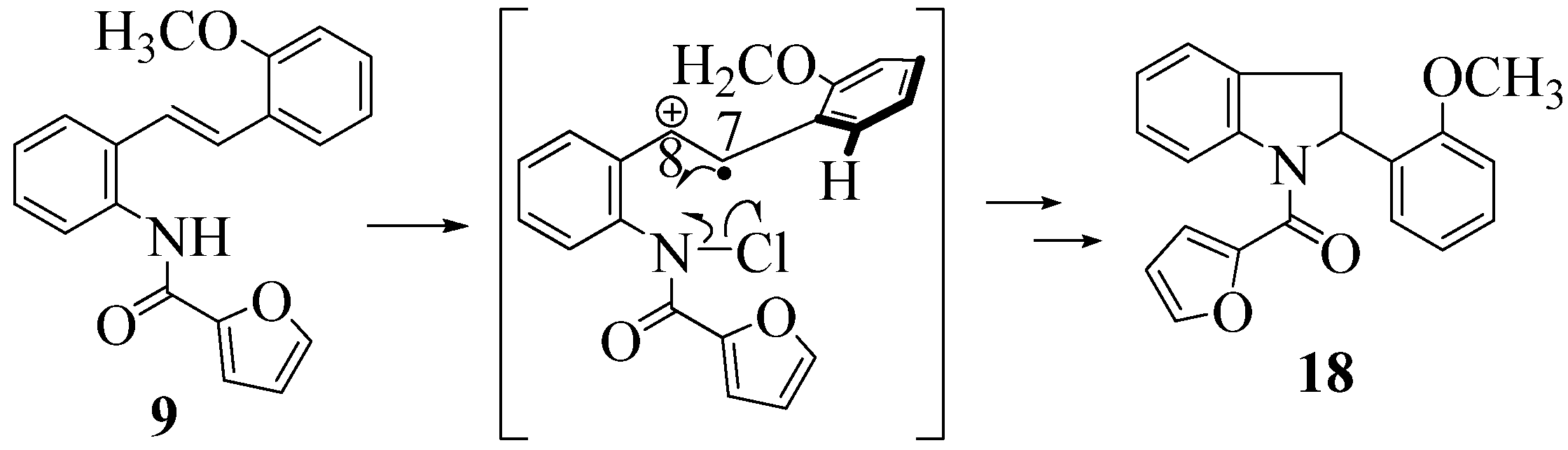
2.1. FeCl3 Promoted Syntheses of Indolines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
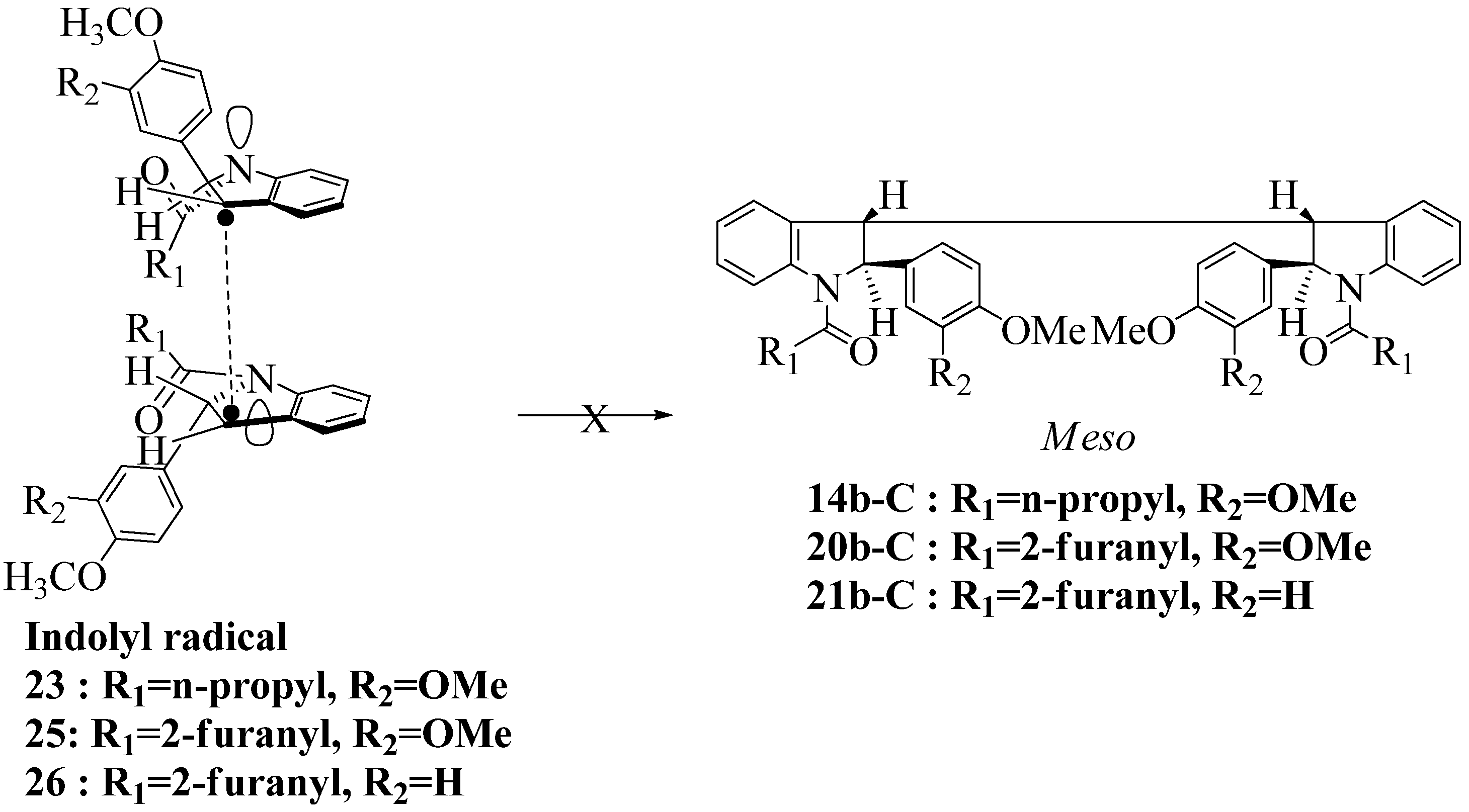{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Stilbene | Indoline (Yield) | Bisindoline (Yield) |
|---|---|---|---|
| 1 |  |  |  |
| 2 |  |  | - |
| 3 |  |  | - |
| 4 |  |  | - |
| 5 |  |  | - |
| 6 |  |  | - |
| 7 |  |  |  |
| 8 |  |  |  |
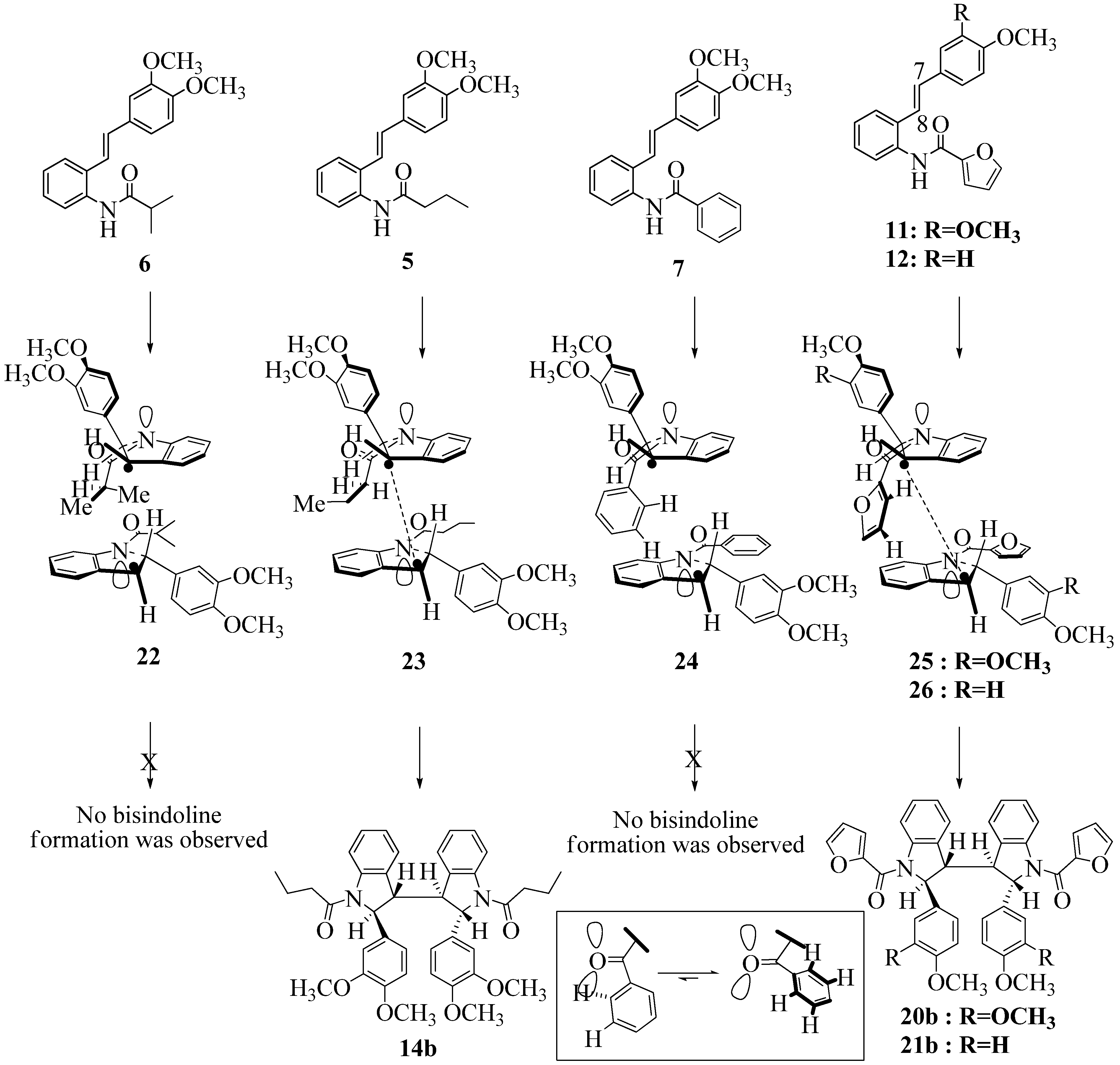
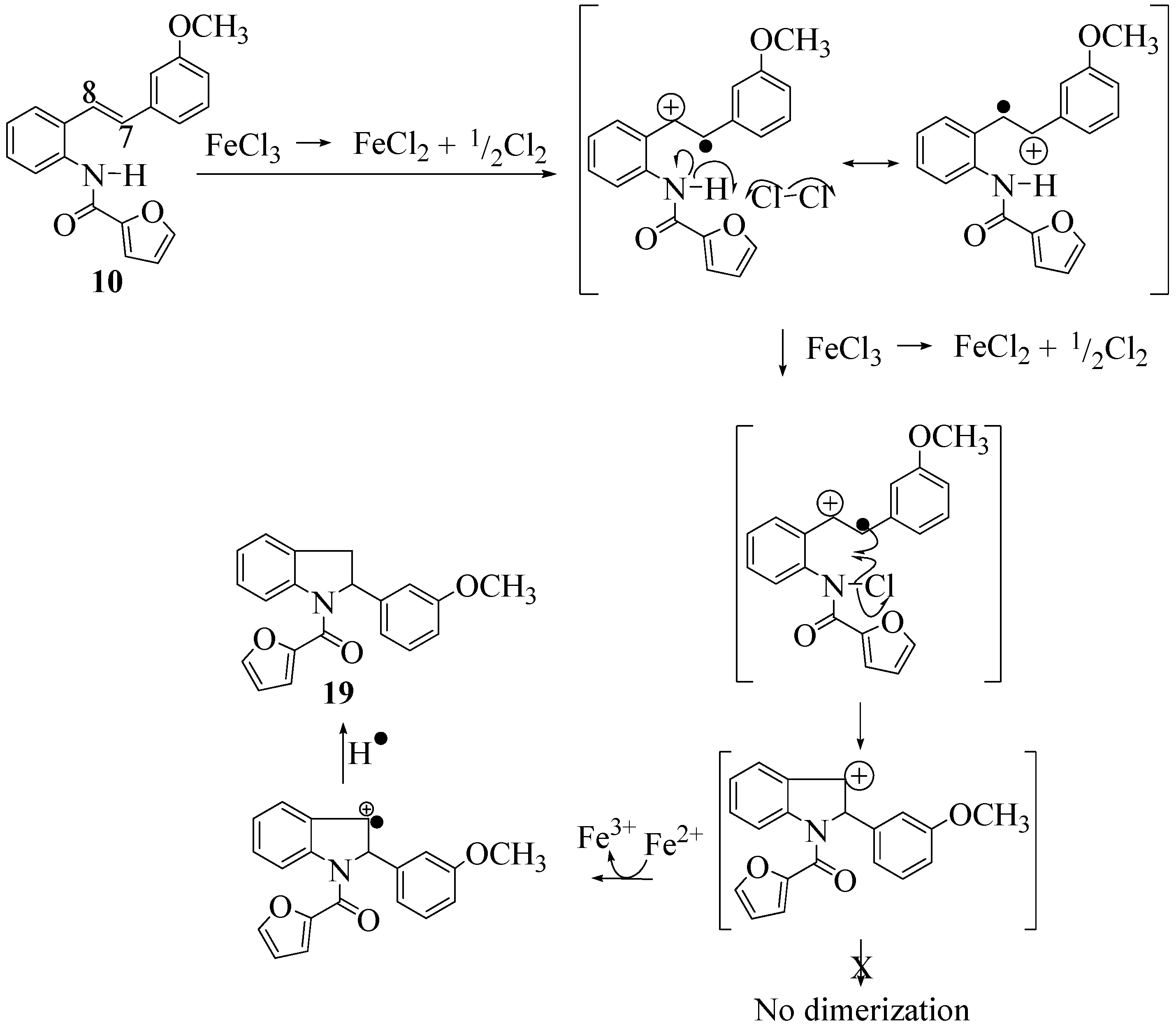
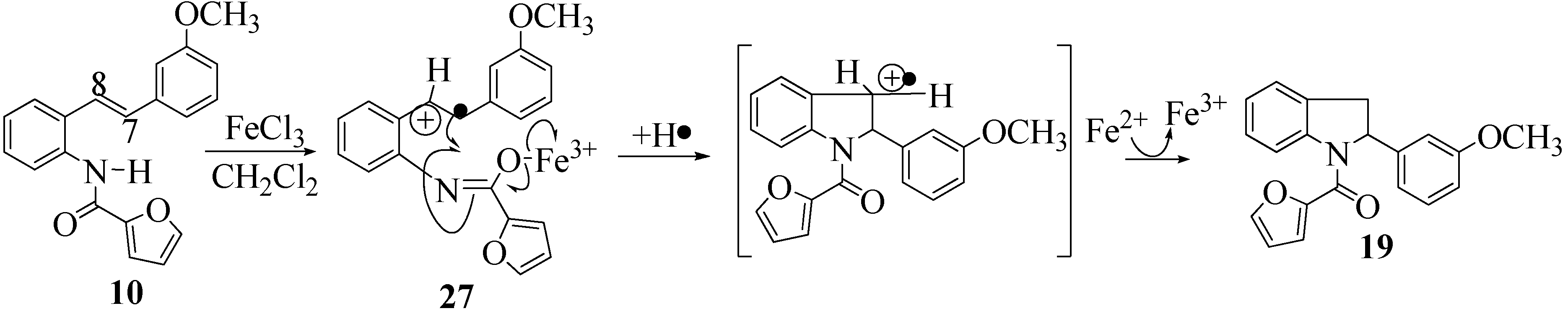
2.2. Effect of Amide Structure on the Radical Cyclization Leading to Indoline Formation

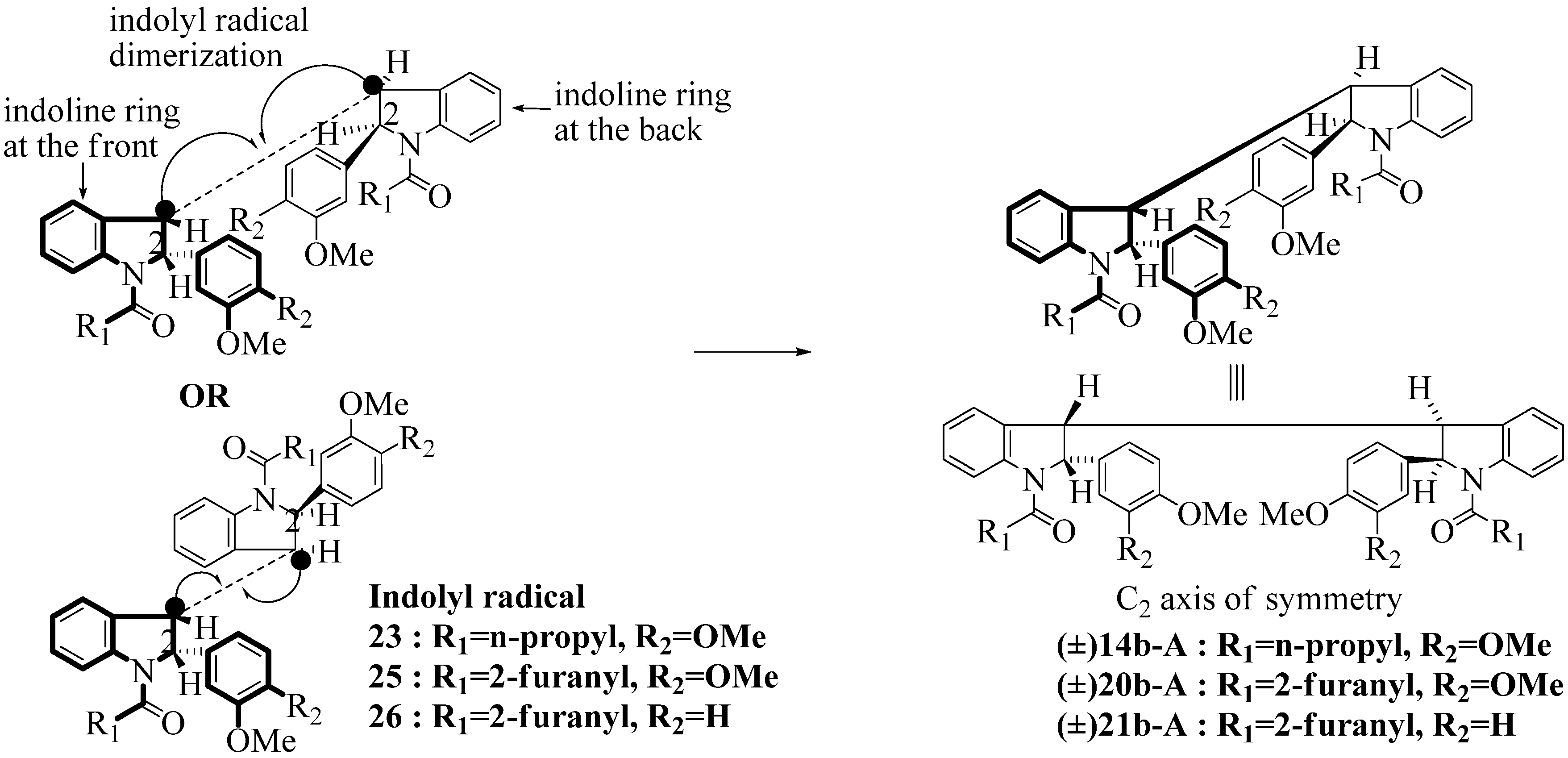
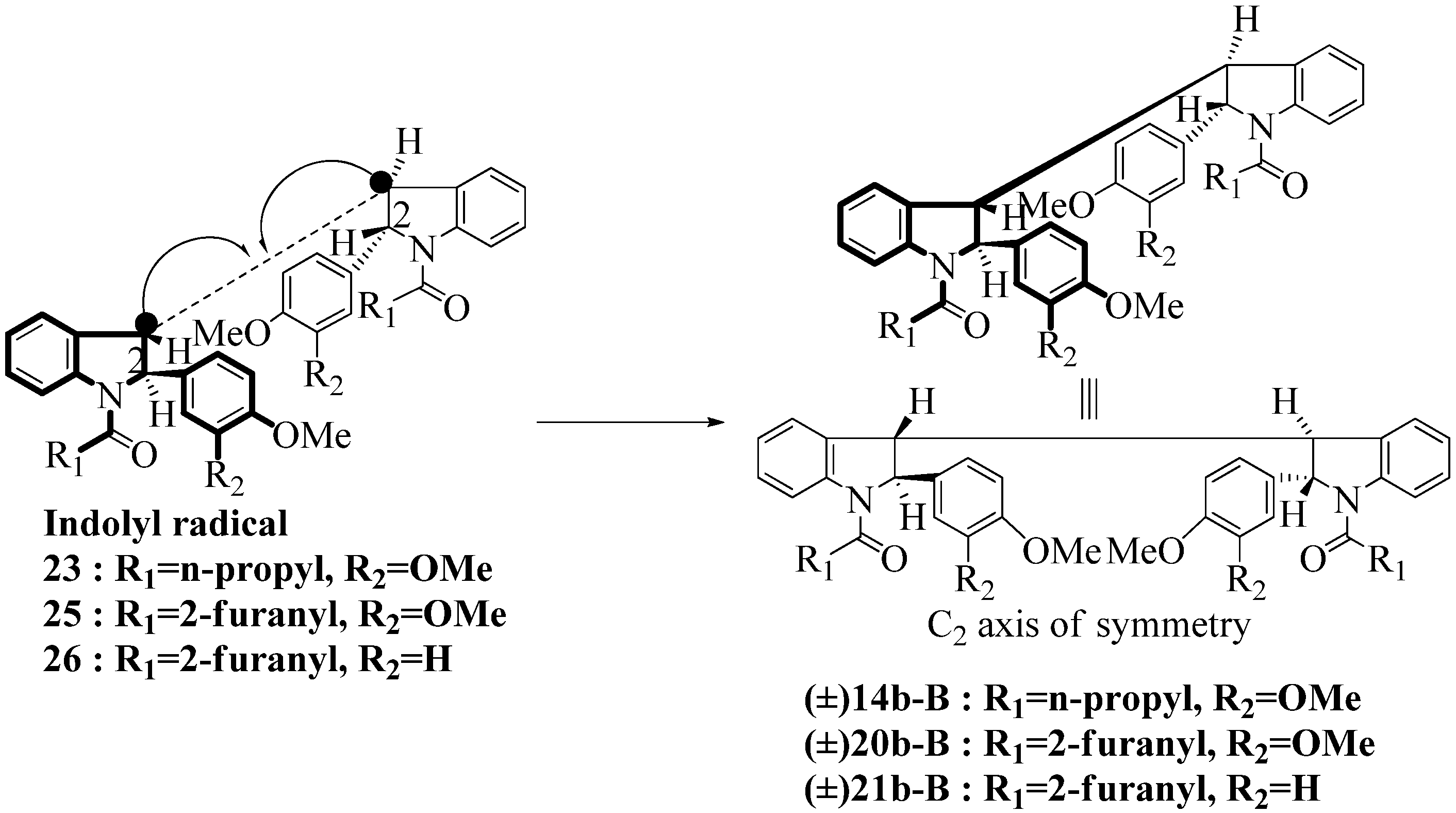
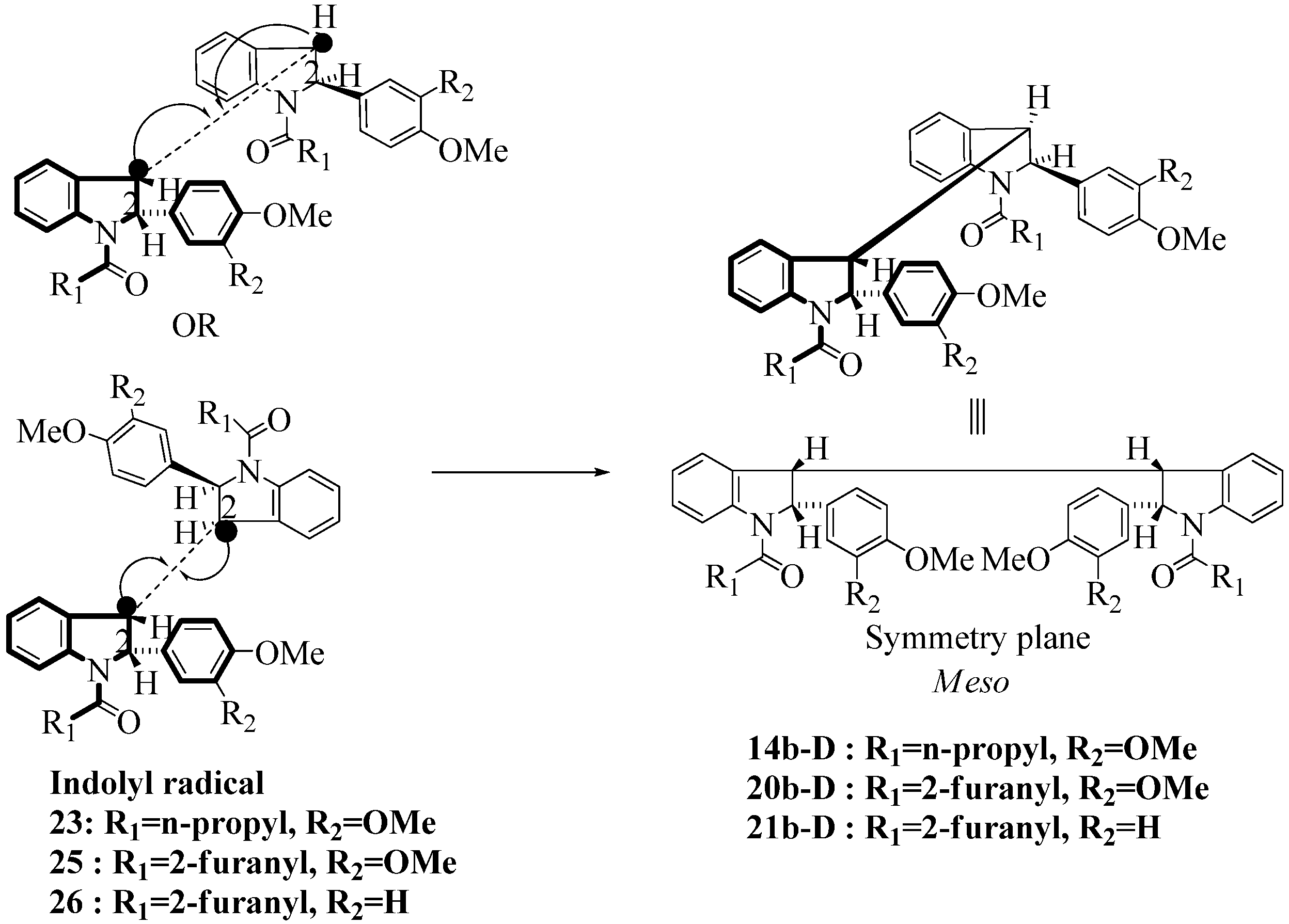
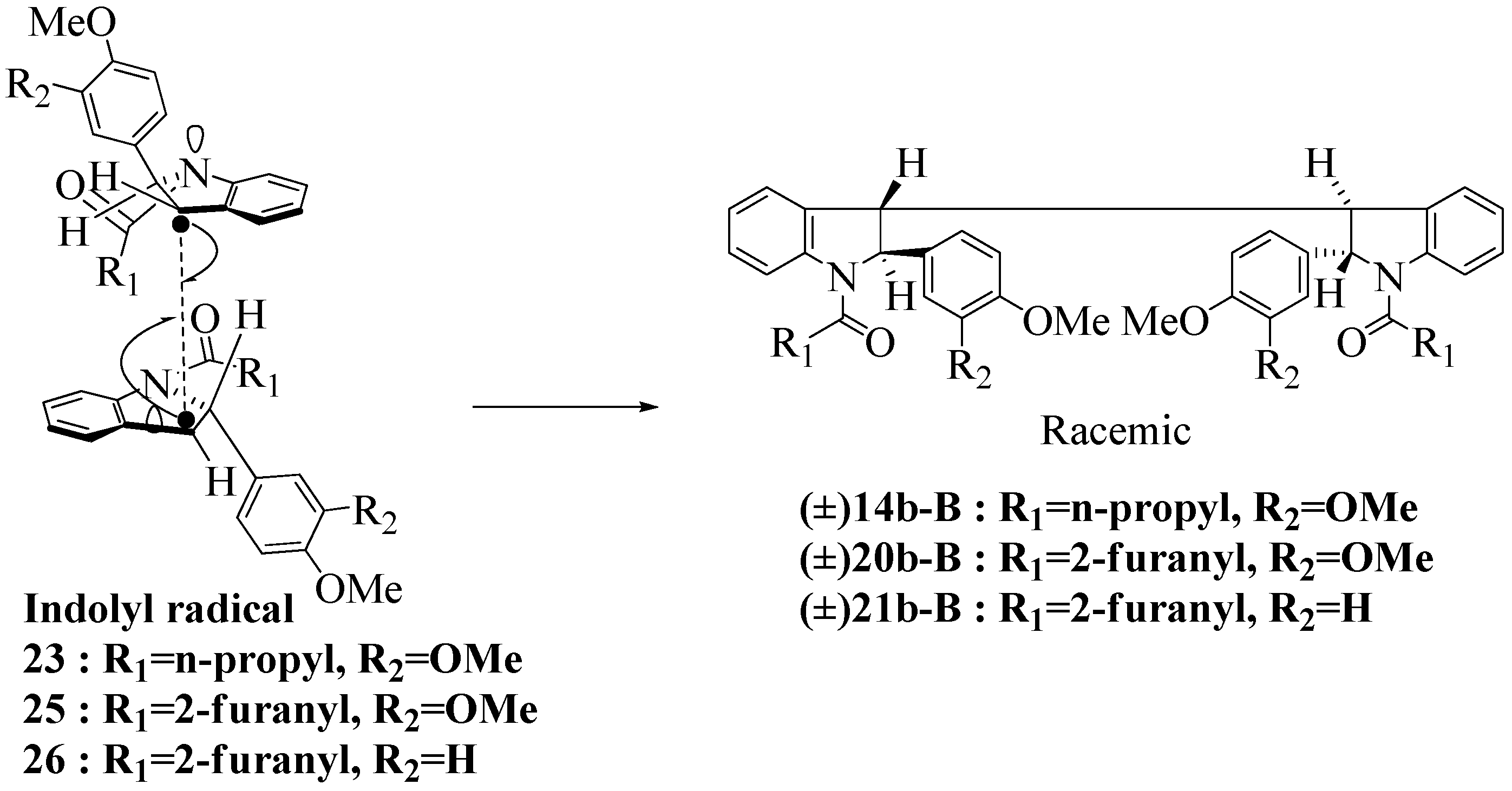
2.3. Dimerization Pathways and PM6 Calculations for the Minor Products
| Stereoisomer | Symmetryelement | Heat of Formation (kcal/mol) | H2-H3 | H2*-H3* | H3-H3* |
|---|---|---|---|---|---|
