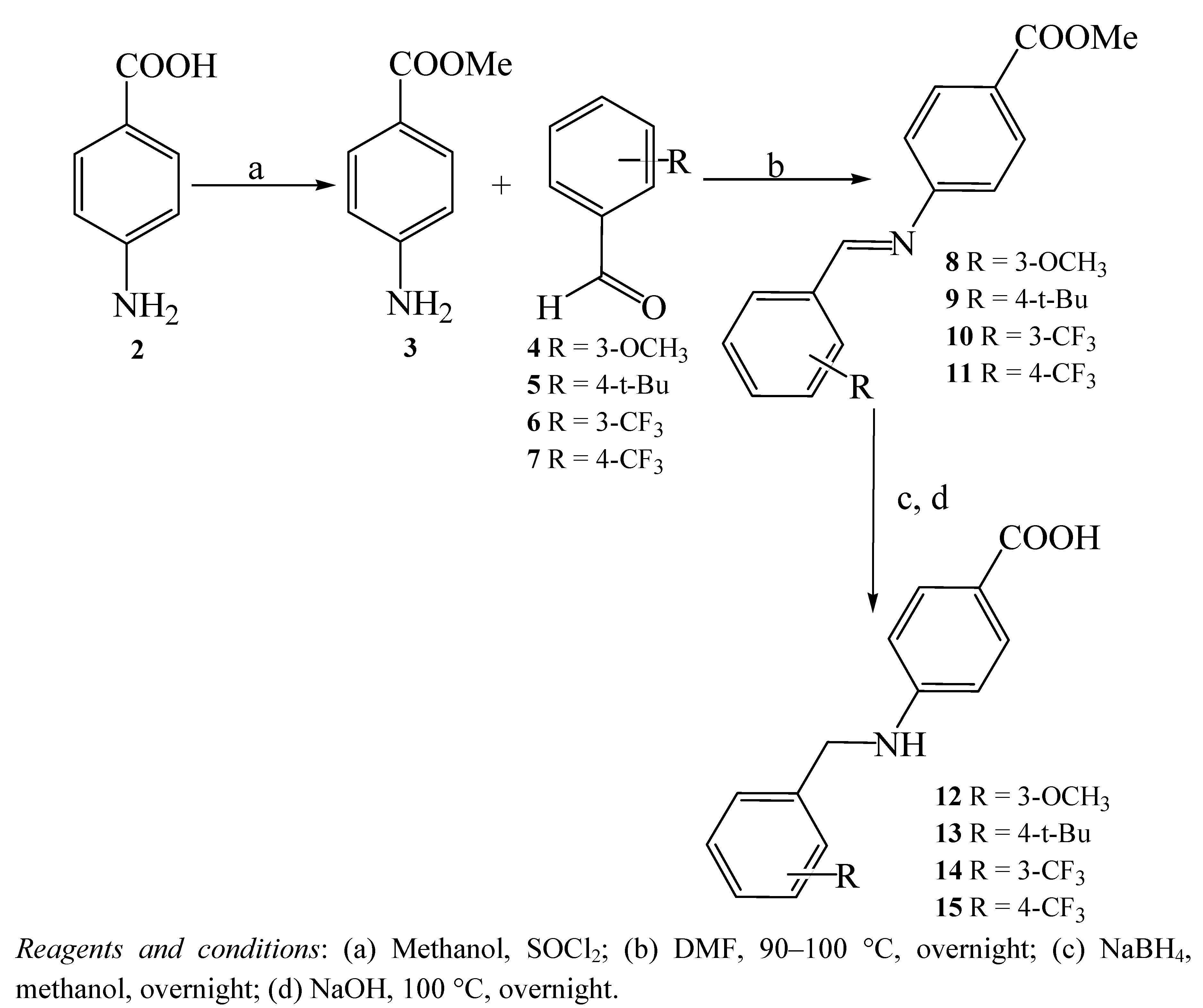
General procedure for the synthesis of 4-aminobenzoic acid derivatives 12–15
4-Aminobenzoic acid (2, 1.37 g, 10 mmol) was dissolved in methanol (100 mL), and then thionyl chloride (200 mmol) was added at 0 °C. The mixture was stirred at room temperature for 20–30 minutes, followed by refluxing at 65–70 °C overnight. Evaporation of the solvent was carried out, followed by neutralization using K2CO3 and extraction with CH2Cl2 (3 × 20 mL). The combined extracts were dried on anhydrous Na2SO4 and filtrated to give 4-amino-benzoic acid methyl ester (3,96%). Subsequently, 3 (1.52 g, 10 mmol) was disolved in DMF (10 mL), then an aldehyde (4–7, 25 mmol) was added. The mixture was heated between 100–150 °C overnight. After removing DMF, methanol (15 mL) was added to the reaction mixture, followed by gradual addition of NaBH4 (4 equivalents) and stirring at room temperature overnight. The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (90:10). Next, desterification was carried out by refluxing with 1M NaOH (2.6 equivalents) at 100 °C overnight. Then, the reaction mixture was neutralized with HCl and extracted with CHCl3 (3 × 20 mL). The combined extracts were dried on anhydrous Na2SO4 and filtered.
