Epitope Prediction Based on Random Peptide Library Screening: Benchmark Dataset and Prediction Tools Evaluation

Abstract
:1. Introduction
2. Results and Discussion
2.1. Datasets Compilation
2.2. Performance Evaluation for the Existing Epitope Prediction Software

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Publication year | Language | Operating System | Service | Notes |
|---|---|---|---|---|---|
| FINDMAP | 2003 | C++ | not stated | no | FINDMAP is a method to acquire information on the 3D structure of the protein by identifying discontinuous epitopes; it maps one mimotope sequence to the protein at a time. |
| SiteLight | 2005 | C++ | Linux | no | SiteLight is a method of predicting the binding site on a 3D structure using random peptide library screening. |
| 3DEX | 2005 | VB | Windows | no | 3DEX allows the analysis of single amino acid of a linear peptide sequence with regard to their spatial neighborhood in the 3D structures of PDB files based on preselectable parameters like distance, string length (frame size) and surface exposure. It maps mimotopes to the protein one by one, one sequence at a time. |
| MIMOP | 2006 | PHP | Independent | Upon request | MIMOP provides an environment for mimotope characterization which integrates two main approaches, MimAlign and MimCons, which deliver to the user mimotope analysis results. |
| MIMOX | 2006 | Perl | Independent | Web | MIMOX has two sections, the first is to derive the consensus sequence, and the second is to map the single sequence to the target protein. |
| Mapitope | 2007 | C++ | Windows | Web | Mapitope is based on that epitope determinants shared by the entire set of peptides are detected. Both web service and source code is available. |
| PepSurf | 2007 | C++ | Linux | Web | PepSurf is an algorithm for mapping a set of affinity-selected peptides to the solved surface of the antigen. Both web service and source code is available. |
| Pepitope | 2007 | C++ | Windows | Web | Pepitope is a combination algorithm of PepSurf and Mapitope, the web service is available freely on the Pepitope server. |
| MEPS | 2007 | Java | Independent | Web | MEPS provides two services, one is to evaluate the likelihood that a given peptide to mimic exposed regions of the protein, and the other one is to generate all peptides of a given length to mimic exposed regions of the protein. |
| Pep-3D-Search | 2008 | VB | Windows | Graphic interface | Pep-3D-Search is an epitope mapping algorithm based on both mimotope and motif analysis. The source code is available freely. |
| EpiSearch | 2009 | not stated | not stated | Web | EpiSearch is an automated detection of conformational epitopes using random peptide library screening. It provides web service freely. |
2.2.1. Criteria and datasets used in methods evaluation




2.2.2. Evaluation through MCC
| MimoID | Antigen | ALL | Mapitope | PepSurf | Pepitope | Pep-3D-Search | EpiSearch | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MCC | PPV | MCC | PPV | MCC | PPV | MCC | PPV | MCC | PPV | |||||
| Antigen-Antibody | ||||||||||||||
| MS00012* | 2OSL_P | 25 | NA | NA | 0.182 | 0.250 | NA | NA | 0.145 | 0.200 | 0.190 | 0.267 | ||
| MS00013* | 3IU3_I | 223 | 0.146 | 0.667 | 0.045 | 0.231 | 0.071 | 0.500 | 0.168 | 0.471 | 0.134 | 0.500 | ||
| MS00029* | 1TET_P | 15 | 0.564 | 1.000 | 0.617 | 1.000 | 0.564 | 1.000 | 0.510 | 0.900 | 0.856 | 1.000 | ||
| MS00030 | 1TET_P | 15 | 0.471 | 1.000 | 0.679 | 1.000 | 0.471 | 1.000 | NA | NA | NA | NA | ||
| MS00048* | 1YY9_A | 624 | -0.066 | 0.000 | -0.003 | 0.000 | -0.033 | 0.000 | 0.049 | 0.400 | -0.005 | 0.000 | ||
| MS00049* | 1N8Z_C | 607 | 0.100 | 0.692 | 0.059 | 0.455 | 0.041 | 1.000 | 0.114 | 0.500 | 0.096 | 0.48 | ||
| MS00052 | 2ADF_A | 196 | 0.074 | 0.429 | 0.164 | 0.556 | —— | —— | 0.157 | 0.359 | NA | NA | ||
| MS00053* | 2ADF_A | 196 | -0.015 | 0.000 | 0.032 | 0.167 | -0.010 | 0.000 | 0.189 | 0.889 | 0.106 | 0.500 | ||