 | C2 axis | −154.22 | 19.15 | 346.41 | 156.58 |
 | C2 axis | −162.78 | 250.79 | 250.79 | 171.63 |
 | Plane (Meso) | −163.46 | 240.71 | 111.62 | 298.43 |
 | Plane (Meso) | −133.96 | 310.52 | 324.60 | 24.01 |
| Stereoisomer | Symmetryelement | Heat of Formation (kcal/mol) | H2-H3 | H2*-H3* | H3-H3* |
|---|---|---|---|---|---|
 | C2 axis | −116.85 | 348.44 | 348.44 | 155.06 |
 | C2 axis | −123.76 | 241.19 | 241.17 | 120.43 |
 | Plane (Meso) | −126.41 | −111.73 | 119.11 | 61.86 |
 | Plane (Meso) | −115.76 | 352.90 | 26.21 | 65.36 |
| Stereoisomer | Symmetryelement | Heat of Formation (kcal/mol) | H2-H3 | H2*-H3* | H3-H3* |
|---|---|---|---|---|---|
 | C2 axis | −48.59 | 340.27 | 340.27 | 115.07 |
 | C2 axis | −55.04 | 247.75 | 247.23 | 172.63 |
 | Plane (Meso) | −56.28 | 240.70 | 111.61 | 298.43 |
 | Plane (Meso) | −43.83 | 12.48 | 26.33 | 69.76 |










3. Experimental
3.1. General
3.2. General Procedure for the FeCl3 Oxidative Coupling
3.2.1. 1-(2-(3,4-Dimethoxyphenyl)indolin-1-yl)butan-1-one (14a)