4-(3-Methoxybenzylamino)-benzoic acid (12). Evaporation of the solvent gave 12 as an off-white powder (88%); mp. 160–161 °C; 1H-NMR (300 MHz, CDCl3) δ 3.68 (s, 3H), 4.26 (s, 2H), 4.72 (br s, 1H), 6.55 (d, J = 8.8 Hz, 2H), 6.77 (dd, J = 8.1, 1.4 Hz, 1H), 6.86–6.89 (m, 2H), 7.21 (t, J = 8.1 Hz, 1H), 7.61 (d, J = 8.8 Hz, 2H), 12.03 (br s, 1H); 13C-NMR (300 MHz, CDCl3) δ 46.3 (1C), 55.4 (1C), 111.7 (1C), 112.5 (1C), 113.3 (1C), 117.6 (1C), 119.8 (1C), 130.0 (2C), 131.6 (2C), 141.7 (1C), 152.9 (1C), 159.9 (1C), 168.0 (1C); IR (thin film) cm-1 3437, 3059, 2940, 1655, 1597, 1489, 1427, 1285, 1177.
4-(4-tert-Butylbenzylamino)-benzoic acid (13). Evaporation of the solvent gave 13 as an off-white powder (83%); mp. 210–211 °C; 1H-NMR (300 MHz, CDCl3) δ 1.27 (s, 9H), 4.36 (s, 2H), 4.80 (br s, 1H), 6.60 (d, J = 8.7 Hz, 2H), 7.26–7.36 (m, 4H), 7.94 (d, J = 8.7 Hz, 2H), 11.87 (br s, 1H); 13C-NMR (300 MHz, CDCl3) δ 31.4 (3C), 34.6 (1C), 47.4 (1C), 111.6 (2C), 117.6 (1C), 125.8 (2C), 127.4 (2C), 132.4 (2C), 135.2 (1C), 150.7 (1C), 152.5 (1C), 172.4 (1C); IR (thin film) cm-1 3426, 3017, 2963, 1670, 1605, 1524, 1478, 1420, 1292.
4-(3-Trifluoromethylbenzylamino)-benzoic acid (14). Evaporation of the solvent gave 14 as an off-white powder (77%); mp. 168–169 °C; 1H-NMR (300 MHz, CDCl3) δ 4.41 (s, 2H), 4.77 (br s, 1H), 6.57 (d, J = 8.6 Hz, 2H), 7.53–7.66 (m, 6H), 11.93 (br s, 1H); 13C-NMR (300 MHz, CDCl3) δ 45.7 (1C), 111.7 (2C), 118.0 (1C), 124.0 (1C), 124.1 (1C), 127.6 (1C), 129.9 (1C), 131.6 (2C), 131.8 (2C), 141.7 (1C), 152.6 (1C), 167.9 (1C); IR (thin film) cm-1 3456, 3062, 2920, 1674, 1605, 1516, 1481, 1424, 1316, 1107.
4-(4-Trifluoromethylbenzylamino)-benzoic acid (15). Evaporation of the solvent gave 15 as a pale yellow powder (93%);mp. 189–190 °C; 1H-NMR (300 MHz, CDCl3) δ 4.41 (s, 2H), 4.79 (br s, 1H), 6.54 (d, J = 8.8 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H), 7.59–7.67 (m, 4H), 12.02 (br s, 1H); 13C-NMR (300 MHz, CDCl3) δ 45.8 (1C), 111.7 (2C), 118.0 (1C), 123.1 (1C), 125.8 (2C), 127.8 (1C), 128.2 (2C), 131.6 (2C), 145.2 (1C), 152.6 (1C), 167.9 (1C); IR (thin film) cm-1 3414, 3042, 2920, 1659, 1605, 1520, 1462, 1424, 1323, 1122.