| MS00054* | 1IQD_C | 156 | 0.093 | 0.142 | -0.006 | 0.091 | 0.113 | 1.000 | 0.023 | 0.130 | 0.129 | 0.360 | ||
| MS00055* | 2GHW_A | 203 | 0.100 | 0.400 | 0.029 | 0.208 | —— | —— | -0.082 | 0.000 | 0.080 | 0.320 | ||
| MS00056 | 2GHW_A | 203 | 0.110 | 0.444 | 0.110 | 0.385 | —— | —— | -0.089 | 0.000 | —— | —— | ||
| MS00057 | 2NY7_G | 317 | 0.004 | 0.100 | 0.026 | 0.222 | —— | —— | 0.006 | 0.097 | 0.062 | 0.300 | ||
| MS00058 | 2NY7_G | 317 | -0.015 | 0.000 | —— | —— | —— | —— | 0.000 | 0.083 | —— | —— | ||
| MS00059* | 2NY7_G | 317 | 0.152 | 0.560 | 0.052 | 0.205 | 0.088 | 0.556 | 0.001 | 0.086 | 0.085 | 0.333 | ||
| MS00099 | 1N8Z_C | 607 | 0.076 | 0.600 | -0.066 | 0.000 | NA | NA | -0.005 | 0.000 | 0.005 | 0.059 | ||
| MS00185* | 1G9M_G | 321 | 0.102 | 0.324 | 0.063 | 0.226 | 0.091 | 0.412 | -0.001 | 0.044 | -0.015 | 0.000 | ||
| MS00186* | 1E6J_P | 210 | 0.021 | 0.167 | 0.158 | 0.478 | 0.036 | 0.333 | 0.119 | 0.275 | 0.114 | 0.364 | ||
| MS00242 | 2OSL_P | 25 | NA | NA | 0.145 | 0.200 | NA | NA | 0.145 | 0.200 | NA | NA | ||
| Protein-Protein | ||||||||||||||
| MS00041* | 1OC0_B | 51 | 0.226 | 0.364 | 0.166 | 0.375 | 0.166 | 0.375 | 0.101 | 0.310 | 0.254 | 0.440 | ||
| MS00047* | 1HX1_B | 114 | 0.028 | 0.238 | 0.114 | 0.360 | —— | —— | -0.022 | 0.167 | 0.190 | 0.480 | ||
| MS00060* | 1WLP_B | 138 | -0.073 | 0.000 | -0.033 | 0.160 | -0.040 | 0.000 | 0.065 | 0.279 | —— | —— | ||
| MS00062* | 1WLP_A | 25 | 0.496 | 0.789 | 0.180 | 0.714 | 0.499 | 1.000 | NA | NA | 0.530 | 1.000 | ||
| MS00139* | 1K4U_S | 62 | 0.247 | 0.778 | 0.217 | 1.000 | 0.247 | 0.778 | 0.391 | 0.611 | —— | —— | ||
| MS00276 | 2GRX_A | 725 | -0.007 | 0.000 | 0.041 | 0.261 | -0.006 | 0.000 | -0.006 | 0.026 | 0.004 | 0.069 | ||
| MS00277* | 2GRX_A | 725 | -0.007 | 0.000 | 0.039 | 0.429 | -0.006 | 0.000 | 0.006 | 0.071 | 0.029 | 0.222 | ||
| MS00278 | 2GSK_A | 590 | -0.008 | 0.000 | 0.070 | 0.545 | -0.003 | 0.000 | 0.038 | 0.263 | -0.018 | 0.000 | ||
| MS00279* | 2GSK_A | 590 | 0.033 | 0.184 | 0.047 | 0.400 | 0.047 | 0.400 | -0.011 | 0.000 | -0.016 | 0.000 | ||
| MS00357* | 1FLT_X | 95 | 0.228 | 0.727 | 0.140 | 0.750 | 0.140 | 0.750 | 0.005 | 0.227 | 0.259 | 0.688 | ||
| MS00384* | 3DOW_B | 12 | 0.527 | 1.000 | 0.764 | 1.000 | 0.527 | 1.000 | NA | NA | NA | NA | ||
| MS00405* | 1SHY_A | 234 | 0.008 | 0.125 | 0.036 | 0.200 | 0.017 | 0.200 | -0.005 | 0.088 | -0.020 | 0.045 | ||
| MS00464 | 1SQ0_A | 214 | 0.077 | 0.444 | 0.021 | 0.188 | NA | NA | 0.037 | 0.194 | 0.080 | 0.333 | ||
| MS00465* | 1SQ0_A | 214 | -0.029 | 0.000 | 0.002 | 0.133 | -0.025 | 0.000 | 0.046 | 0.231 | 0.071 | 0.357 | ||
| MS00671* | 1D4V_B | 163 | -0.046 | 0.000 | 0.117 | 0.381 | -0.021 | 0.000 | -0.039 | 0.000 | -0.017 | 0.083 | ||
| MS00976* | 3BT1_A | 135 | 0.240 | 0.786 | -0.073 | 0.114 | —— | —— | 0.240 | 0.593 | 0.025 | 0.240 | ||
| MS00984* | 1EER_A | 166 | 0.078 | 0.455 | 0.006 | 0.250 | -0.033 | 0.000 | 0.089 | 0.500 | 0.001 | 0.231 | ||
| MS01004 | 1MQ8_B | 177 | -0.031 | 0.000 | -0.030 | 0.000 | NA | NA | -0.014 | 0.069 | NA | NA | ||
| MS01036* | 3EZE_B | 85 | 0.071 | 0.400 | 0.331 | 0.917 | 0.230 | 0.857 | 0.393 | 0.850 | 0.313 | 0.909 | ||
| MS01037 | 3EZE_B | 85 | -0.109 | 0.000 | 0.336 | 0.727 | NA | NA | 0.408 | 0.750 | 0.198 | 0.550 | ||
| MS01038 | 3EZE_B | 85 | 0.288 | 0.543 | 0.364 | 0.704 | —— | —— | 0.375 | 0.731 | 0.086 | 0.429 | ||
| MS01061* | 1MQ8_B | 177 | -0.051 | 0.000 | -0.020 | 0.000 | -0.041 | 0.000 | 0.009 | 0.081 | -0.030 | 0.000 | ||
| MS01062 | 1MQ8_B | 177 | -0.030 | 0.000 | -0.062 | 0.000 | —— | —— | 0.024 | 0.135 | -0.041 | 0.000 | ||
| MS01063 | 1MQ8_B | 177 | -0.027 | 0.000 | -0.039 | 0.000 | —— | —— | -0.018 | 0.065 | -0.031 | 0.000 | ||
| MS01105/10/15* | 1II4_A | 155 | 0.059 | 0.385 | 0.163 | 0.545 | 0.095 | 0.571 | 0.233 | 0.523 | 0.274 | 0.750 | ||
| MS01154* | 1HX1_A | 400 | -0.006 | 0.000 | 0.010 | 0.111 | -0.004 | 0.000 | 0.040 | 0.156 | 0.029 | 0.154 | ||
