3.1. General
Optical rotations were measured with a Jasco P-1020 digital polarimeter.
1H- and
13C-NMR spectra were recorded with JMN A500 and ECP 600 FT NMR spectrometers with Me
4Si as the internal standard for solutions in CDCl
3 and CD
3OD. MALDI-TOFMS was recorded on an Applied Biosystems Voyager DE RP mass spectrometer. High-resolution mass spectra were recorded on a JEOL JMS-700 under FAB conditions. TLC was performed on Silica Gel 60 F254 (E. Merck) with detection by quenching of UV fluorescence and by charring with 10% H
2SO
4. Column chromatography was carried out on Silica Gel 60 (E. Merck). The compounds 3,4,6-Tri-
O-acetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-
D-galactopyranosyl-(1→4)- [2,3,4-tri-
O-acetyl-α-
D-fucopyranosyl-(1→3)]- 6-
O-acetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-
D-glucopyranosyl trichloroacetimidate
(8) [
6], 2-(trimethylsilyl)ethyl 2,3,4,6-tetra-
O-acetyl-β-
D-galactopyranosyl-(1→4)-6-
O-benzyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-
D-glucopyranoside (
12) [
3], phenyl 2,3,4-tri-
O-benzyl-1-thio-β-
D-fucopyranoside (
13) [
6], 2-(Trimethylsilyl)ethyl 4,6-
O-benzyl-idene-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-
D-glucopyranoside (
16) [
6], 2-(trimethyl-silyl)ethyl 6-
O-benzyl-3-
O-chloroacetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-
D-gluco-pyranoside (
19) [
6] and 2-(trimethylsilyl)ethyl 6-
O-benzyl-2-deoxy-2-phthalimide-β-
D-gluco-pyranoside (
22) [
18] were prepared as reported. Benzylceramide
9 was prepared by the conventional four-steps procedure [
15] from phytosphingosine, which was purchased from Degussa (The Netherlands).
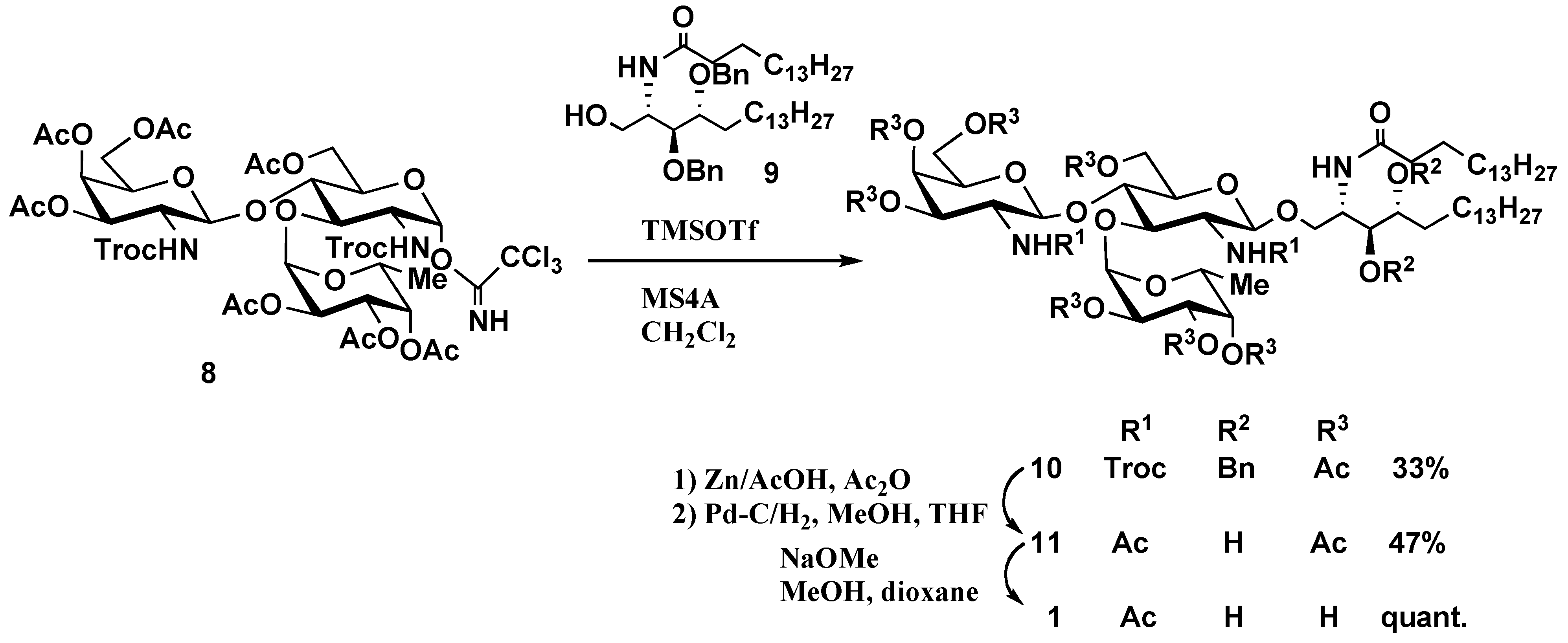
3,4,6-Tri-O-acetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-galactopyranosyl-(1→4)- [2,3,4-tri-O-acetyl-α-D-fucopyranosyl-(1→3)]-6-O-acetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonyl-amino)-β-D-glucopyranosyl-(1→1)-(2S,3S,4R)-3,4-di-O-benzyl-2-hexadecanamido-octadecane-3,4-di-ol (10). Four Å molecular sieves (250 mg) were added to a solution of 8 (21 mg, 16.5 μmol) and (2S,3S,4R)-3-O-benzoyl-2-hexadecanamido-4-octa-decene-1,3-diol 9 (24 mg, 30.0 μmol) in dry CH2Cl2 (0.5 mL) and the mixture was stirred for 16 h at room temperature, then cooled to 0 °C. TMSOTf (3 μL, 0.01 mmol) was added, and the mixture was stirred for 1 h at 0 °C, then neutralized with Et3N. The solids were filtered off and washed with CHCl3. The combined filtrate and washings were successively washed with water, dried (MgSO4), and concentrated. The product was purified by silica gel column chromatography using 3:1 toluene-EtOAc as eluent to give 10 (10 mg, 33%). [α]D23 = +18.2°(c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 4.94 (d, 1H, J1,2=3.7Hz, H-1 of fuc), 4.50 (br. d, 1H, H-1 of GlcNAc), 4.34 (br. s, 1H, H-1, of GalNAc). MALDI-TOFMS: Calcd for C86H129Cl6N3O27Na [M+Na]+: m/z 1868.7 Found: 1869.4.