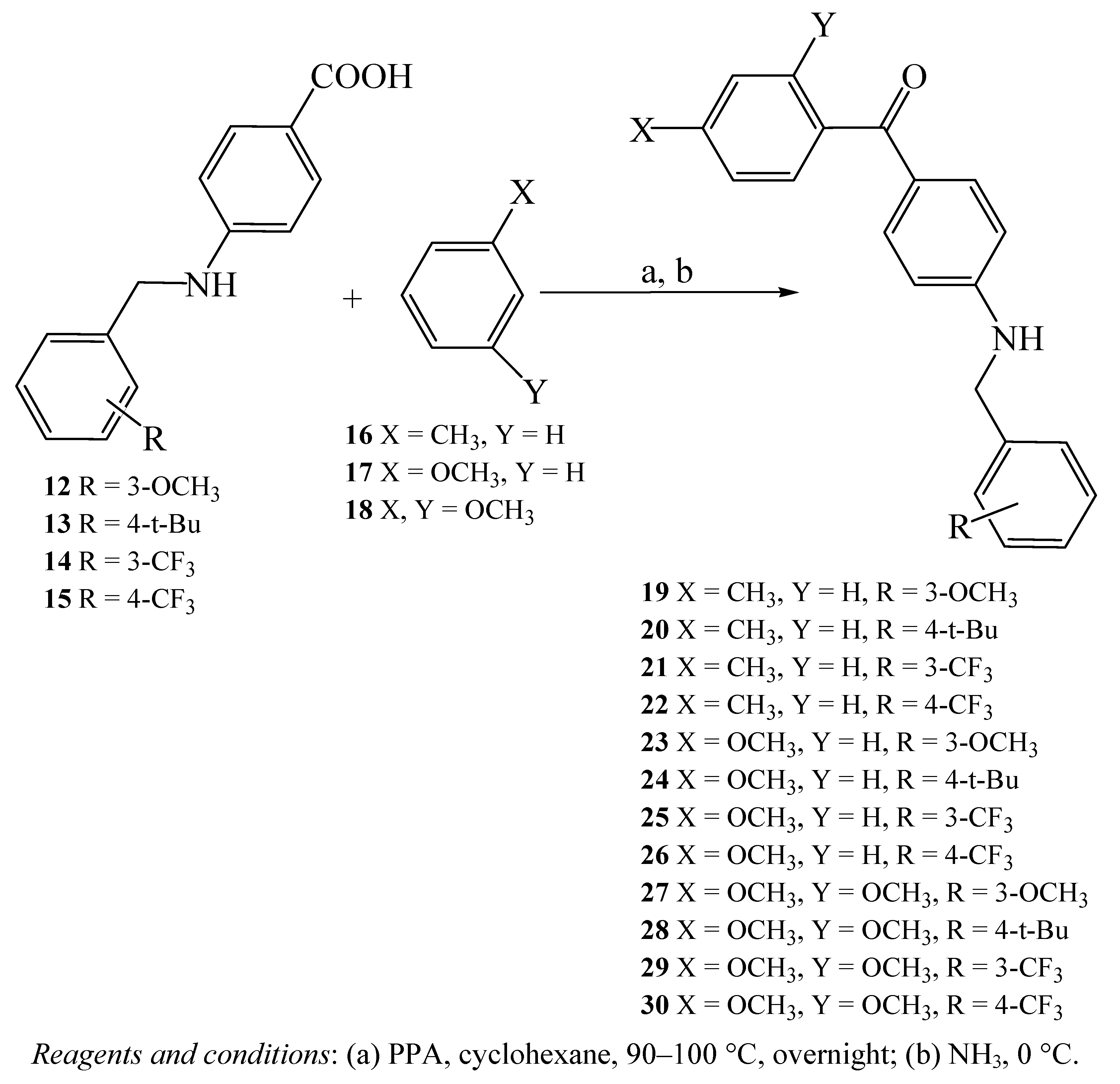
General procedure for the synthesis of benzylamino-methanones 19–30
4-Aminobenzoic acid derivative 12–15 (2 mmol) was dissolved in cyclohexane (10 mL), and polyphosphoric acid (6.5 g) was added. Then a benzene derivative 16–18 (20 mmol) was added. The mixture was stirred carefully at 90–110 °C overnight and then poured on crushed ice. The solution was carefully made alkaline with 25% ammonia and then extracted with CHCl3 (3 × 20 mL). The combined extracts were dried on anhydrous Na2SO4 and filtered.