| MS01190* | 1G1S_D | 28 | 0.386 | 0.750 | 0.388 | 0.636 | —— | —— | NA | NA | NA | NA | ||
| MS01191 | 1G1S_D | 28 | 0.386 | 0.750 | 0.274 | 0.556 | —— | —— | NA | NA | NA | NA | ||
| MS01192 | 1G1S_D | 28 | NA | NA | NA | NA | —— | —— | NA | NA | NA | NA | ||

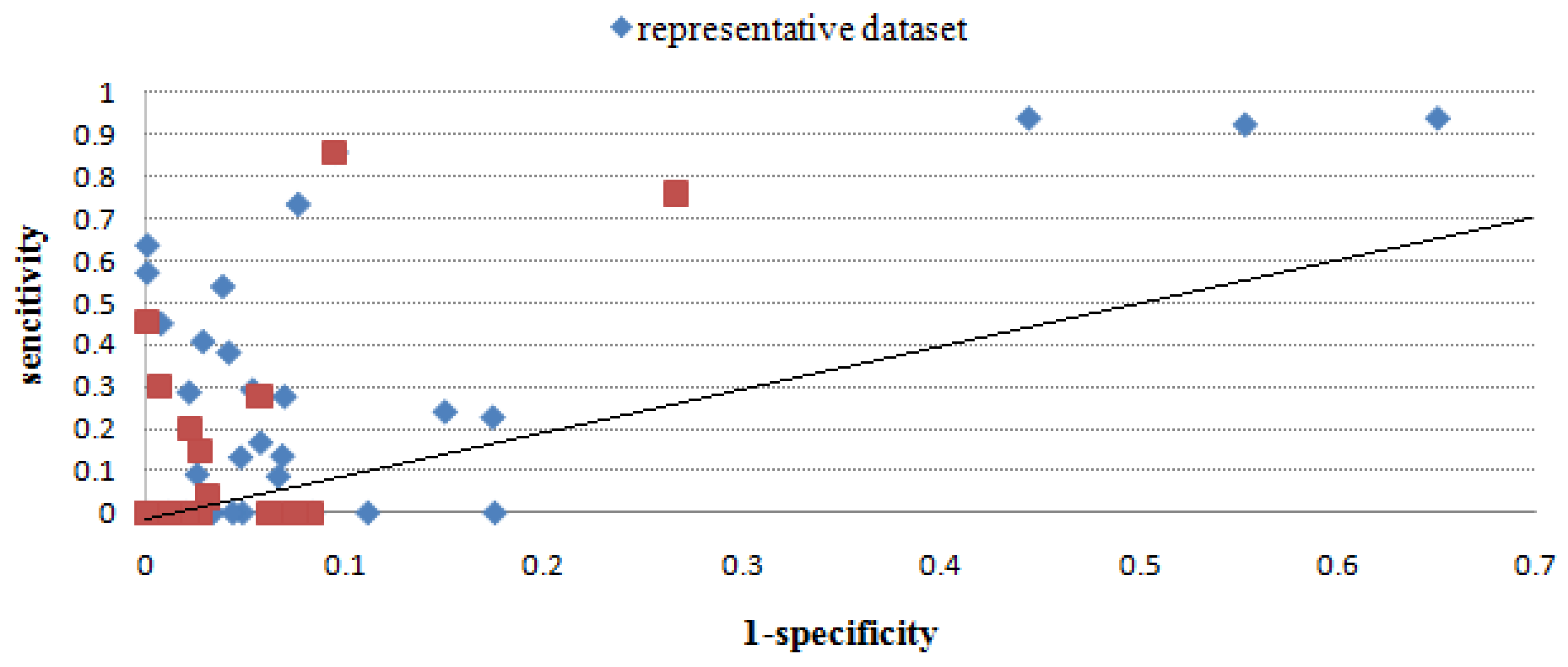
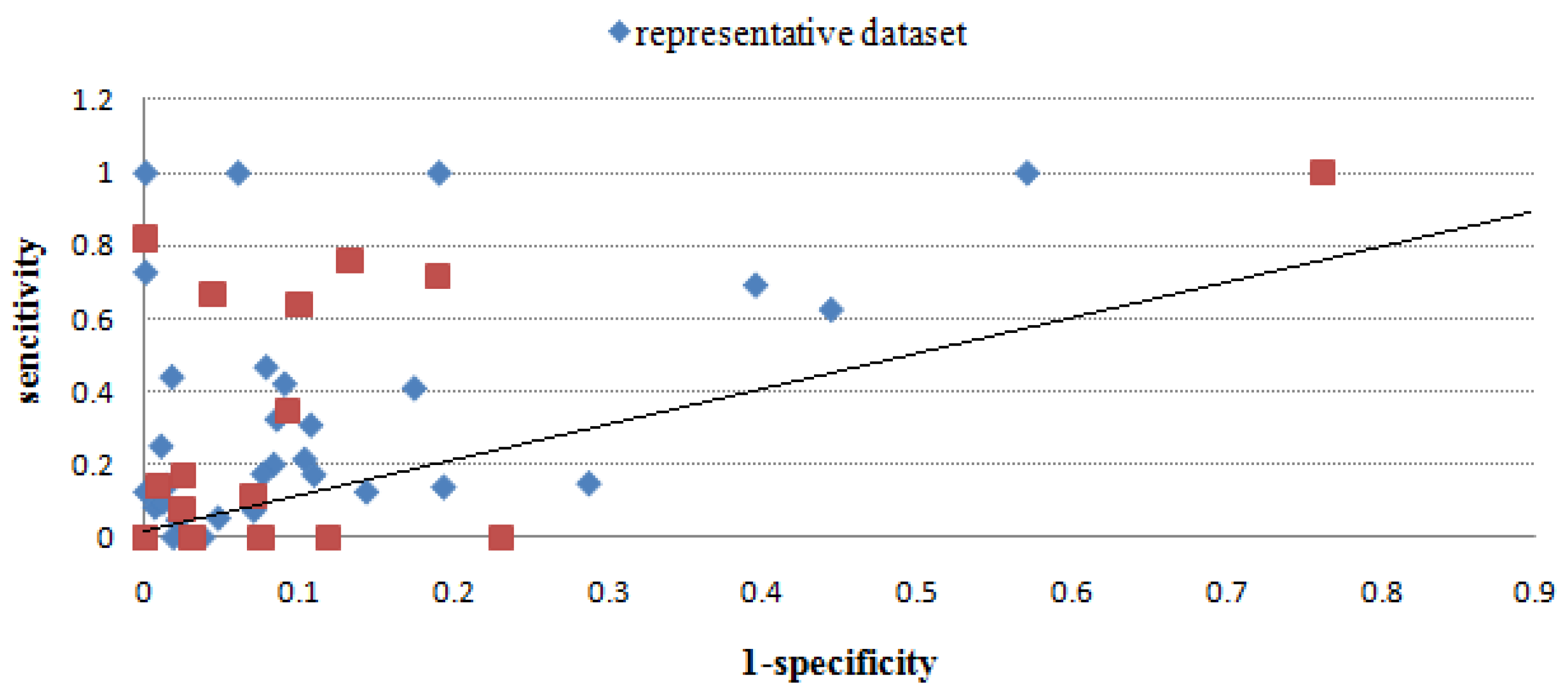
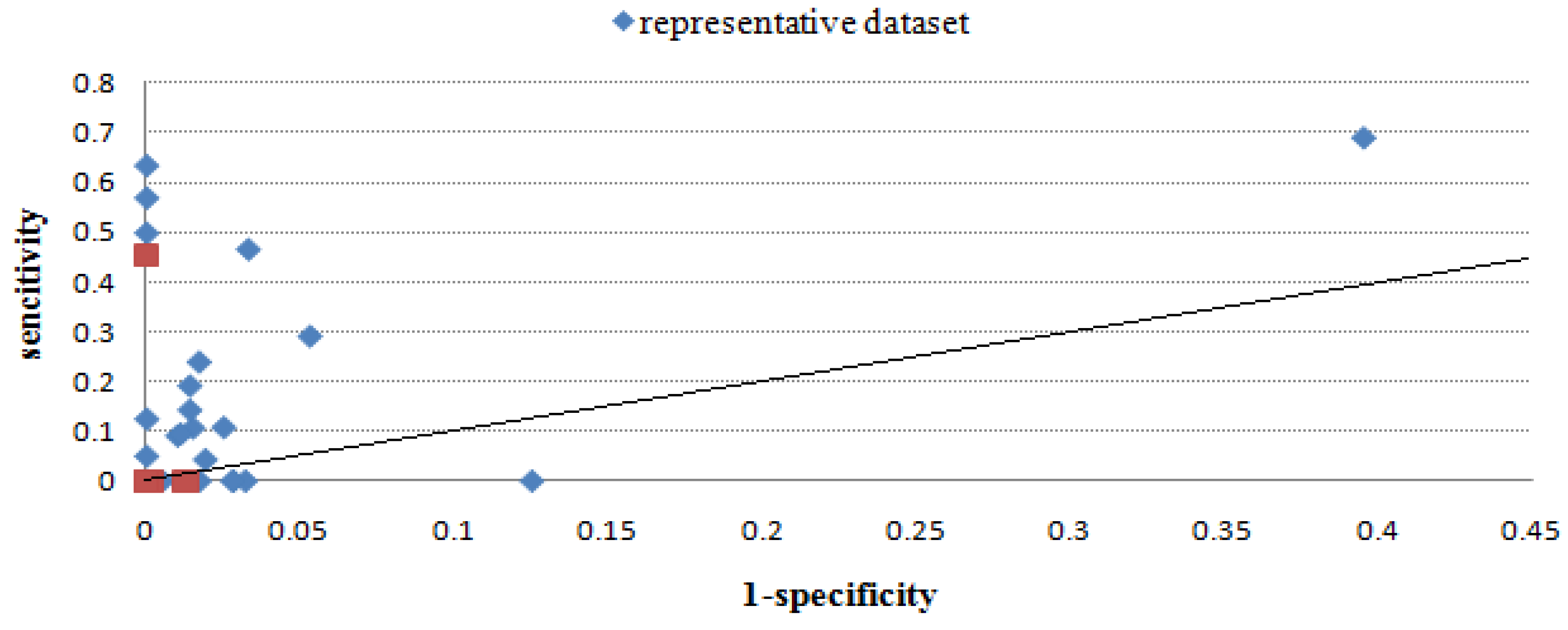
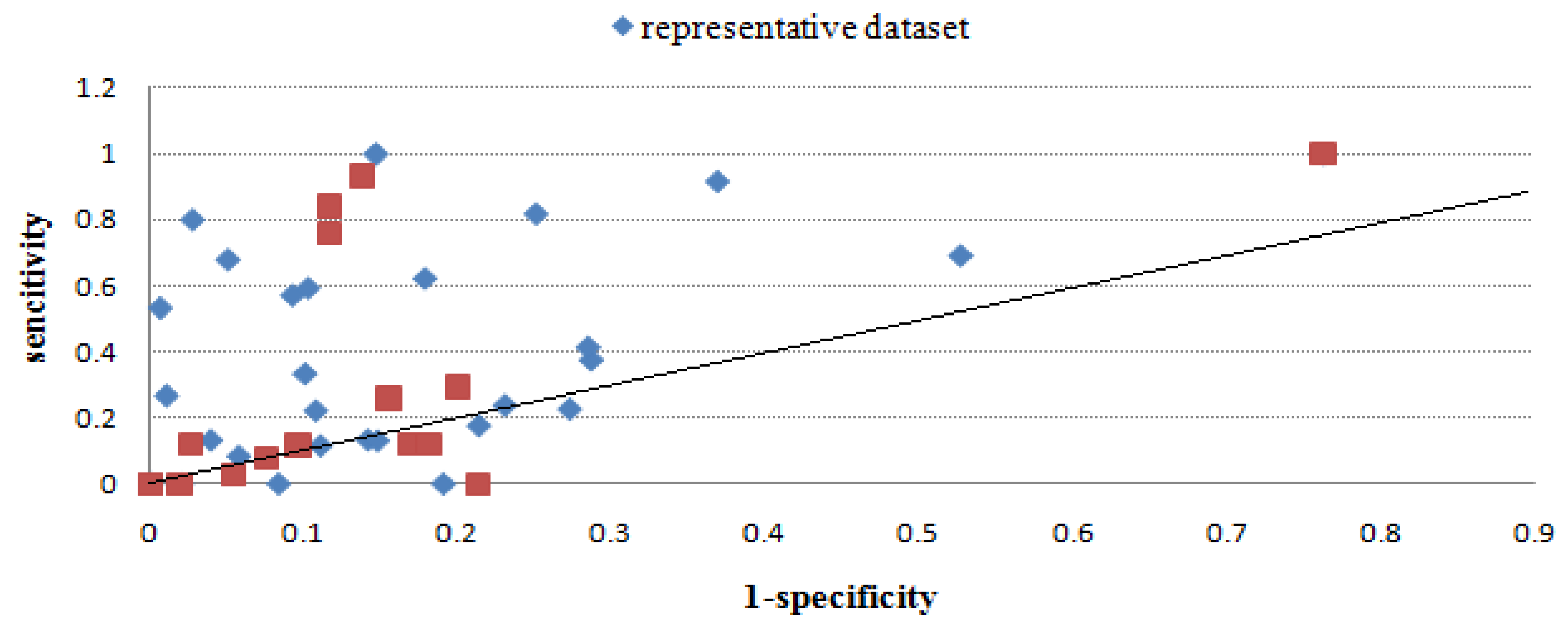
2.2.3. Evaluation through sensitivity/1-specificity
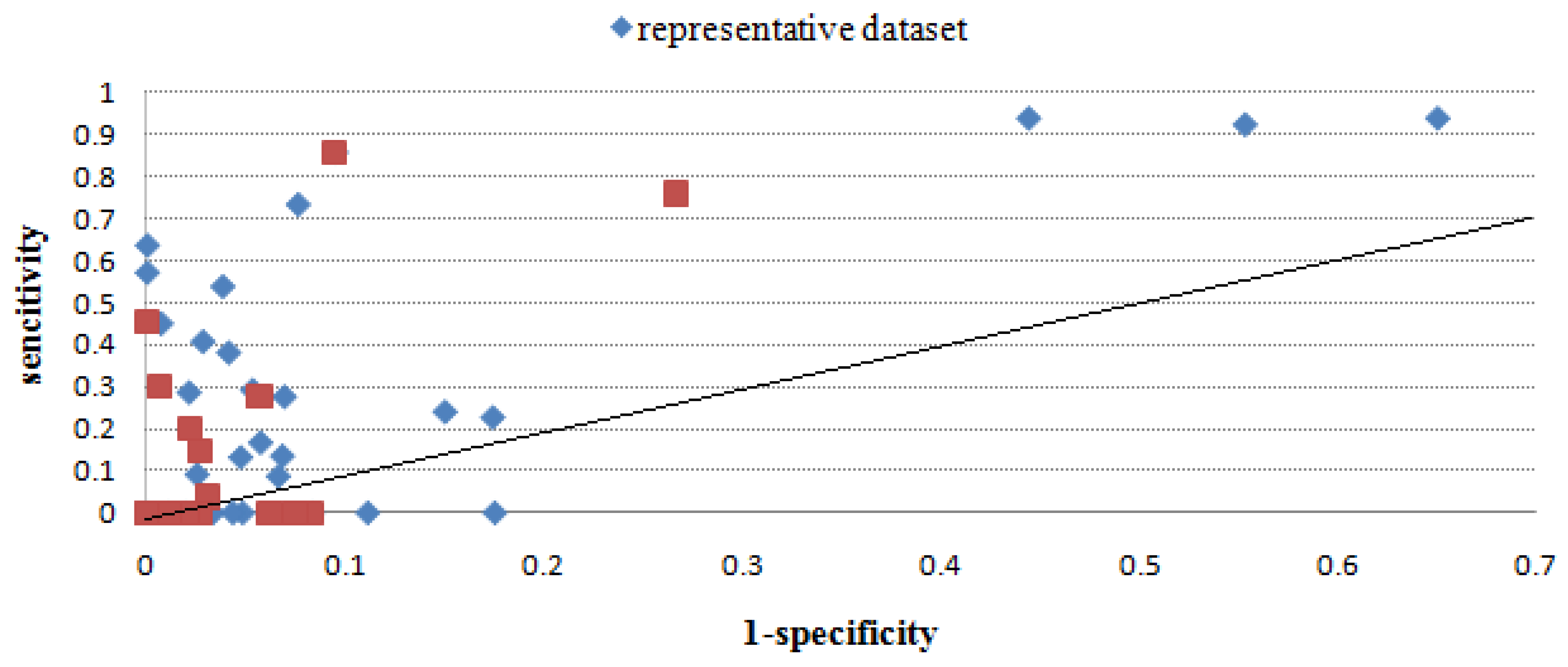
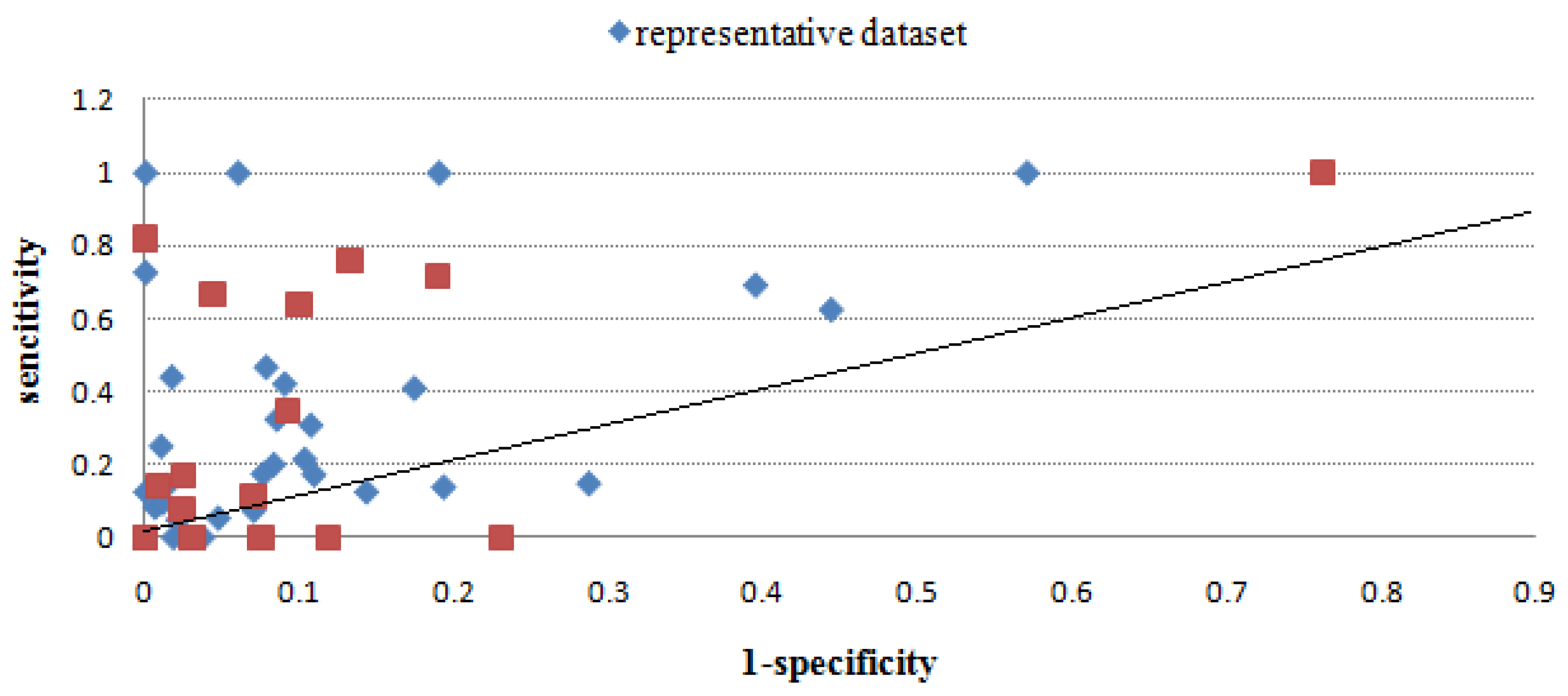
| MimoID | Antigen | EPI | Mapitope | PepSurf | Pepitope | Pep-3D-Search | EpiSearch | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sen | Spe | Sen | Spe | Sen | Spe | Sen | Spe | Sen | Spe | |||
| Antigen-Antibody | ||||||||||||
| MS00012* | 2OSL_P | 4 | NA | NA | 1.000 | 0.429 | NA | NA | 1.000 | 0.238 | 1.000 | 0.400 |
| MS00013* | 3IU3_I | 28 | 0.286 | 0.979 | 0.214 | 0.897 | 0.107 | 0.985 | 0.571 | 0.908 | 0.357 | 0.949 |
| MS00029* | 1TET_P | 11 | 0.636 | 1.000 | 0.727 | 1.000 | 0.636 | 1.000 | 0.818 | 0.750 | 1.000 | 1.000 |
| MS00030 | 1TET_P | 11 | 0.455 | 1.000 | 0.818 | 1.000 | 0.455 | 1.000 | NA | NA | NA | NA |
| MS00048* | 1YY9_A | 15 | 0.000 | 0.952 | 0.000 | 0.982 | 0.000 | 0.984 | 0.267 | 0.990 | 0.000 | 0.959 |
| MS00049* | 1N8Z_C | 20 | 0.450 | 0.993 | 0.250 | 0.990 | 0.050 | 1.000 | 0.800 | 0.973 | 0.660 | 0.978 |
| MS00052 | 2ADF_A | 15 | 0.200 | 0.978 | 0.667 | 0.956 | —— | —— | 0.933 | 0.862 | NA | NA |
| MS00053* | 2ADF_A | 15 | 0.000 | 0.967 | 0.200 | 0.917 | 0.000 | 0.983 | 0.533 | 0.994 | 0.333 | 0.972 |
| MS00054* | 1IQD_C | 16 | 0.938 | 0.350 | 0.125 | 0.857 | 0.125 | 1.000 | 0.375 | 0.714 | 0.562 | 0.886 |
| MS00055* | 2GHW_A | 29 | 0.276 | 0.931 | 0.172 | 0.891 | —— | —— | 0.000 | 0.810 | 0.276 | 0.902 |
| MS00056 | 2GHW_A | 29 | 0.276 | 0.943 | 0.345 | 0.908 | —— | —— | 0.000 | 0.787 | —— | —— |
| MS00057 | 2NY7_G | 26 | 0.038 | 0.969 | 0.077 | 0.976 | —— | —— | 0.115 | 0.904 | 0.231 | 0.952 |
| MS00058 | 2NY7_G | 26 | 0.000 | 0.973 | —— | —— | —— | —— | 0.077 | 0.924 | —— | —— |
| MS00059* | 2NY7_G | 26 | 0.538 | 0.962 | 0.308 | 0.893 | 0.192 | 0.986 | 0.115 | 0.890 | 0.346 | 0.938 |
| MS00099 | 1N8Z_C | 20 | 0.300 | 0.993 | 0.000 | 0.969 | NA | NA | 0.000 | 0.981 | 0.050 | 0.973 |
| MS00185* | 1G9M_G | 15 | 0.733 | 0.924 | 0.467 | 0.922 | 0.467 | 0.967 | 0.133 | 0.859 | 0.000 | 0.922 |
| MS00186* | 1E6J_P | 11 | 0.091 | 0.975 | 1.000 | 0.940 | 0.091 | 0.990 | 1.000 | 0.854 | 0.727 | 0.930 |
| MS00242 | 2OSL_P | 4 | NA | NA | 1.000 | 0.238 | NA | NA | 1.000 | 0.238 | NA | NA |
| Protein-Protein | ||||||||||||
| MS00041* | 1OC0_B | 13 | 0.923 | 0.447 | 0.692 | 0.605 | 0.692 | 0.605 | 0.692 | 0.474 | 0.846 | 0.632 |
| MS00047* | 1HX1_B | 22 | 0.227 | 0.826 | 0.409 | 0.826 | —— | —— | 0.227 | 0.728 | 0.545 | 0.859 |
| MS00060* | 1WLP_B | 29 | 0.000 | 0.917 | 0.138 | 0.807 | 0.000 | 0.972 | 0.414 | 0.716 | —— | —— |
| MS00062* | 1WLP_A | 16 | 0.938 | 0.556 | 0.625 | 0.556 | 0.500 | 1.000 | NA | NA | 0.562 | 1.000 |
| MS00139* | 1K4U_S | 24 | 0.292 | 0.947 | 0.125 | 1.000 | 0.292 | 0.947 | 0.917 | 0.632 | —— | —— |
| MS00276 | 2GRX_A | 36 | 0.000 | 0.980 | 0.167 | 0.975 | 0.000 | 0.987 | 0.028 | 0.946 | 0.056 | 0.961 |
| MS00277* | 2GRX_A | 36 | 0.000 | 0.981 | 0.083 | 0.994 | 0.000 | 0.987 | 0.083 | 0.943 | 0.111 | 0.980 |
| MS00278 | 2GSK_A | 42 | 0.000 | 0.989 | 0.143 | 0.991 | 0.000 | 0.998 | 0.119 | 0.974 | 0.000 | 0.947 |
| MS00279* | 2GSK_A | 42 | 0.167 | 0.943 | 0.095 | 0.989 | 0.095 | 0.989 | 0.000 | 0.980 | 0.000 | 0.958 |
| MS00357* | 1FLT_X | 21 | 0.381 | 0.959 | 0.143 | 0.986 | 0.143 | 0.986 | 0.238 | 0.770 | 0.524 | 0.932 |
| MS00384* | 3DOW_B | 7 | 0.571 | 1.000 | 1.000 | 1.000 | 0.571 | 1.000 | NA | NA | NA | NA |
| MS00405* | 1SHY_A | 27 | 0.087 | 0.934 | 0.174 | 0.924 | 0.043 | 0.981 | 0.130 | 0.853 | 0.043 | 0.900 |