2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-galactopyranosyl-(1→4)- [2,3,4-tri-O-acetyl-α-D-fucopyranosyl-(1→3)]-2-acetamido-6-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→1)-(2S,3S,4R)-hexa-decanamido-octadecane-3,4-di-ol (11). To a solution of 10 (31mg, 16.8 μmol) in acetic anhydride (2 mL) and AcOH (2 mL) was added zinc powder (100 mg). The reaction mixture was stirred for 16 h at room temperature. After completion of the reaction, the solids were filtered off and the filtrate was concentrated with toluene. The solution of the product and Pd/C (10%, 100 mg) in 1:1 MeOH/THF (2.0 mL) was stirred for 16 h at room temperature under H2, then filtered and concentrated. The product was purified by silica gel column chromatography using 2:1 toluene acetone as eluent to give 11 as an amorphous powder (11 mg, 47%). [α]D23 = +5.3°(c 0.7, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 5.15 (d, 1H, J1,2=3.7Hz, H-1 of fuc), 4.33 (br. d, 1H, H-1 of GlcNAc), 4.30 (br. s, 1H, H-1, of GalNAc). MALDI-TOFMS: Calcd for C70H119N3O25Na [M+Na]+: m/z 1424.8 Found: 1424.5.
2-Acetamido-2-deoxy-β-D-galactopyranosyl-(1→4)-[α-D-fucopyranosyl-(1→3)]-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→1)-(2S,3S,4R)–hexadecanamido-octadecane-3,4-di-ol (1). To a solution of 11 (11 mg, 7.8 μmol) in MeOH (2 mL) was added dioxane (2 mL) and NaOMe (25 mg) at 40 °C. The mixture was stirred for 2 h and then neutralized with Amberlite IR 120 [H+]. The mixture was filtered and concentrated. The product was purified by Sephadex LH-20 column chromatography in 1:1 CHCl3-MeOH to give 1 as white solid (10 mg, quant.). [α]D25 +17.0 (c=0.06, 1:1 CHCl3-MeOH).1H-NMR (500 MHz, 1:1 CDCl3-CD3OD): δ 5.13 (d, 1H, J=3.7Hz, H-1 of Fuc), 4.62 (d, 1H, J =8.3Hz, H-1 of GlcNAc), 4.32 (d, 1H, d, 1H, J =8.0Hz, H-1 of GalNAc). MALDI-TOFMS: Calcd for C56H105N3O18Na: m/z 1130.7 Found: 1130.4 [M+Na]+. HR-FABMS: Calcd for C56H105N3O18Na: m/z 1130.7291. Found m/z 1130.7257 [M+Na]+.
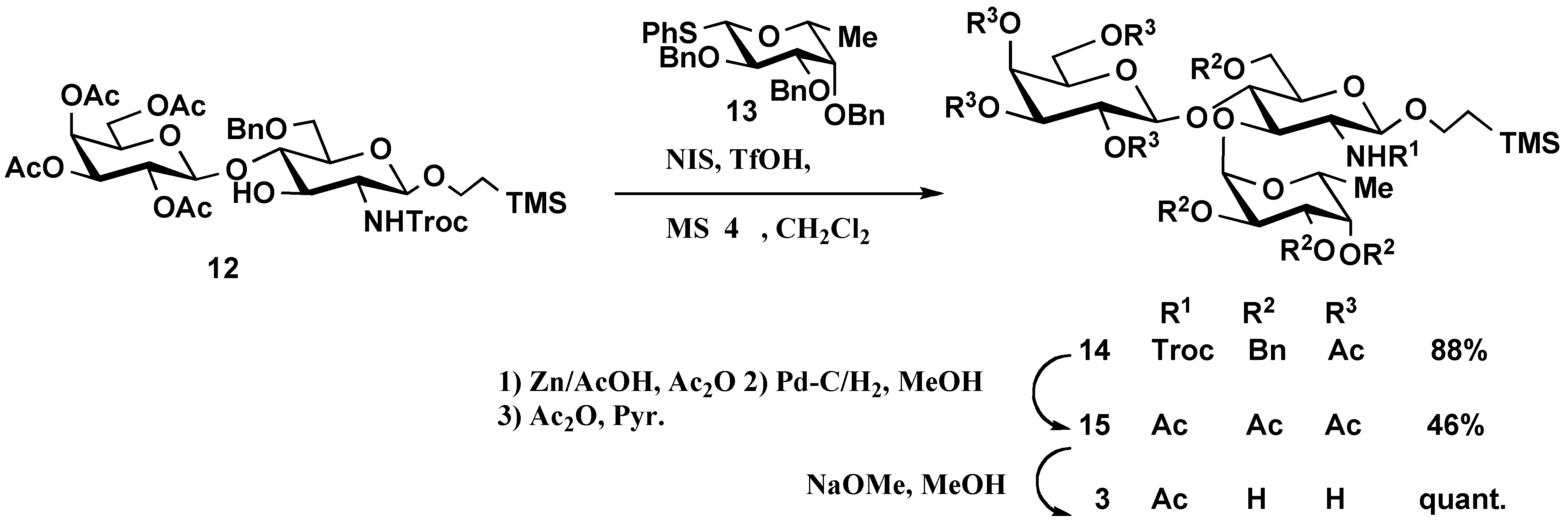
2-(Trimethylsilyl)ethyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)- [2,3,4-tri-O-benzyl-α-D-fucopyranosyl-(1→3)]-6-O-benzyl-2-deoxy-2-(2,2,2-trichloroethoxy- carbonylamino)-β-D-gluco-pyranoside (14). To a solution of 12 (99 mg, 0.11 mmol) and 13 (89 mg, 0.17 mmol) in dry CH2Cl2 (1.5 mL) was added powdered MS 4Å (200 mg), and the mixture was stirred for 2 h at room temperature, then cooled to -60 °C. NIS (57 mg, 0.03 mmol) and TfOH (1.5 μL, 0.01 mmol) were added to the mixture, which was stirred for 3 h at -60 °C, then neutralized with Et3N. The solids were filtered off and washed with CHCl3. The combined filtrate and washings were successively washed with aq Na2S2O3 and water, dried (MgSO4), and concentrated. The product was purified by silica gel column chromatography using 3:1 hexane-EtOAc as eluent to give 14 (128 mg, 88%). [α]D24 +20.4 (c 0.7, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 7.28–7.14 (m, 20H, 4Ph), 5.74 (d, 1H, NH), 5.26 (d, 1H, H-4 of Gal), 5.11 (t, 1H, H-2 of Gal), 4.92–4.84 (m, 2H, H-3 of Gal, benzylmethylene), 4.85 (d, 1H, J1,2=3.7 Hz, H-1 of Fuc), 4.75–4.54 (m, 7H, benzylmethylene × 5, CH2CCl3), 4.40 (d, 1H, J1,2=7.3 Hz, H-1 of GlcNAc), 4.38 (d, 1H, J1,2=7.9 Hz, H-1 of Gal), 4.27 (dd, 2H, benzylmethylene x 2), 4.06–4.02 (m, 2H, H-6a of GlcNAc, H-5 of Fuc), 3.97–3.75 (m, 9H, H-2, H-3, H-6b of GlcNAc, H-5, H-6 of Gal, H-2, H-3 of Fuc, CH2CH2Si(CH3)3), 3.66–3.62 (m, 3H, H-4, H-5 of GlcNAc, H-4 of Fuc), 3.38–3.32 (m, 1H, CH2CH2Si(CH3)3), 2.07–1.87 (m, 12H, CH3CO × 4), 1.09 (d, 3H, H-6 of Fuc), 0.88–0.73 (m, 2H, CH2CH2Si(CH3)3), –0.09 (s, 9H, Si(CH3)3); 13C-NMR (125 MHz, CDCl3): δ 170.2, 170.0, 154.2, 139.0, 138.9, 138.7, 138.3, 128.4, 128.23, 128.20, 127.8, 127.74, 127.65, 127.59, 127.5, 127.3, 100.2 (C-1 of GlcNAc), 100.1 (C-1 of Gal), 99.3 (C-1 of Fuc), 95.8, 79.5, 77.8, 76.4, 76.3, 75.6, 74.8, 74.6, 74.1, 73.5, 73.2, 72.9, 71.0, 70.4, 69.0, 67.4, 66.8, 66.7, 61.1, 53.6, 29.7, 20.8, 20.62, 20.57, 18.2, 16.5, –1.4 (Si(CH3)3); MALDI-TOFMS: Calcd for C62H78Cl3NO20SiNa: m/z 1312.4 Found: 1312.9 [M+Na]+.