3.2.2. 1,1'-2,2'- bis(3,4-Dimethoxyphenyl)- 3,3'- biindoline-1,1'-diyl dibutan-1-one ((±)14b)
3.2.3. 1-(2-(3,4-dimethoxyphenyl)indolin-1-yl)-2-methylpropan-1-one (15)
3.2.4. (2-(3,4-Dimethoxyphenyl)indolin-1-yl)(phenyl)methanone (16)
3.2.5. Cyclohexyl(2-(3,4-dimethoxyphenyl)indolin-1-yl)methanone (17)
3.2.6. Furan-2-yl(2-(2-methoxyphenyl)indolin-1-yl)methanone (18)
3.2.7. Furan-2-yl(2-(3-methoxyphenyl)indolin-1-yl)methanone (19)
3.2.8. (2-(3,4-Dimethoxyphenyl)indolin-1-yl)(furan-2-yl)methanone (20a)
3.2.9. (2,2'- bis(3,4-Dimethoxyphenyl)-3,3'-biindoline-1,1'-diyl)-bis(furan-2-ylmethanone) ((±)20b)
3.2.10. Furan-2-yl(2-(4-methoxyphenyl)indolin-1-yl)methanone (21a)
3.2.11. (2,2'- bis(4-methoxyphenyl)- 3,3'- biindoline-1,1'-diyl) bis(furan-2-ylmethanone) ((±)21b)
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
References and Notes
- Fuwa, H.; Sasaki, M. Synthesis of 2-substituted indoles and indolines via Suzuki-Miyaura coupling/5-endo-trig cyclization strategies. J. Org. Chem. 2009, 74, 212–221. [Google Scholar] [CrossRef]