[4-(3-Methoxybenzylamino)phenyl]-p-tolyl-methanone (19). The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (80:20) to give pure 19 as a yellow powder (22%); Rf = 0.52 (CHCl3-MeOH, 98:2); mp. 123–124 °C; 1H-NMR (300 MHz, CDCl3) δ 2.32 (s, 3H), 3.78 (s, 3H), 4.37 (s, 2H), 4.65 (br s, 1H), 6.60 (d, J = 7.7 Hz, 2H), 6.80–6.94 (m, 2H), 7.22–7.28 (m, 4H), 7.62 (d, J = 7.2 Hz, 2H), 7.71 (d, J = 7.7 Hz, 2H); 13C-NMR (300 MHz, CDCl3) δ 21.6 (1C), 47.7 (1C), 55.3 (1C), 111.6 (2C), 112.8 (1C), 113.1 (1C), 119.6 (1C), 126.9 (1C), 127.7 (2C), 129.9 (2C), 130.6 (1C), 132.9 (2C), 136.2 (1C), 140.0 (1C), 141.9 (1C), 151.7 (1C), 160.0 (1C), 195.1 (1C); IR (thin film) cm-1 3356, 3017, 2963, 1636, 1593, 1528, 1468, 1262, 1150; MS (ESI, positive mode) m/z [M+H]+ 332.16451 (C22H22NO2 requires 332.15723); Anal. Calcd for C22H21NO2: C 79.73, H 6.39, N 4.23, found: C 79.68, H 6.41, N 4.21.
[4-(4-tert-Butylbenzylamino)phenyl]-p-tolyl-methanone (20). The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (85:15) to give pure 20 as a yellow powder (23%); Rf = 0.57 (CHCl3-MeOH, 98:2); mp. 140–141 °C; 1H-NMR (300 MHz, CDCl3) δ 1.23 (s, 9H), 2.32 (s, 3H), 3.83 (s, 2H), 4.12 (br s, 1H), 6.65 (d, J = 8.6 Hz, 2H), 7.10 (d, J = 7.8 Hz, 2H), 7.16–7.30 (m, 4H), 7.62–7.75 (m, 4H); 13C-NMR (300 MHz, CDCl3) δ 22.8 (1C), 29.5 (3C), 32.0 (1C), 39.1 (1C), 121.1 (1C), 123.7 (1C), 126.8 (1C), 128.3 (2C), 128.9 (2C), 129.0 (2C), 129.4 (2C), 130.2 (2C), 130.8 (1C), 132.9 (2C), 143.0 (1C), 150.7 (1C), 194.2 (1C); IR (thin film) cm-1 3422, 3337, 3024, 2963, 1636, 1586, 1501, 1439, 1319; MS (ESI, positive mode) m/z [M+H]+ 358.21016 (C25H28NO requires 358.20926); Anal. Calcd for C25H27NO: C 83.99, H 7.61, N 3.92, found: C 83.87, H 7.58, N 3.97.
p-Tolyl-[4-(3-trifluoromethylbenzylamino)phenyl]-methanone (21). The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (80:20) to give pure 21 as a reddish-orange oil (39%); Rf = 0.70 (CHCl3-MeOH, 98:2); 1H-NMR (300 MHz, CDCl3) δ 2.38 (s, 3H), 4.46 (s, 2H), 4.78 (br s, 1H), 6.58 (d, J = 5.2 Hz, 2H), 7.23 (d, J = 9.2 Hz, 2H), 7.43–7.74 (m, 8H); 13C-NMR (300 MHz, CDCl3) δ 21.5 (1C), 47.3 (1C), 111.6 (2C), 111.8 (1C), 113.7 (1C), 125.1 (1C), 127.6 (1C), 128.6 (2C), 129.2 (2C), 129.8 (1C), 132.9 (2C), 134.3 (1C), 136.2 (1C), 138.8 (1C), 141.9 (1C), 143.5 (1C), 151.4 (1C), 195.1 (1C); IR (thin film) cm-1 3356, 3024, 2924, 1651, 1593, 1527, 1439; MS (ESI, positive mode) m/z [M+H]+ 370.14133 (C22H19F3NO requires 370.13405).