2-(Trimethylsilyl)ethyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)- [2,3,4-tri-O-acetyl-α-D-fucopyranosyl-(1→3)]-6-O-acetyl-2-acetamido-2-deoxy-β-D-glucopyranoside (15). To a solution of 14 (107 mg, 0.08 mmol) in acetic anhydride (6 mL) and AcOH (6 mL) was added zinc powder (150 mg). The reaction mixture was stirred for 12 h at 40 °C. After completion of the reaction, the solids were filtered off and the filtrate was concentrated with toluene. The solution of the product and Pd/C (10%, 100 mg) in MeOH (2.0 mL) was stirred for 16 h at room temperature under H2, then filtered and concentrated. The residue was acetylated with acetic anhydride (2 mL) in pyridine (3 mL) for 16 h at room temperature. The reaction mixture was poured into ice-water and extracted with CHCl3. The extract was washed sequentially with 5% HCl, aq NaHCO3 and water, dried (MgSO4), and concentrated. The product was purified by silica gel column chromatography using 5:1 toluene-acetone as eluent to give 15 (37 mg, 46%) as an amorphous powder. [α]D24 +11.8 (c 0.7, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 6.47 (d, 1H, NH), 5.36 (d, 1H, H-4 of Gal), 5.23–5.21 (m, 2H, H-2 of Gal, H-4 of Fuc), 5.10–5.00 (m, 3H, H-3 of Gal, H-2, H-3 of Fuc), 5.08 (d, 1H, J1,2=3.7 Hz, H-1 of Fuc), 4.57 (dd, 1H, H-6a of Gal), 4.52 (d, 1H, J1,2=7.9 Hz, H-1 of Gal),4.42 (dd, 1H, H-5 of Fuc), 4.37 (d, 1H, J1,2=7.9 Hz, H-1 of GlcNAc), 4.33 (dd, 1H, H-6b of Gal), 4.24 (br d, 1H, H-2 of GlcNAc), 4.09 (d, 2H, H-6 of GlcNAc), 3.93–3.85 (m, 4H, H-3, H-4 of GlcNAc, H-5 of Gal, CH2CH2Si(CH3)3), 3.69(s, 1H, H-5 of GlcNAc), 2.18–1.81 (m, 27H, CH3CO × 9), 1.13 (d, 3H, H-6 of Fuc), 0.98–0.80 (m, 2H, CH2CH2Si(CH3)3), –0.02 (s, 9H, Si(CH3)3); 13C-NMR (125 MHz, CDCl3): δ 170.9, 170.7, 170.5, 170.3, 170.2, 170.0, 169.9, 169.5, 99.5 (C-1 of Gal), 99.4 (C-1 of GlcNAc), 97.3 (C-1 of Fuc), 73.4, 72.5, 71.8, 71.3, 71.2, 69.9, 68.8, 68.2, 67.9, 66.7, 66.3, 65.7, 64.9, 61.0, 48.5, 22.9, 21.0, 20.9, 20.7, 20.6, 20.52, 20.47, 17.9, 15.7, –1.5 (Si(CH3)3); MALDI-TOFMS: Calcd for C41H63NO23SiNa: m/z 988.3 Found: 988.4 [M+Na]+.
2-(Trimethylsilyl)ethyl β-D-galactopyranosyl-(1→4)-[α-D-fucopyranosyl-(1→3)]-2-acetamido-2-deoxy-β-D-glucopyranoside (3). To a solution of 15 (36 mg, 0.04 mmol) in MeOH (5 mL) NaOMe (25 mg) was added at 40 °C. The mixture was stirred for 2 h and then neutralized with Amberlite IR 120 [H+]. The mixture was filtered and concentrated. The product was purified by Sephadex LH-20 column chromatography in 1 : 1 CHCl3-MeOH to give 5 as white solid (24 mg, quant.). [α]D24 +14.4 (c 0.3, CH3OH); 1H-NMR (500 MHz, CD3OD): δ 5.12 (d, 1H, J1,2=4.3 Hz, H-1 of Fuc), 4.42 (d, 1H, J1,2=7.9 Hz, H-1 of GlcNAc), 4.32 (d, 1H, J1,2=7.9 Hz, H-1 of Gal); 13C=NMR (125 MHz, CD3OD): δ 173.4, 104.1 (C-1 of GlcNAc), 102.2 (C-1 of Gal), 102.1 (C-1 of Fuc), 74.8, 73.6, 72.7, 71.5, 70.90, 70.86, 68.5, 67.9, 62.9, 61.1, 56.7, 30.7, 23.5, 18.8, 16.8, –1.3 (Si(CH3)3); MALDI-TOFMS: Calcd for C25H47NO15SiNa: m/z 652.3 Found: 652.6 [M+Na]+. HR-FABMS: Calcd for C25H47NO15SiNa: m/z 652.2613. Found m/z 652.2642 [M+Na]+.