| MS00464 | 1SQ0_A | 27 | 0.148 | 0.973 | 0.111 | 0.930 | NA | NA | 0.259 | 0.845 | 0.259 | 0.925 |
| MS00465* | 1SQ0_A | 27 | 0.000 | 0.957 | 0.074 | 0.930 | 0.000 | 0.968 | 0.222 | 0.893 | 0.185 | 0.952 |
| MS00671* | 1D4V_B | 19 | 0.000 | 0.889 | 0.421 | 0.910 | 0.000 | 0.972 | 0.000 | 0.917 | 0.105 | 0.847 |
| MS00976* | 3BT1_A | 27 | 0.407 | 0.972 | 0.148 | 0.713 | —— | —— | 0.593 | 0.898 | 0.222 | 0.824 |
| MS00984* | 1EER_A | 38 | 0.132 | 0.953 | 0.053 | 0.953 | 0.000 | 0.984 | 0.132 | 0.961 | 0.079 | 0.922 |
| MS01004 | 1MQ8_B | 17 | 0.000 | 0.919 | 0.000 | 0.925 | NA | NA | 0.118 | 0.831 | NA | NA |
| MS01036* | 3EZE_B | 25 | 0.240 | 0.850 | 0.440 | 0.983 | 0.240 | 0.983 | 0.680 | 0.950 | 0.400 | 0.983 |
| MS01037 | 3EZE_B | 25 | 0.000 | 0.917 | 0.640 | 0.900 | NA | NA | 0.840 | 0.883 | 0.440 | 0.850 |
| MS01038 | 3EZE_B | 25 | 0.760 | 0.733 | 0.760 | 0.867 | —— | —— | 0.760 | 0.883 | 0.240 | 0.867 |
| MS01061* | 1MQ8_B | 17 | 0.000 | 0.825 | 0.000 | 0.963 | 0.000 | 0.875 | 0.176 | 0.787 | 0.000 | 0.925 |
| MS01062 | 1MQ8_B | 17 | 0.000 | 0.925 | 0.000 | 0.769 | —— | —— | 0.294 | 0.800 | 0.000 | 0.875 |
| MS01063 | 1MQ8_B | 17 | 0.000 | 0.938 | 0.000 | 0.881 | —— | —— | 0.118 | 0.819 | 0.000 | 0.919 |
| MS01105/10/15* | 1II4_A | 37 | 0.135 | 0.932 | 0.324 | 0.915 | 0.108 | 0.975 | 0.622 | 0.822 | 0.486 | 0.949 |
| MS01154* | 1HX1_A | 21 | 0.000 | 0.989 | 0.048 | 0.979 | 0.000 | 0.995 | 0.333 | 0.900 | 0.190 | 0.942 |
| MS01190* | 1G1S_D | 7 | 0.857 | 0.905 | 1.000 | 0.810 | —— | —— | NA | NA | NA | NA |
| MS01191 | 1G1S_D | 7 | 0.857 | 0.905 | 0.714 | 0.810 | —— | —— | NA | NA | NA | NA |
| MS01192 | 1G1S_D | 7 | NA | NA | NA | NA | —— | —— | NA | NA | NA | NA |
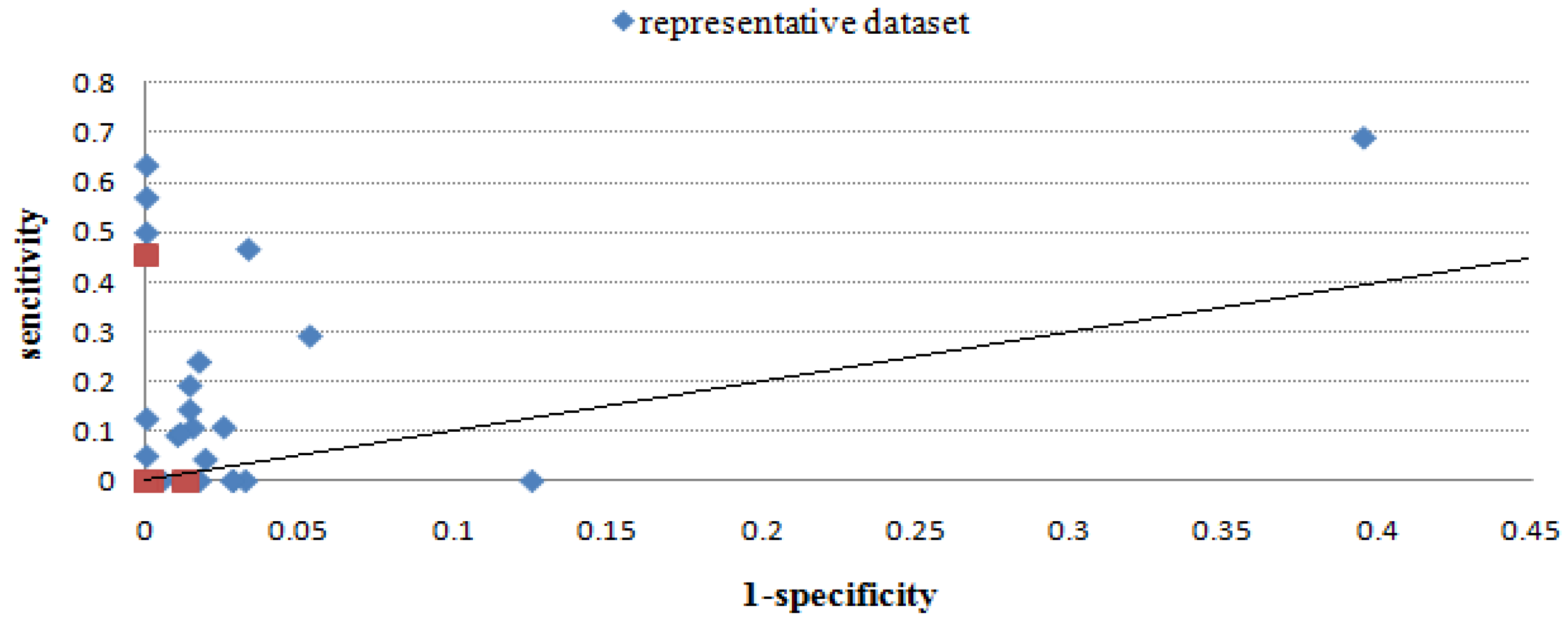

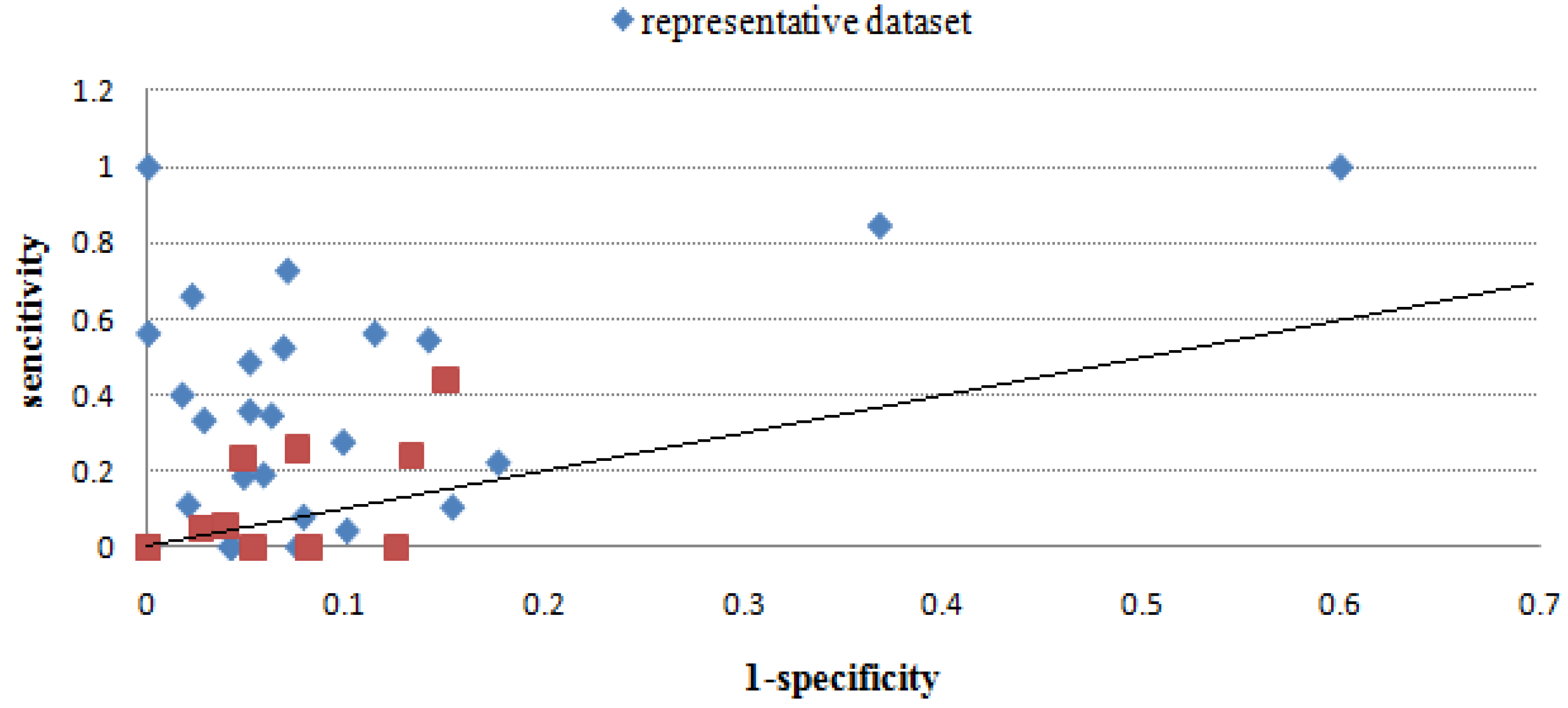
2.2.4. Overall performance evaluation
| Statistics | Mapitope | PepSurf | Pepitope | Pep-3D-Search | EpiSearch |
|---|---|---|---|---|---|
| Antigen-Antibody | |||||
| sensitivity | 0.326 | 0.434 | 0.212 | 0.455 | 0.426 |
| specificity | 0.931 | 0.869 | 0.990 | 0.804 | 0.905 |
| PPV | 0.407 | 0.334 | 0.580 | 0.273 | 0.345 |
| MCC | 0.120 | 0.134 | 0.143 | 0.085 | 0.141 |
| Protein-Protein | |||||
| sensitivity | 0.254 | 0.305 | 0.149 | 0.333 | 0.241 |
| specificity | 0.895 | 0.889 | 0.956 | 0.842 | 0.907 |
| PPV | 0.311 | 0.409 | 0.330 | 0.288 | 0.317 |
| MCC | 0.105 | 0.127 | 0.099 | 0.099 | 0.099 |
| Benchmark dataset (Representative dataset) | |||||
| sensitivity | 0.280(0.320) | 0.339(0.326) | 0.172(0.174) | 0.368(0.387) | 0.289(0.342) |
| specificity | 0.908(0.890) | 0.892(0.901) | 0.968(0.965) | 0.841(0.845) | 0.921(0.922) |
| PPV | 0.346(0.377) | 0.384(0.398) | 0.419(0.429) | 0.284(0.322) | 0.329(0.378) |
| MCC | 0.112(0.127) | 0.129(0.126) | 0.116(0.112) | 0.092(0.101) | 0.112(0.139) |

3. Materials and Methods
3.1. Construction of the Datasets
| Mimo_ID | PDB_ID | Template | Target | Library(1) | Ref (2) |
|---|---|---|---|---|---|
| Antigen-Antibody | |||||
| MS00012* | 2OSL | B-lymphocyte antigen CD20 | Anti-CD20 monoclonal antibody rituximab | 13 × 9 | 16705086 |
| MS00013* | 3IU3 | Interleukin-2 receptor subunit alpha | Anti-CD25 monoclonal antibody basiliximab | 6 × 9 | 17440057 |
| MS00029* | 1TET | Heat-labile enterotoxin B chain | Anti-LTP-B monoclonal antibody TE33 | 10 × 9 | 16273596 |
| MS00030 | 1TET | Heat-labile enterotoxin B chain | Anti-LTP-B monoclonal antibody TE33 | 5 × 11 | 16273596 |
| MS00048* | 1YY9 | Epidermal growth factor receptor | Cetuximab | 4 × 12 | 16288119 |
| MS00049* | 1N8Z | Receptor tyrosine-protein kinase erbB-2 | Trastuzumab | 5 × 12 | 15210798 |
| MS00052 | 2ADF | von Willebrand factor | Anti-vWF monoclonal antibody 82D6A3 | 2 × 15 | 12855771 |
| MS00053* | 2ADF | von Willebrand factor | Anti-vWF monoclonal antibody 82D6A3 | 3 × 8 | 12855771 |