p-Tolyl-[4-(4-trifluoromethylbenzylamino)phenyl]-methanone (22). The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (80:20) to give pure 22 as a dark-yellow powder (96%); Rf =0.49 (CHCl3-MeOH, 98:2); mp. 115–116 °C; 1H-NMR (300 MHz, CDCl3) δ 2.35 (s, 3H), 4.49 (s, 2H), 4.82 (br s, 1H), 6.60 (dd, J = 9.0, 1.9 Hz, 2H), 7.27 (t, J = 8.7 Hz, 2H), 7.42 (d, J = 6.2 Hz, 2H), 7.60 (dd, J = 9.0, 1.9 Hz, 2H), 7.66–7.78 (m, 4H); 13C-NMR (300 MHz, CDCl3) δ 21.6 (1C), 47.3 (1C), 111.7 (2C), 127.0 (1C), 127.7 (1C), 128.8 (2C), 129.4 (2C), 130.2 (2C), 130.5 (2C), 130.6 (1C), 130.7 (2C), 131.1 (1C), 132.9 (1C), 142.0 (1C), 152.0 (1C), 195.1 (1C); IR (thin film) cm-1 3356, 3021, 2920, 1647, 1597, 1528, 1451; MS (ESI, positive mode) m/z [M+H]+ 370.14133 (C22H19F3NO requires 370.13405); Anal. Calcd for C22H18F3NO: C 71.53, H 4.91, N 3.79, found: C 71.48, H 4.95, N 3.67.
[4-(3-Methoxybenzylamino)phenyl]-(4-methoxyphenyl)-methanone (23).The residue, after evaporation of the solvent, was purified by column chromatography eluting with CHCl3/MeOH (99:1) to give pure 23 as an orange oil (28%); Rf = 0.44 (CHCl3-MeOH, 98:2); 1H-NMR (300 MHz, CDCl3) δ 3.73 (s, 3H), 3.85 (s, 3H), 4.37 (s, 2H), 4.63 (br s, 1H), 6.59 (d, J = 8.7 Hz, 2H), 6.78–6.95 (m, 6H), 7.65–7.74 (m, 4H); 13C-NMR (300 MHz, CDCl3) δ 47.6 (1C), 55.3 (1C), 55.7 (1C), 111.3 (2C), 113.3 (2C), 120.3 (1C), 127.0 (1C), 129.0 (1C), 129.9 (1C), 130.8 (1C), 131.5 (1C), 132.0 (2C), 132.8 (2C), 140.1 (1C), 151.6 (1C), 160.0 (1C), 162.4 (1C), 194.3 (1C); IR (thin film) cm-1 3352, 3005, 2925, 1636, 1597, 1528, 1458, 1316, 1258, 1169; MS (ESI, positive mode) m/z [M+H]+ 348.14725 (C22H22NO3 requires 348.15214).
[4-(4-tert-Butylbenzylamino)phenyl]-(4-methoxyphenyl)-methanone (24).The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (80:20) to give pure 24 as a red oil (60%); Rf = 0.52 (CHCl3-MeOH, 98:2); 1H-NMR (300 MHz, CDCl3) δ 1.26 (s, 9H), 3.84 (s, 3H), 3.89 (s, 2H), 4.10 (br s, 1H), 6.65 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.6 Hz, 2H), 7.11 (d, J = 8.2 Hz, 1H), 7.20 (d, J = 7.6 Hz, 2H), 7.29 (t, J = 7.6 Hz, 2H), 7.58–7.66 (m, 2H), 7.78 (d, J = 8.6 Hz, 2H); 13C-NMR (300 MHz, CDCl3) δ 31.4 (3C), 37.7 (1C), 38.2 (1C), 55.5 (1C), 123.7 (2C), 125.7 (1C), 126.7 (1C), 128.1 (1C), 128.4 (2C), 128.9 (2C), 131.1 (1C), 131.4 (2C), 133.9 (1C), 135.4 (1C), 138.6 (1C), 149.2 (1C), 149.6 (1C), 162.5 (1C), 194.5 (1C); IR (thin film) cm-1 3476, 3364, 3005, 2963, 1620, 1601, 1508, 1458, 1420, 1254; MS (ESI, positive mode) m/z [M+H]+ 374.19873 (C25H28NO2 requires 374.20418).