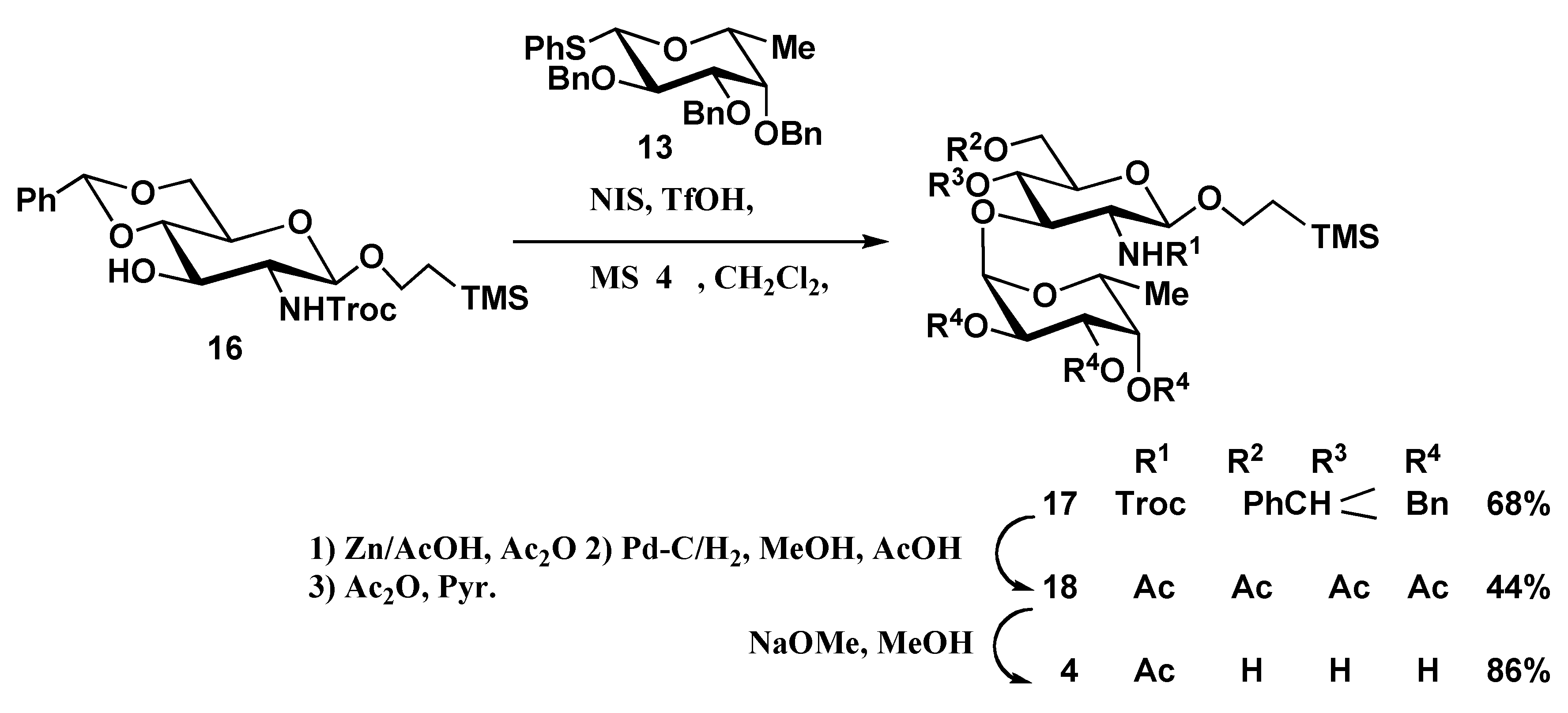
2-(Trimethylsilyl)ethyl 2,3,4-tri-O-benzyl-α-D-fucopyranosyl-(1→3)-4,6-O-benzylidene-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranoside (17). To a solution of 16 (329 mg, 0.61 mmol) and 13 (479 mg, 0.91 mmol) in dry CH2Cl2 (1.5 mL) was added powdered 4Å MS (800 mg), and the mixture was stirred for 2 h at room temperature, then cooled to -60 °C. NIS (307 mg, 1.37 mmol) and TfOH (16 μL, 0.18 mmol) were added to the mixture, which was stirred for 3 h at -60 °C, then neutralized with Et3N. The solids were filtered off and washed with CHCl3. The combined filtrate and washings were successively washed with aq Na2S2O3 and water, dried (MgSO4), and concentrated. The product was purified by silica gel column chromatography using 7:1 hexane-EtOAc as eluent to give 17 (394 mg, 68%). [α]D24 +18.9 (c 2.4, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 7.41–6.89 (m, 20H, 4Ph), 5.49 (br s, 1H, H-1 of Fuc), 5.31 (s, 1H, OCHPh), 5.14 (br s, 1H, NH), 4.86–4.75 (m, 3H, benzylmethylene × 2, CH2CCl3), 4.64 (d, 1H, J1,2=8.6 Hz, H-1 of GlcNAc), 4.60 (d, 1H, CH2CCl3), 4.48 (t, 2H, benzylmethylene × 2), 4.36–4.29 (m, 2H, benzylmethylene × 2), 4.25–4.18 (m, 2H, H-3, H-6a of GlcNAc), 3.96–3.85 (m, 3H, CH2CH2Si(CH3)3, H-2, H-5 of Fuc), 3.81–3.67 (m, 3H, H-4, H-6b of GlcNAc, H-3 of Fuc), 3.52–3.40 (m, 4H, CHCH2Si(CH3)3, H-2, H-5 of GlcNAc, H-4 of Fuc), 1.01 (d, 3H, H-6 of Fuc), 0.94–0.79 (m, 2H, CH2CH2Si(CH3)3), –0.07 (s, 9H, Si(CH3)3); 13C-NMR (125 MHz, CDCl3): δ 153.7, 138.8, 138.4, 138.2, 136.9, 129.3, 128.5, 128.34, 128.27, 128.1, 128.0, 127.5, 127.3, 127.1, 126.2, 101.6, 100.8 (C-1 of GlcNAc), 97.0 (C-1 of Fuc), 95.4, 82.5, 78.6, 75.1, 74.8, 74.4, 73.6, 73.3, 71.4, 68.7, 67.6, 67.0, 65.8, 57.0, 29.6, 18.2, 16.7, –1.5 (Si(CH3)3); MALDI-TOFMS: Calcd for C48H58Cl3NO11SiNa [M+Na]+: m/z 980.3. Found: 980.1.