| MS00054* | 1IQD | Coagulation factor VIII | Anti-coagulation factor VIII monoclonal antibody BO2C11 | 27 × 12 | 12676786 |
| MS00055* | 2GHW | Spike glycoprotein | Anti-spike glycoprotein monoclonal antibody 80R | 18 × 15 | 16630634 |
| MS00056 | 2GHW | Spike glycoprotein | Anti-spike glycoprotein monoclonal antibody 80R | 9 × 16, 9 × 15, 19 × 14, 4 × 13 | 16630634 |
| MS00057 | 2NY7 | Surface protein gp120 | Anti-gp120 monoclonal antibody b12 | 1 × 12, 1 × 15 | 16940148 |
| MS00058 | 2NY7 | Surface protein gp120 | Anti-gp120 monoclonal antibody b12 | 1 × 6, 1 × 12, 1 × 13, 1 × 16, 1 × 18, 2 × 14, 2 × 20, 2 × 22 8 × 15, 13 × 21 | 16940148 |
| MS00059* | 2NY7 | Surface protein gp120 | Anti-gp120 monoclonal antibody b12 | 1 × 10, 1 × 13, 17 × 14 | 16940148 |
| MS00099 | 1N8Z | Receptor tyrosine-protein kinase erbB-2 | Trastuzumab | 2 × 12 | 15536075 |
| MS00185* | 1G9M | Envelope glycoprotein gp120 | Anti-gp120 monoclonal antibody 17b | 10 × 12, 1 × 10 | 14596802 |
| MS00186* | 1E6J | Capsid protein p24 | Anti-p42 monoclonal antibody 13b5 | 14 × 14, 2 × 7 | 14596802 |
| MS00242 | 2OSL | B-lymphocyte antigen CD20 | Anti-CD20 monoclonal antibody rituximab | 7 × 12 | 16814270 |
| Protein-Protein | |||||
| MS00041* | 1OC0 | Vitronectin | Plasminogen activator inhibitor 1 | 8 × 13, 1 × 7, 1 × 11 | 16813566 |
| MS00047* | 1HX1 | BAG family molecular chaperone regulator 1 | Heat shock cognate 71 kDa protein | 8 × 15 | 7649995 |
| MS00060* | 1WLP | Neutrophil cytosol factor 1 | Cytochrome b-245 | 2 × 8, 31 × 9 | 7592831 |
| MS00062* | 1WLP | Cytochrome b-245 light chain | Neutrophil cytosol factor 1 | 4 × 5, 3 × 9, 1 × 10, 1 × 8 | 7624379 |
| MS00139* | 1K4U | Neutrophil cytosol factor 1 | Neutrophil cytosol factor 2 | 28 × 9, 2 × 10, 4 × 12, 2 × 6, 1 × 8 | 8663333 |
| MS00276 | 2GRX | Ferrichrome-iron receptor | Protein tonB | 12 × 12 | 16414071 |
| MS00277* | 2GRX | Ferrichrome-iron receptor | Protein tonB | 6 × 9 | 16414071 |
| MS00278 | 2GSK | Vitamin B12 transporter btuB | Protein tonB | 2 × 12 | 16414071 |
| MS00279* | 2GSK | Vitamin B12 transporter btuB | Protein tonB | 6 × 9 | 16414071 |
| MS00357* | 1FLT | Vascular endothelial growth factor receptor 1 | Vascular endothelial growth factor A | 4 × 7 | 17401149 |
| MS00384* | 3DOW | Calreticulin | Gamma-aminobutyric acid receptor-associated protein | 5 × 12 | 17916189 |
| MS00405* | 1SHY | Hepatocyte growth factor | Hepatocyte growth factor receptor | 2 × 12, 1 × 13 | 17947467 |
| MS00464 | 1SQ0 | von Willebrand factor | Platelet glycoprotein Ib alpha chain | 2 × 11 | 18363340 |
| MS00465* | 1SQ0 | von Willebrand factor | Platelet glycoprotein Ib alpha chain | 3 ×11 | 18363340 |
| MS00671* | 1D4V | Tumor necrosis factor ligand superfamily member 10 | Tumor necrosis factor receptor superfamily member 10B | 13 × 9 | 20156289 |
| MS00976* | 3BT1 | Urokinase-type plasminogen activator | Urokinase plasminogen activator surface receptor | 19 × 15 | 8041758 |
| MS00984* | 1EER | Erythropoietin | Erythropoietin receptor | 1 × 10 | 8662529 |
| MS01004 | 1MQ8 | Integrin alpha-L beta-2 | Intercellular adhesion molecule 1 | 1 × 14 | 8953648 |
| MS01036* | 3EZE | Phosphocarrier protein HPr | Phosphoenolpyruvate-protein phosphotransferase | 11 × 6 | 9350871 |
| MS01037 | 3EZE | Phosphocarrier protein HPr | Phosphoenolpyruvate-protein phosphotransferase | 9 × 10 | 9350871 |
| MS01038 | 3EZE | Phosphocarrier protein HPr | Phosphoenolpyruvate-protein phosphotransferase | 6 × 15 | 9350871 |
| MS01061* | 1MQ8 | Integrin alpha-L beta-2 | Intercellular adhesion molecule 1 | 12 × 9, 1 × 8 | 11532073 |
| MS01062 | 1MQ8 | Integrin alpha-L beta-2 | Intercellular adhesion molecule 1 | 1 × 9, 7 × 16 | 12963036 |
| MS01063 | 1MQ8 | Integrin alpha-L beta-2 | Intercellular adhesion molecule 1 | 1 × 16 | 12963036 |
| MS01105/10/15*(3) | 1II4 | Heparin-binding growth factor 2 | Fibroblast growth factor receptor 2 | 30 × 7 | 12032665 |