- Prediger, I.; Weiss, T.; Reiser, O. Facile access to 2-arylindolines and 2-arylindoles by microwave-assisted tandem radical cyclization. Synthesis 2008, 2008, 2191–2198. [Google Scholar] [CrossRef]
- Anas, S.; Kagan, H.B. Routes toward enantiopure 2-substituted indolines: An overview. Tetrahedron: Asymmetry 2009, 20, 2193–2199. [Google Scholar] [CrossRef]
- Bailey, W.F.; Luderer, M.R.; Mealy, M.J. Preparation of differentially 1,3-disubstituted indolines by intramolecular carbolithiation. Tetrahedron Lett. 2003, 44, 5303–5305. [Google Scholar] [CrossRef]
- Kuwano, R.; Sato, K.; Kurokawa, T.; Karube, D.; Ito, Y. Catalytic asymmetric hydrogenation of heteroaromatic compounds, indoles. J. Am. Chem. Soc. 2000, 122, 7614–7615. [Google Scholar] [CrossRef]
- Leroi, C.; Bertin, D.; Dufils, P.-E.; Gigmes, D.; Marque, S.; Tordo, P.; Couturier, J.-L.; Guerret, O.; Ciufolini, M.A. Alkoxyamine-mediated radical synthesis of indolinones and indolines. Org. Lett. 2003, 5, 4943–4945. [Google Scholar]