(4-Methoxy-phenyl)-[4-(3-trifluoromethyl-benzylamino)-phenyl]-methanone (25). The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (80:20) to give pure 25 as a red oil (33%); Rf = 0.60 (CHCl3-MeOH, 98:2); 1H-NMR (300 MHz, CDCl3) δ 3.82 (s, 3H), 4.46 (s, 2H), 4.73 (br s, 1H), 6.60 (dd, J = 6.9, 1.8 Hz, 2H), 6.93 (dd, J = 6.9, 1.8 Hz, 2H), 7.45–7.61 (m, 4H), 7.64–7.74 (m, 4H); 13C-NMR (300 MHz, CDCl3) δ 47.2 (1C), 55.5 (1C), 111.7 (2C), 113.4 (2C), 120.4 (1C), 123.9 (1C), 124.4 (1C), 127.5 (1C), 129.0 (1C), 129.3 (1C), 130.6 (1C), 130.9 (1C), 131.4 (2C), 132.0 (2C), 139.7 (1C), 151.2 (1C), 162.5 (1C), 194.3 (1C); IR (thin film) cm-1 3348, 3032, 2932, 1636, 1601, 1528, 1455, 1327, 1169; MS (ESI, positive mode) m/z [M+H]+ 386.13624 (C22H19F3NO2 requires 386.12896).
(4-Methoxyphenyl)-[4-(4-trifluoromethylbenzylamino)phenyl]-methanone (26). The residue, after evaporation of the solvent, was purified by column chromatography eluting with CH2Cl2/EtOH (98:2) to give pure 26 as a yellow oil (22%); Rf = 0.56 (CHCl3-MeOH, 98:2); 1H-NMR (300 MHz, CDCl3) δ 3.71 (s, 3H), 4.41 (s, 2H), 4.70 (br s, 1H), 6.50 (dd, J = 9.5, 4.0 Hz, 2H), 6.89 (dd, J = 9.3, 3.2 Hz, 2H), 7.20 (t, J = 5.0 Hz, 2H), 7.38 (dd, J = 9.3, 3.2 Hz, 2H), 7.52 (t, J = 5.0 Hz, 2H), 7.64 (dd, J = 9.5, 4.0 Hz, 2H); 13C-NMR (300 MHz, CDCl3) δ 47.4 (1C), 55.9 (1C), 111.6 (1C), 111.9 (2C), 113.6 (2C), 122.5 (1C), 126.0 (2C), 127.7 (2C), 129.2 (1C), 131.5 (2C), 132.9 (2C), 142.8 (1C), 151.3 (1C), 157.1 (1C), 162.7 (1C), 194.4 (1C); IR (thin film) cm-1 3345, 3035, 2963, 1636, 1597, 1531, 1462, 1323, 1165; MS (ESI, positive mode) m/z [M+H]+ 386.13624 (C22H19F3NO2 requires 386.12896).
(2,4-Dimethoxyphenyl)-[4-(3-methoxybenzylamino)phenyl]-methanone (27).The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (75:25) to give 27 pure as a pink powder (53%); Rf = 0.33 (CHCl3-MeOH, 98:2); mp. 126–127 °C; 1H-NMR (300 MHz, CDCl3) δ 3.68 (s, 3H), 3.77 (s, 3H), 3.83 (s, 3H), 4.34 (s, 2H), 4.66 (br s, 1H), 6.48–6.55 (m, 4H), 6.78–6.91 (m, 4H), 7.25 (t, J = 7.7 Hz, 1H), 7.99 (d, J = 8.7 Hz, 2H); 13C-NMR (300 MHz, CDCl3) δ 47.6 (1C), 55.3 (1C), 55.5 (1C), 55.7 (1C), 98.6 (1C), 104.2 (1C), 111.5 (2C), 112.8 (1C), 113.1 (1C), 119.6 (1C), 122.6 (1C), 127.7 (1C), 129.9 (1C), 131.1 (1C), 132.7 (2C), 140.1 (1C), 151.9 (1C), 158.8 (1C), 160.0 (1C), 162.3 (1C), 194.0 (1C); IR (thin film) cm-1 3348, 3005, 2936, 1636, 1597, 1489, 1458, 1316, 1262, 1161; MS (ESI, positive mode) m/z [M+H]+ 378.15691 (C23H24NO4 requires 378.16271); Anal. Calcd for C23H23NO4: C 73.19, H 6.14, N 3.71, found: C 73.25, H 6.21, N 3.68.