| MS01154* | 1HX1 | Heat shock cognate 71 kDa protein | BAG family molecular chaperone regulator 1 | 8 × 12 | 11121403 |
| MS01190* | 1G1S | P-selectin glycoprotein ligand 1 | P-selectin | 5 × 17 | 12393589 |
| MS01191 | 1G1S | P-selectin glycoprotein ligand 1 | P-selectin | 2 × 15 | 12393589 |
| MS01192 | 1G1S | P-selectin glycoprotein ligand 1 | P-selectin | 1 × 13, 1 × 18 | 12393589 |
3.2. Algorithm Evaluation
4. Conclusions
Authors’ Contribution
Additional Materials
Supplementary File 1
Supplementary File 2
Acknowledgements
References and Notes
- Peters, B.; Sidney, J.; Bourne, P.; Bui, H.H.; Buus, S.; Doh, G.; Fleri, W.; Kronenberg, M.; Kubo, R.; Lund, O.; Nemazee, D.; Ponomarenko, J.V.; Sathiamurthy, M.; Schoenberger, S.P.; Stewart, S.; Surko, P.; Way, S.; Wilson, S.; Sette, A. The design and implementation of the immune epitope database and analysis resource. Immunogenetics 2005, 57, 326–336. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H. Antigenicity and immunogenicity of synthetic peptides. Biologicals 2001, 29, 209–213. [Google Scholar] [CrossRef]
- Barlow, D.J.; Edwards, M.S.; Thornton, J.M. Continuous and discontinuous protein antigenic determinants. Nature 1986, 322, 747–748. [Google Scholar] [CrossRef]
- Irving, M.B.; Pan, O.; Scott, J.K. Random-peptide libraries and antigen-fragment libraries for epitope mapping and the developmentof vaccines and diagnostics. Curr. Opin. Chem. Biol. 2001, 5, 314–324. [Google Scholar] [CrossRef]
- Westwood, O.M.R.; Hay, F.C. Epitope Mapping: A Practical Approach; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Gomara, M.J.; Haro, I. Synthetic peptides for the immunodiagnosisof human diseases. Curr. Med. Chem. 2007, 14, 531–546. [Google Scholar] [CrossRef]
- Camacho, C.J.; Vajda, S. Protein docking along smooth association pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 10636–10641. [Google Scholar] [CrossRef]
- Rus, J.J.; Burnett, R.M. Type-specific epitope locations revealed by X-ray crystallographic study of adenovirus type 5 hexon. Mol. Ther. 2000, 1, 3–4. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 2008, 9, 514. [Google Scholar] [CrossRef]
- Novotny, J.; Handschumacher, M.; Haber, E.; Bruccoleri, R.E.; Carlson, W.B.; Fanning, D.W.; Smith, J.A.; Rose, G.D. Antigenic determinants in proteins coincide with surface regions accessible to large probes (antibody domains). Proc. Natl. Acad. Sci. USA 1989, 83, 226–230. [Google Scholar]
- Hopp, T.P.; Woods, K.P. Prediction of Protein Antigenetic Determinants from Amino Acid Sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3824–3828. [Google Scholar] [CrossRef]
- Emini, E.A.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of hepatitis avirus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar]
- Garnier, J.; Osguthorpe, D.J.; Robson, B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 1978, 120, 97–120. [Google Scholar] [CrossRef]
- Pellequer, J.L.; Westhof, E.; Van Regenmortel, M.H. Correlation between the location of antigenic sites and the prediction of turns in proteins. Immunol. Lett. 1993, 36, 83–99. [Google Scholar] [CrossRef]
- Levitt, M. Conformational preferences of amino acids in globular proteins. Biochemistry 1978, 17, 4277–4285. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry 1974, 13, 211–222. [Google Scholar] [CrossRef]
- Blythe, M.J.; Flower, D.R. Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci. 2005, 14, 246–248. [Google Scholar]
- Larsen, J.E.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins 2003, 52, 80–87. [Google Scholar] [CrossRef]
- Greenbaum, J.A.; Andersen, P.H.; Blythe, M.; Bui, H.H.; Cachau, R.E.; Crowe, J.; Davies, M.; Kolaskar, A.S.; Lund, O.; Morrison, S.; Mumey, B.; Ofran, Y.; Pellequer, J.L.; Pinilla, C.; Ponomarenko, J.V.; Raghava, G.P.; van, Regenmortel, M.H.; Roggen, E.L.; Sette, A.; Schlessinger, A.; Sollner, J.; Zand, M.; Peters, B. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J. Mol. Recognit. 2007, 20, 75–82. [Google Scholar] [CrossRef]