2-(Trimethylsilyl)ethyl 2,3,4-tri-O-acetyl-α-D-fucopyranosyl-(1→3)-2-acetamido-4,6-di-O-acetyl-2-deoxy-β-D-glucopyranoside (18). To a solution of 17 (113 mg, 0.12 mmol) in acetic anhydride (7 mL) and AcOH (7 mL) was added zinc powder (150 mg). The reaction mixture was stirred for 12 h at 40 °C. After completion of the reaction, the solids were filtrered off and the filtrate was concentrated with toluene. The solution of the product and Pd/C (10%, 150 mg) in 3:1 MeOH-AcOH (2.0 mL) was stirred for 12 h at room temperature under H2, then filtered and concentrated. The residue was acetylated with acetic anhydride (6 mL) in pyridine (10 mL) for 12 h at room temperature. The reaction mixture was poured into ice-water and extracted with CHCl3. The extract was washed sequentially with 5% HCl, aq NaHCO3 and water, dried (MgSO4), and concentrated. The product was purified by silica gel column chromatography using 5:1 toluene-acetone as eluent to give 18 (35 mg, 44%)as an amorphous powder. [α]D24 +74.9 (c 0.3, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 5.85 (d, 1H, NH), 5.25 (d, 1H, J1,2=3.7 Hz, H-1 of Fuc), 5.26–5.23 (m, 2H, H-3, H-4 of Fuc), 5.08 (dd, 1H, H-2 of Fuc), 5.06 (d, 1H, J1,2=7.9 Hz H-1 of GlcNAc), 4.97 (dd, 1H, H-4 of GlcNAc), 4.67 (t, 1H, H-3 of GlcNAc), 4.25 (dd, 1H, H-5 of Fuc), 4.14 (dd, 1H, H-6a of GlcNAc), 3.99 (dd, 1H, H-6b of GlcNAc), 3.92–3.87 (m, 1H, CH2CH2Si(CH3)3), 3.61–3.51 (m, 2H, H-5 of GlcNAc, CH2CH2Si(CH3)3), 3.09–3.04 (m, 1H, H-2 of GlcNAc), 1.08 (d, 3H, H-6 of Fuc), 0.96–0.83 (m, 2H, CH2CH2Si(CH3)3), -0.02 (s, 9H, Si(CH3)3); 13C-NMR (125 MHz, CDCl3): δ 170.7, 170.5, 98.1 (C-1 of GlcNAc), 95.8 (C-1 of Fuc), 73.7, 72.3, 71.5, 71.0, 67.6, 67.4, 67.2, 64.9, 62.5, 58.1, 23.8, 20.9, 20.8, 20.6, 18.1, 16.1 -1.4 (Si(CH3)3); MALDI-TOFMS: Calcd for C29H47NO15SiNa [M+Na]+: m/z 700.3 Found: 700.5.
2-(Trimethylsilyl)ethyl α-D-fucopyranosyl-(1→3)-2-acetamido-2-deoxy-β-D-glucopyranoside (4). Compound 4 was prepared from 18 (24 mg, 0.035 mmol) by the same method described for preparation of 3. The product was purified by Sephadex LH-20 column chromatography in 1:1 CHCl3-MeOH to give 4 as white solid (14 mg, 86%). [α]D24 +52.9 (c 0.1, CH3OH); 1H-NMR (500 MHz, CD3OD): δ 4.93 (d, 1H, J1,2=3.1 Hz, H-1 of Fuc), 4.30 (d, 1H, J1,2=8.5 Hz H-1 of GlcNAc); 13C-NMR (125 MHz, CD3OD): δ 173.3, 103.6 (C-1 of GlcNAc), 102.2 (C-1 of Fuc), 86.3, 79.5, 77.4, 73.7, 72.5, 71.6, 70.8, 68.5, 67.9, 62.6, 60.2, 55.9, 30.7, 23.4, 18.8, 16.9, -1.26 (Si(CH3)3); MALDI-TOFMS: Calcd for C19H37NO10SiNa: m/z 490.2 Found: 490.6 [M+Na]+. HR-FABMS: Calcd for C19H37NO10SiNa: m/z 490.2084. Found m/z 490.2072 [M+Na]+.
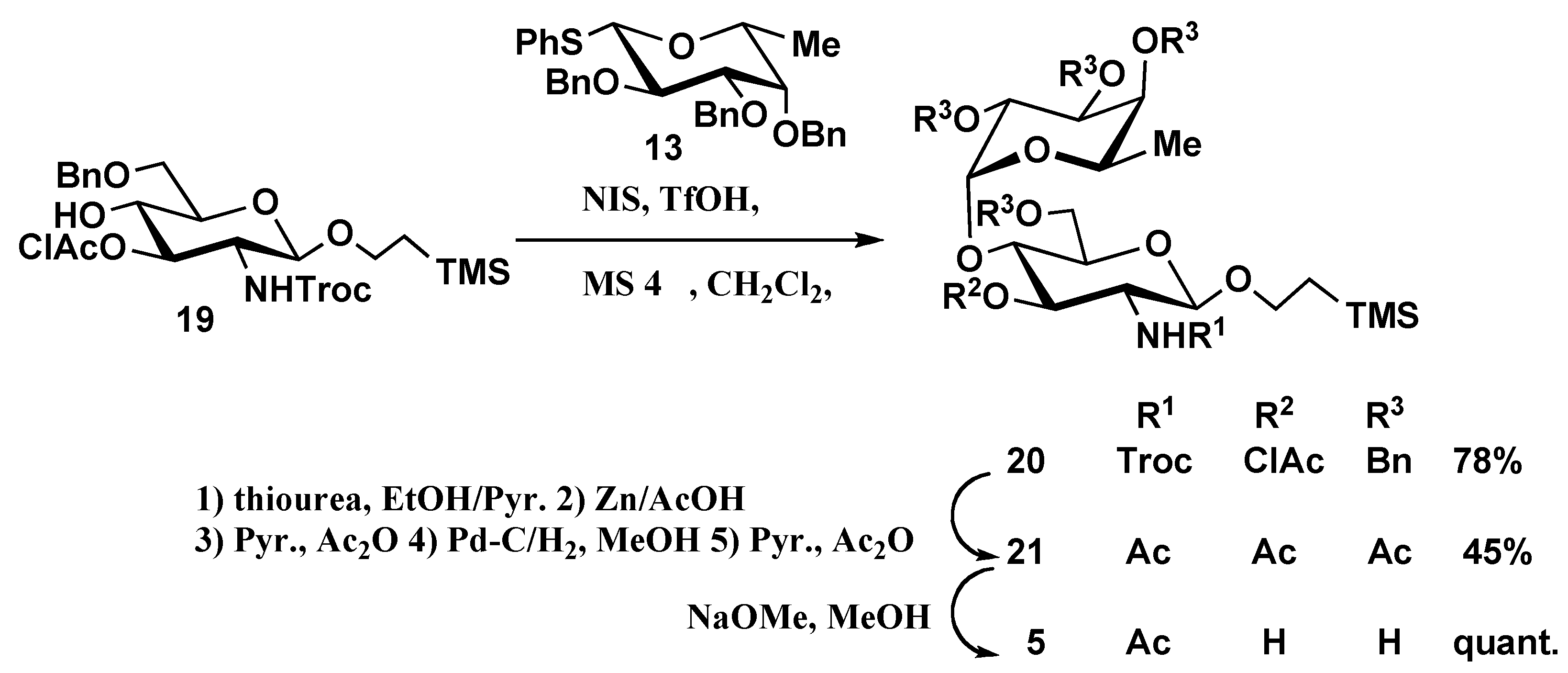
2-(Trimethylsilyl)ethyl 2,3,4-tri-O-benzyl-α-D-fucopyranosyl-(1→4)-6-O-benzyl-3-O-chloroacetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranoside(20). Compound 20 was prepared from 19 (226 mg, 0.36 mmol) and 13 (383 mg, 0.73 mmol) by the same method described for preparation of 17. The product was purified by silica gel column chromatography using10:1 hexane-EtOAc as eluent to give 20 as syrup (296 mg, 78%). [α]D24 +11.4 (c 4.0, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 7.37–7.10 (m, 20H, 4 Ph), 5.49 (d, 1H, NH), 5.22 (t, 1H, H-3 