- Muñiz, K. Advancing palladium-catalyzed C-N bond formation: Bisindoline construction from successive amide transfer to internal alkenes. J. Am. Chem. Soc. 2007, 129, 14542–14543. [Google Scholar] [CrossRef]
- Zhu, Q.; Huang, H.; Shi, D.; Shen, Z.; Xia, C. An efficient synthesis of chiral diamines with rigid backbones: Application in enantioselective michael addition of malonates to nitroalkenes. Org. Lett. 2009, 11, 4536–4539. [Google Scholar] [CrossRef]
- Movassaghi, M.; Schmidt, M.A. Concise total synthesis of (-)-calycanthine, (+)-chimonanthine, and (+)-folicanthine. Angew. Chem. Int. Ed. 2007, 46, 3725–3728. [Google Scholar] [CrossRef]
- Thomas, N.F.; Velu, S.S.; Weber, J.-F.F.; Lee, K.C.; Hadi, A.H.A.; Richomme, P.; Rondeau, D.; Noorbatcha, I.; Awang, K. A tandem highly stereoselective FeCl3-promoted synthesis of a bisindoline: Synthetic utility of radical cations in heterocyclic construction. Tetrahedron 2004, 60, 11733–11742. [Google Scholar] [CrossRef]
- Kam, T.-S.; Low, Y.-Y. We thank Prof. Kam and Mr Low (Dept. of Chem., University of Malaya) for crystal data of the bisindoline 4b. A CIF file is included in the supplementary data submitted.
- Thomas, N.F.; Kee, C.-H.; Ariffin, A.; Awang, K.; Weber, J.-F.F.; Lim, C.-G.; Mukhtar, M.R.; A Hadi, A.H. The subtle co-catalytic intervention of benzophenone in radical cation mediated cyclization - an improved synthesis of 2-(3', 4'-dimethoxyphenyl) indoline. Heterocycles 2008, 75, 1097–1108. [Google Scholar] [CrossRef]
- Kee, C.H.; Ariffin, A.; Awang, K.; Takeya, K.; Morita, H.; Inayat Hussain, S.; Chan, K.M.; Wood, P.J.; Threadgill, M.D.; Lim, C.G.; et al. Challenges associated with the synthesis of unusual o-carboxamido stilbenes by the Heck protocol: Intriguing substituent effects, their toxicological and chemopreventive implications. Org. Biomol. Chem. 2010, 8, 5646–5660. [Google Scholar] [CrossRef]
- Crich, D.; Shirai, M.; Brebion, F.; Rumthao, S. Enantioselective alkene radical cations reactions. Tetrahedron 2006, 62, 6501–6518. [Google Scholar] [CrossRef]