- Westhof, E.; Altschuh, D.; Moras, D.; Bloomer, A.C.; Mondragon, A.; Klug, A.; Van Regenmortel, M.H. Correlation between segmentalmobility and the location of antigenic determinants in proteins. Nature 1984, 311, 123–126. [Google Scholar] [CrossRef]
- Tainer, J.A.; Getzoff, E.D.; Alexander, H.; Houghten, R.A.; Olson, A.J.; Lerner, R.A.; Hendrickson, W.A. The reactivity of anti-peptide antibodiesis a function of the atomic mobility of sites in a protein. Nature 1984, 312, 127–134. [Google Scholar] [CrossRef]
- Thornton, J.M.; Edwards, M.S.; Taylor, W.R.; Barlow, D.J. Location of 'continuous' antigenic determinants in the protruding regions of proteins. Embo J. 1986, 5, 409–413. [Google Scholar]
- Amit, A.G.; Mariuzza, R.A.; Phillips, S.E.; Poljak, R.J. Three-dimensional structure of an antigen-antibody complex at 2.8 Å resolution. Science 1986, 233, 747–753. [Google Scholar]
- Halperin, I.; Wolfson, H. SiteLight: Binding-site prediction using phage display libraries. Protein Sci. 2003, 12, 1344–1359. [Google Scholar] [CrossRef]
- Mumey, B.M. A new method for mapping discontinuous antibody epitopes to reveal structural features of proteins. J. Comput. Biol. 2003, 10, 555–567. [Google Scholar] [CrossRef]
- Kulkarni-Kale, U.; Bhosle, S.; Kolaskar, A.S. CEP: a conformational epitope prediction server. Nucleic Acids Res. 2005, 33, W168–W171. [Google Scholar] [CrossRef]
- Andersen, P.H.; Nielsen, M.; Lund, O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006, 15, 2558–2567. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: novel expressionvectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Tong, A.H.Y.; Drees, B.; Nardelli, G.; Bader, G.D.; Brannetti, B.; Castagnoli, L.; Evangelista, M.; Ferracuti, S.; Nelson, B.; Paoluzi, S.; Quondam, M.; Zucconi, A.; Hogue, C.W.; Fields, S.; Boone, C.; Cesareni, G. A combinedexperimental and computational strategy to define proteininteraction networks for peptide recognition modules. Science 2002, 295, 321–324. [Google Scholar]
- Thom, G.; Cockroft, A.C.; Buchanan, A.G.; Candotti, C.J.; Cohen, E.S.; Lowne, D.; Monk, P.; Shorrock-Hart, C.P.; Lutz, J.; Ralph, R. Minter Probing aprotein-protein interaction by in vitro evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 7619–7624. [Google Scholar]
- Wang, L.F.; Yu, M. Epitope identification and discoveryusing phage display libraries: Applications in vaccine developmentand diagnostics. Curr. Drug Targets 2004, 5, 1–15. [Google Scholar] [CrossRef]
- Thie, H.; Meyer, T.; Schirrmann, T.; Hust, M.; Dubel, S. Phage display derived therapeutic antibodies. Curr. Pharm. Biotechnol. 2008, 9, 439–446. [Google Scholar] [CrossRef]
- Geysen, H.M.; Rodda, S.J.; Mason, T.J. A priori delineation of a peptide which mimics a discontinuous antigenic determinant. Mol. Immunol. 1986, 23, 709–715. [Google Scholar] [CrossRef]
- Moreau, V.; Granier, C.; Villard, S.; Laune, D.; Molina, F. Discontinuous epitope prediction based on mimotope analysis. Bioinformatics 2006, 22, 1088–1095. [Google Scholar] [CrossRef]
- Huang, J.; Gutteridge, A.; Honda, W.; Kanehisa, M. MIMOX: a web tool for phage display based epitope mapping. BMC Bioinformatics 2006, 10, 1–10. [Google Scholar]
- Schreiber, A.; Humbert, M.; Benz, A.; Dietrich, U. 3D-Epitope-Explorer (3DEX): Localization of Conformational Epitopes within Three-Dimensional Structures of Proteins. J. Comput. Chem. 2005, 26, 879–887. [Google Scholar] [CrossRef]
- Bublil, E.M.; Freund, N.T.; Mayrose, I.; Penn, O.; Roitburd-berman, A.; Rubinstein, N.D.; Pupko, T.; Gershoni, J.M. Stepwise Prediction of Conformational Discontinuous B-Cell Epitopes Using the Mapitope Algorithm. Bioinformatics 2007, 304, 294–304. [Google Scholar]