of GlcNAc), 4.89 (d, 1H, benzylmethylene), 4.87 (d, 1H, J1,2=3.7 Hz, H-1 of Fuc), 4.76–4.53 (m, 9H, H-4 of Fuc, benzylmethylene × 6, CH2CCl3), 4.50 (d, 1H, J1,2=7.3 Hz, H-1 of GlcNAc), 4.45 (d, 1H, benzylmethylene), 3.92–3.79 (m, 5H, H-4, 6a of GlcNAc, H-2, 5 of Fuc, CH2CH2Si(CH3)3), 3.75–3.62 (m, 5H, H-2, H-6b of GlcNAc, H-3 of Fuc, ClCH2CO), 3.51–3.46 (m, 2H, CH2CH2Si(CH3)3, H-5 of GlcNAc), 0.97 (d, 3H, H-6 of Fuc), 0.9820.83 (m, 2H, CH2CH2Si(CH3)3), –0.09 (s, 9H, Si(CH3)3); 13C-NMR (125 MHz, CDCl3): δ 167.1, 154.1, 138.5, 138.2, 138.1, 128.6, 128.43, 128.36, 128.3, 128.1, 127.62, 127.55, 127.47, 127.37, 100.1(C-1 of Fuc), 98.8(C-1 of GlcNAc), 95.5, 78.9, 77.7, 75.6, 74.9, 74.8, 74.6, 74.2, 74.0, 73.3, 73.2, 68.7, 67.6, 67.1, 55.5, 40.8, 18.1, 16.6, –1.32 (Si(CH3)3); MALDI-TOFMS: Calcd for C50H61Cl4NO12SiNa [M+Na]+: m/z 1058.3 Found: 1059.2.
2-(Trimethylsilyl)ethyl 2,3,4-tri-O-acetyl-α-D-fucopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranoside (21). To a solution of 20 (296 mg, 0.29 mmol) in EtOH (2.5 mL) was added pyridine (1.5 mL) and thiourea (173 mg, 2.32 mmol). The reaction mixture was stirred for 6 h at 80 °C. The mixture was diluted with CHCl3, washed with aq 5%HCl, aq NaHCO3 and brine, dried (MgSO4) and concentrated. The solution of the residue in AcOH (2 mL) was added zinc powder (350 mg). The reaction mixture was stirred for 12 h at 60 °C. After completion of the reaction, the solids were filtered off and the filtrate was concentrated with toluene. The residue was acetylated with acetic anhydride (4 mL) in pyridine (7 mL). The reaction mixture was poured into ice-water and extracted with CHCl3. The extract was washed sequentially with 5% HCl, aq. NaHCO3 and water, dried (MgSO4), and concentrated. The solution of the product in MeOH (1.5 mL) and THF (0.5 mL) was hydrogenolysed under hydrogen in the presence of 10% Pd/C (150 mg) for 16 h at room temperature, then filtered and concentrated. The residue was acetylated with acetic anhydride (3 mL) in pyridine (5 mL). The reaction mixture was poured into ice-water and extracted with CHCl3. The extract was washed sequentially with 5% HCl, aq NaHCO3 and water, dried (MgSO4), and concentrated. The product was purified by silica gel column chromatography using 5:1 toluene-acetone as eluent to give 21 as an amorphous powder (86 mg, 45%). [α]D24 +49.6 (c 0.5, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 5.81 (d, 1H, NH), 5.34 (d, 1H, J1,2=3.7 Hz, H-1 of Fuc), 5.22–5.05 (m, 4H, H-3 of GlCNAc, H-2,H-3, H-4 of Fuc), 4.58 (d, 1H, J1,2=7.9 Hz H-1 of GlcNAc), 4.46 (dd, 1H, H-6a of GlcNAc), 4.08–4.01 (m, 2H, H-6b of GlcNAc, H-5 of Fuc), 3.95 (t, 1H, H-4 of GlcNAc), 3.88–3.83 (m, 1H, CH2CH2Si(CH3)3), 3.78 (dd, 1H, H-2 of GlcNAc), 3.61–3.57 (m, 1H, H-5 of GlcNAc), 3.53–3.48 (m, 1H, CH2CH2Si(CH3)3), 2.11–1.82 (m, 18H, CH3CO × 6), 1.04 (d, 3H, H-6 of Fuc), 0.91–0.78 (m, 2H, CH2CH2Si(CH3)3), –0.07 (s, 9H, Si(CH3)3); 13C-NMR (125 MHz, CDCl3): δ 171.0, 170.7, 170.5, 170.3, 170.1, 170.0, 99.8 (C-1 of GlcNAc), 96.0 (C-1 of Fuc), 75.5, 72.1, 71.8, 70.9, 67.3, 67.2, 66.9, 65.5, 62.7, 54.6, 29.6, 23.1, 20.9, 20.8, 20.7, 20.6, 20.5, 17.8, 15.8, –1.5 (Si(CH3)3); MALDI-TOFMS: Calcd for C29H47NO15SiNa [M+Na]+: m/z 700.3 Found: 700.9.