- Ahmad, K.; Thomas, N.F.; Mukhtar, M.R.; Noorbatcha, I.; Faizal Weber, J.-F.; Nafiah, M.A.; Velu, S.S.; Takeya, K.; Morita, H.; Lim, C.-G.; et al. A FeCl3-promoted highly atropodiastereoselective cascade reaction: Synthetic utility of radical cations in indolostilbene construction. Tetrahedron 2009, 65, 1504–1516. [Google Scholar]
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2006. [Google Scholar]
- Stewart, J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Stewart, J. Mopac 2009; Stewart Computational Chemistry: Colorado Springs, CO, USA, 2009. Available online: http://OpenMOPAC.net.
- Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Iron-catalyzed reactions in organic synthesis. Chem. Rev. 2004, 104, 6217–6254. [Google Scholar] [CrossRef]
- Díaz, D.D.; Miranda, P.O.; Padrón, J.I.; Martín, V.S. Recent uses of Iron (III) chloride in organic synthesis. Curr. Org. Chem. 2006, 10, 457–476. [Google Scholar] [CrossRef]
- Sarhan, A.A.O.; Bolm, C. Iron (III) chloride in oxidative C-C coupling reactions. Chem. Soc. Rev. 2009, 38, 2730–2744. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kee, C.H.; Ariffin, A.; Awang, K.; Noorbatcha, I.; Takeya, K.; Morita, H.; Lim, C.G.; Thomas, N.F. Cyclization vs. Cyclization/Dimerization in o-Amidostilbene Radical Cation Cascade Reactions: The Amide Question. Molecules 2011, 16, 7267-7287. https://doi.org/10.3390/molecules16097267
Kee CH, Ariffin A, Awang K, Noorbatcha I, Takeya K, Morita H, Lim CG, Thomas NF. Cyclization vs. Cyclization/Dimerization in o-Amidostilbene Radical Cation Cascade Reactions: The Amide Question. Molecules. 2011; 16(9):7267-7287. https://doi.org/10.3390/molecules16097267
Chicago/Turabian StyleKee, Chin Hui, Azhar Ariffin, Khalijah Awang, Ibrahim Noorbatcha, Koichi Takeya, Hiroshi Morita, Chuan Gee Lim, and Noel Francis Thomas. 2011. "Cyclization vs. Cyclization/Dimerization in o-Amidostilbene Radical Cation Cascade Reactions: The Amide Question" Molecules 16, no. 9: 7267-7287. https://doi.org/10.3390/molecules16097267