- Mayrose, I.; Shlomi, T.; Rubinstein, N.D.; Gershoni, J.M.; Ruppin, E.; Sharan, R.; Pupko, T. Epitope mapping using combinatorial phage-display libraries: a graph-based algorithm. Nucleic Acids Res. 2007, 35, 69–78. [Google Scholar] [CrossRef]
- Huang, Y.X.; Bao, Y.L.; Guo, S.Y.; Wang, Y.; Zhou, C.G.; Li, Y.X. Pep-3D-Search: a method for B-cell epitope prediction based on mimotope analysis. BMC Bioinformatics 2008, 9, 538. [Google Scholar] [CrossRef]
- Mayrose, I.; Penn, O.; Erez, E.; Rubinstein, N.D.; Shlomi, T.; Freund, N.T.; Bublil, E.M.; Ruppin, E.; Sharan, R.; Gershoni, J.M.; Martz, E.; Pupko, T. Pepitope: epitope mapping from affinity-selected peptides. Bioinformatics 2007, 23, 3244–3246. [Google Scholar] [CrossRef]
- Negi, S.S.; Braun, W. Automated detection of conformational epitopes using phage display peptide sequences. Bioinform. Biol. C 2009, 3, 71–81. [Google Scholar]
- Enshell-Seijffers, D.; Denisov, D.; Groisman, B.; Smelyanski, L.; Meyuhas, R.; Gross, G.; Denisova, G.; Gershoni, J.M. The mapping and reconstitution of a conformational discontinuous B-cell epitope of HIV-1. J. Mol. Biol. 2003, 334, 87–101. [Google Scholar] [CrossRef]
- Castrignanò, T.; D'Onorio De Meo, P.; Carrabino, D.; Orsini, M.; Floris, M.; Tramontano, A. The MEPS server foridentifying protein conformational epitopes. BMC Bioinformatics 2007, 8, S6. [Google Scholar]
- Ru, B.; Huang, J.; Dai, P.; Li, S.; Xia, Z.; Ding, H.; Lin, H.; Guo, F.B.; Wang, X. MimoDB: A New Repository for Mimotope Data Derived from Phage Display Technology. Molecules 2010, 15, 8279–8288. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Hubbard, S.J. NACCESS Computer Program. Available online: http://www.bioinf.manchester.ac.uk/naccess/ (accessed on 2 February 2011).
- Tsodikov, O.V.; Record, M.T.J.; Sergeev, Y.V. A novel computer program for fast exact calculation of accessible and molecular surface areas and average surface curvature. J. Comput. Chem. 2002, 23, 600–609. [Google Scholar] [CrossRef]
- Ponomarenko, J.V.; Bourne, P.E. Antibody-protein interactions: benchmark datasets and predictiontools evaluation. BMC Struct. Biol. 2007, 7, 64. [Google Scholar] [CrossRef]
- Huang, J.; Honda, W. CED: a conformational epitope database. BMC Immunol. 2006, 7, 7. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Papangelopoulos, M.; Zajonc, D.M.; Peters, B.; Sette, A.; Bourne, P.E. IEDB-3D: structural data within the immune epitope database. Nucleic Acids Res. 2010, 39, D1164–D1170. [Google Scholar] [Green Version]
- Sobolev, V.; Eyal, E.; Gerzon, S.; Potapov, V.; Babor, M.; Prilusky, J.; Edelman, M. PACE: a suite of tools for protein structure predictionand analysis based on complementarity and environment. Nucleic Acids Res. 2005, 33, W39–W43. [Google Scholar] [CrossRef]
- Shide, L.; Dandan, Z.; Chi, Z.; Martin, Z. Prediction of antigenic epitopes on protein surfaces by consensus scoring. BMC Bioinformatics 2009, 10, 302. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, P.; Chen, W.; Huang, Y.; Wang, H.; Ma, Z.; Lv, Y. Epitope Prediction Based on Random Peptide Library Screening: Benchmark Dataset and Prediction Tools Evaluation. Molecules 2011, 16, 4971-4993. https://doi.org/10.3390/molecules16064971
Sun P, Chen W, Huang Y, Wang H, Ma Z, Lv Y. Epitope Prediction Based on Random Peptide Library Screening: Benchmark Dataset and Prediction Tools Evaluation. Molecules. 2011; 16(6):4971-4993. https://doi.org/10.3390/molecules16064971
Chicago/Turabian StyleSun, Pingping, Wenhan Chen, Yanxin Huang, Hongyan Wang, Zhiqiang Ma, and Yinghua Lv. 2011. "Epitope Prediction Based on Random Peptide Library Screening: Benchmark Dataset and Prediction Tools Evaluation" Molecules 16, no. 6: 4971-4993. https://doi.org/10.3390/molecules16064971
APA StyleSun, P., Chen, W., Huang, Y., Wang, H., Ma, Z., & Lv, Y. (2011). Epitope Prediction Based on Random Peptide Library Screening: Benchmark Dataset and Prediction Tools Evaluation. Molecules, 16(6), 4971-4993. https://doi.org/10.3390/molecules16064971