2-(Trimethylsilyl)ethyl α-D-fucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranoside (5). Compound 5 was prepared from 21 (86 mg, 0.13 mmol) by the same method described for preparation of 3. The product was purified by Sephadex LH-20 column chromatography in 1:1 CHCl3-MeOH to give 5 as white solid (61 mg, quant.). [α]D24 +33.9 (c 0.3, CH3OH); 1H-NMR (500 MHz, CD3OD): δ 4.98 (d, 1H, J1,2=3.7 Hz, H-1 of Fuc), 4.32 (d, 1H, J1,2=7.9 Hz H-1 of GlcNAc); 13C-NMR (125 MHz, CD3OD): δ 173.5, 103.5 (C-1 of Fuc), 102.0 (C-1 of GlcNAc), 82.2, 76.9, 75.9, 73.5, 71.7, 70.6, 68.6, 67.9, 62.5, 56.8, 30.7, 23.0, 18.8, 16.7, –1.3 (Si(CH3)3); MALDI-TOFMS: Calcd for C19H37NO10SiNa: m/z 490.2 Found: 491.0 [M+Na]+. HR-FABMS: Calcd for C19H37NO10SiNa: m/z 490.2084. Found m/z 490.2062 [M+Na]+.
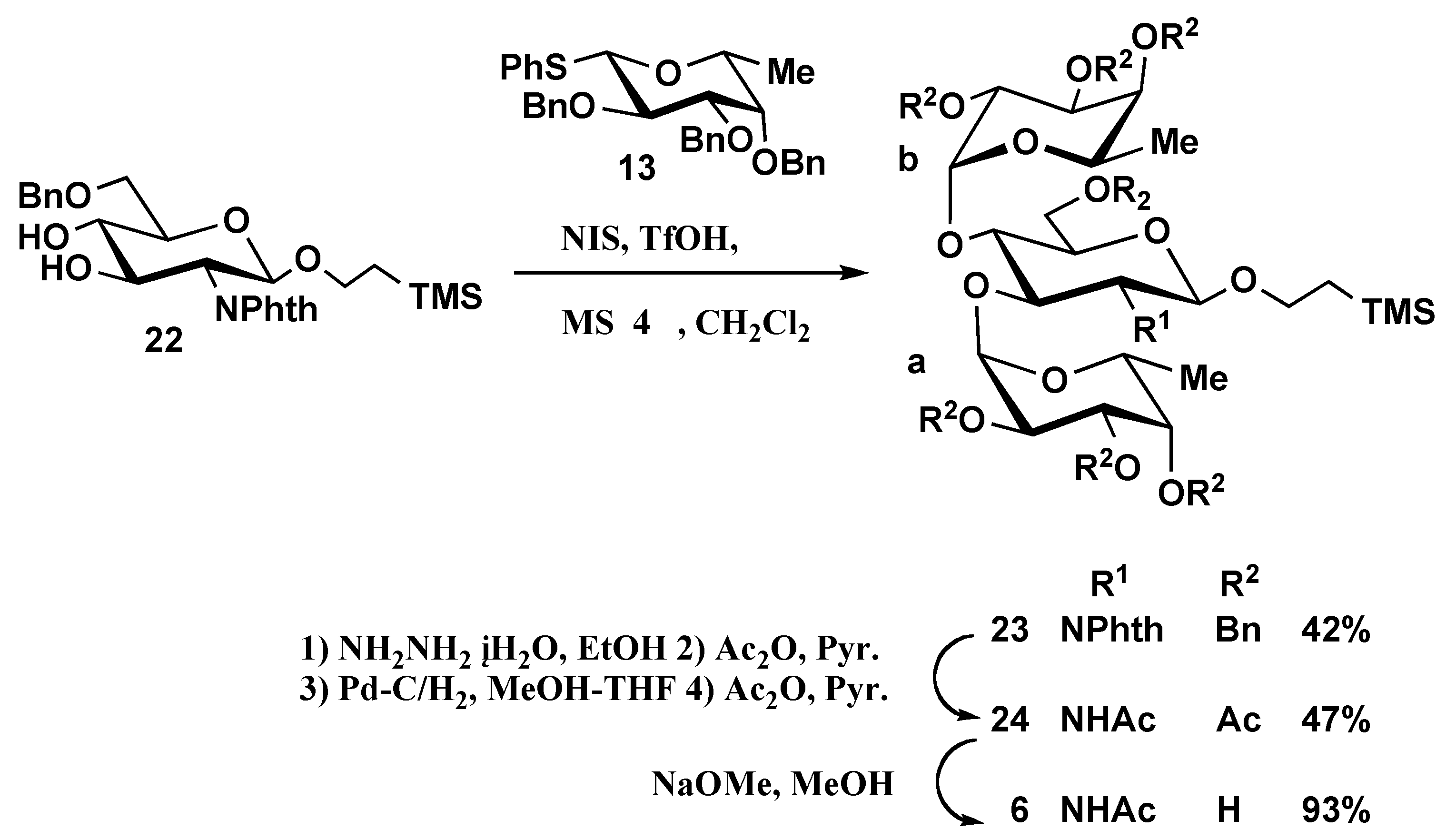
2-(Trimethylsilyl)ethyl 2,3,4-tri-O-benzyl-α-D-fucopyranosyl-(1→3)- [2,3,4-tri-O-benzyl-α-D-fuco-pyranosyl-(1→4)]-6-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (23). Compound 23 was prepared from 22 (89 mg, 0.18 mmol) and 13 (757 mg, 1.44 mmol) by the same method described for preparation of 14. The product was purified by silica gel column chromatography using 10:1 hexane- EtOAc as eluent to give 23 as syrup (99 mg, 42%). [α]D24 +48.3 (c 1.2, CHCl3); 1H-NMR (600 MHz, CDCl3): δ 7.90–7.26 (m, 39H, NPhth, 8 Ph), 6.20 (d, 1H, J1,2=3.6 Hz, H-1 of Fuc b), 5.19 (d, 2H, J1,2=8.5 Hz H-1 of GlcNAc, J1,2=4.4 Hz H-1 of Fuc a), 5.07 (d, 1H, benzylmethylene), 5.01 (dd, 1H, H-3 of GlcNAc), 4.93–4.88(m, 3H, benzylmethylene × 3), 4.79–4.66 (m, 8H, benzylmethylene × 8), 4.62–4.58 (m, 2H, benzylmethylene × 2), 4.41 (dd, 1H, H-2 of GlcNAc), 4.18–4.13 (m, 2H, H-4 of GlcNAc, H-2 of Fuc b), 4.10 (dd, 1H,H-3 of Fuc a), 4.07–3.99 (m, 2H, CH2CH2Si(CH3)3, H-5 of Fuc b), 3.97–3.84 (m, 6H, H-5, H-6 of GlcNAc, H-2, H-5 of Fuc a, H-3 of Fuc b), 3.63–3.57 (m, 2H, H-4 of Fuc b, CH2CH2Si(CH3)3), 3.47(br.d, 1H, H-4 of Fuc a), 1.19 (d, 3H, H-6 of Fuc b) 0.95 (d, 3H, H-6 of Fuc a,), 0.93–0.81 (m, 2H, CH2CH2Si(CH3)3), –0.01 (s, 9H, Si(CH3)3); 13C-NMR (150 MHz, CDCl3): δ 139.1, 138.7, 138.52, 138.50, 138.3, 133.8, 128.3, 128.23, 128.18, 128.12, 128.05, 128.03, 127.93, 127.87, 127.7, 127.6, 127.5, 127.42, 127.37, 127.35, 127.28, 127.23, 127.1, 123.1, 169.3, 9, 97.6 (C-1 of GlcNAc), 97.5 (C-1 of Fuc a), 95.0 (C-1 of Fuc b), 78.8, 78.7, 78.4, 78.1, 78.0, 76.4, 76.3, 74.9, 74.6, 73.5, 73.3, 73.1, 73.0, 72.9, 72.8, 69.7, 67.7, 67.0, 66.7, 56.3, 29.7, 17.8, 16.6, 15.8, –1.5 (Si(CH3)3); MALDI-TOFMS: Calcd for C80H89NO15SiNa [M+Na]+: m/z 1354.6 Found: 1354.8.
2-(Trimethylsilyl)ethyl 2,3,4-tri-O-acetyl-α-D-fucopyranosyl-(1→3)- [2,3,4-tri-O-acetyl-α-D-fuco-pyranosyl-(1→4)]- 2-acetamido-6-O-acetyl-2-deoxy-β-D-glucopyranoside (24). To a solution of 23 (60 mg, 0.05 mmol in EtOH (10 mL)) was added hydrazine monohydrate (3.3 mL, 0.07 mmol). The reaction mixture was refluxed for 3 h, then concentrated. The residue was acetylated with Ac2O (3 mL) in pyridine (5 mL). The mixture was poured into ice-water and extracted with CHCl3. The extract was washed sequentially with 5% HCl, aq NaHCO3 and water, dried (MgSO4), and concentrated. The solution of the product in MeOH (1 mL) and THF (1 mL) was hydrogenolysed under hydrogen in the presence of 10% Pd/C (100 mg) for 15 h at room temperature, then filtered and concentrated. The residue was acetylated with acetic anhydride (5 mL) in pyridine (7 mL). The reaction mixture was poured into ice-water and extracted with CHCl3. The extract was washed sequentially with 5% HCl, aq NaHCO3 and water, dried (MgSO4), and concentrated. The product was purified by silica gel column chromatography using 9:1 toluene-acetone as eluent to give 24 as syrup (19 mg, 47%). [α]D24 +75.2 (c 0.5 CHCl3); 1H-NMR (500 MHz, CDCl3): δ 6.43 (d, 1H, NH), 5.28 (dd, 1H, H-3 of Fuc b), 5.23–5.18 (m, 3H, H-3, H-4 of Fuc a, H-4 of Fuc b), 5.13 (d, 1H, J1,2=3.7 Hz, H-1 of Fuc b), 5.08–5.03 (m, 2H, H-3 of GlcNAc, H-2 of Fuc b), 5.04 (d, 1H, J1,2=9.8 Hz H-1 of GlcNAc), 4.66 (d, 1H, J1,2=3.7 Hz H-1 of Fuc a), 4.52 (dd, 1H, H-6a of GlcNAc), 4.43–4.36 (m, 2H, H-6b of GlcNAc, H-5 of Fuc b), 4.13–4.08 (m, 2H, H-2 of GlcNAc, H-5 of Fuc a), 3.96 (t, 1H, H-5 of GlcNAc), 3.91–3.85 (m, 1H, CH2CH2Si(CH3)3), 3.60–3.55 (m, 2H, H-4 of GlcNAc, H-2 of Fuc a), 3.46–3.40 (m, 1H, CH2CH2Si(CH3)3), 2.13–1.87 (m, 21H, CH3CO) 1.10 (d, 3H, H-6 of Fuc b) 1.07 (d, 3H, H-6 of Fuc a), 0.95–0.80 (m, 2H, CH2CH2Si(CH3)3), –0.03 (s, 9H, Si(CH3)3); 13C-NMR (125 MHz, CDCl3): δ 170.5, 170.43, 170.36, 170.31, 169.9, 169.5, 169.3, 98.5 (C-1 of Fuc a), 98.2 (C-1 of Fuc b), 97.6 (C-1 of GlcNAc), 76.9, 76.8, 76.7, 74.1, 74.0, 73.7, 73.4, 71.2, 70.9, 68.3, 68.00, 67.97, 67.3, 66.5, 65.8, 65.7, 64.5, 49.3, 29.7, 23.4, 21.4, 20.9, 20.7, 20.6, 18.0, 16.0, 15.8, 15.7, 14.1, –1.5 (Si(CH3)3); MALDI-TOFMS: Calcd for C39H61NO21SiNa [M+Na]+: m/z 930.3 Found: 930.5.
2-(Trimethylsilyl)ethyl α-D-fucopyranosyl-(1→3)-[ α-D-fucopyranosyl-(1→4)]- 2-acetamido-2-deoxy-β-D-glucopyranoside (6). Compound 6 was prepared from 24 (16 mg, 0.03 mmol) by the same method described for preparation of 3. The product was purified by Sephadex LH-20 column chromatography in 1:1 CHCl3-MeOH to give 6 as white solid (10 mg, 93%). [α]D24 +72.2 (c 0.2 CH3OH); 1H-NMR (600 MHz, CD3OD): δ 4.97 (d, 1H, J1,2=3.9 Hz H-1 of Fuc b), 4.66 (d, 1H, J1,2=2.8 Hz H-1 of Fuc a), 4.38 (d, 1H, J1,2=6.1 Hz H-1 of GlcNAc), –0.09 (s, 9H, Si(CH3)3); 13C-NMR (150 MHz, CD3OD): δ 172.8, 101.8 (C-1 of GlcNAc), 101.6 (C-1 of Fuc a), 100.1 (C-1 of Fuc b), 79.3, 78.3, 74.8, 73.7, 73.5, 71.4, 70.1, 69.8, 68.5, 68.3, 67.6, 63.1, 54.7, 27.0, 23.1, 18.9, 16.7, –1.3 (Si(CH3)3); MALDI-TOFMS: Calcd for C25H47NO14SiNa: m/z 636.3 Found: 636.7 [M+Na]+. HR-FABMS: Calcd for C25H47NO14SiNa: m/z 636.2664. Found m/z 636.2681 [M+Na]+.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