[4-(4-tert-Butylbenzylamino)phenyl]-(2,4-dimethoxyphenyl)-methanone (28). The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (80:20) to give pure 28 as a brown powder (82%); Rf = 0.45 (CHCl3-MeOH, 98:2); mp. 138–139 °C; 1H-NMR (300 MHz, CDCl3) δ 1.27 (s, 9H), 3.70 (s, 3H), 3.77 (s, 3H), 3.89 (s, 2H), 4.08 (br s, 1H), 6.50 (d, J = 8.6 Hz, 2H), 6.58 (d, J = 8.3 Hz, 2H), 7.08 (d, J = 8.6 Hz, 2H), 7.24–7.31 (m, 3H), 7.57 (d, J = 8.3 Hz, 1H), 7.64 (d, J = 5.5 Hz, 1H); 13C-NMR (300 MHz, CDCl3) δ 31.4 (3C), 34.5 (1C), 37.6 (1C), 55.5 (1C), 55.6 (1C), 98.8 (1C), 104.5 (1C), 114.5 (1C), 122.5 (1C), 125.7 (2C), 128.0 (2C), 128.8 (1C), 131.1 (1C), 131.3 (2C), 133.8 (2C), 135.5 (1C), 149.6 (1C), 158.9 (1C), 162.5 (1C), 194.4 (1C); IR (thin film) cm-1 3480, 3364, 3005, 2963, 1623, 1602, 1505, 1462, 1410, 1312, 1277, 1211; MS (ESI, positive mode) m/z [M+H]+ 404.22034 (C26H30NO3 requires 404.21474); Anal. Calcd for C26H29NO3: C 77.39, H 7.24, N 3.47, found: C 77.42, H 7.30, N 3.51.
(2,4-Dimethoxyphenyl)-[4-(3-trifluoromethylbenzylamino)phenyl]-methanone (29).The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (75:25) to give pure 29 as an olive-green oil (32%); Rf = 0.58 (CHCl3-MeOH, 98:2); 1H-NMR (300 MHz, CDCl3)δ 3.71 (s, 3H), 3.82 (s, 3H), 4.43 (s, 2H), 4.83 (br s, 1H), 6.46–6.55 (m, 4H), 7.25 (dd, J = 7.1, 2.0 Hz, 1H), 7.42–7.58 (m, 4H), 7.67 (d, J = 8.8 Hz, 2H); 13C-NMR (300 MHz, CDCl3) δ 47.2 (1C), 55.5 (1C), 55.7 (1C), 98.9 (1C), 104.2 (1C), 111.6 (2C), 122.5 (1C), 123.9 (1C), 124.0 (1C), 124.3 (1C), 124.4 (1C), 128.1 (1C), 129.3 (1C), 130.6 (1C), 131.1 (1C), 132.6 (2C), 139.7 (1C), 151.6 (1C), 158.9 (1C), 162.4 (1C), 194.1 (1C); IR (thin film) cm-1 3337, 3009, 2936, 1636, 1597, 1528, 1505, 1458, 1327, 1161; MS (ESI, positive mode) m/z [M+H]+ 416.14680 (C23H21F3NO3 requires 416.13953).
(2,4-Dimethoxyphenyl)-[4-(4-trifluoromethylbenzylamino)phenyl]-methanone (30).The residue, after evaporation of the solvent, was purified by column chromatography eluting with cyclohexane/EtOAc (75:25) to give pure 30 as a dark-brown oil (50%); Rf = 0.44 (CHCl3-MeOH, 98:2); 1H-NMR (300 MHz, CDCl3)δ 3.72 (s, 3H), 3.82 (s, 3H), 4.45 (s, 2H), 4.76 (br s, 1H), 6.48 (dd, J = 7.9, 1.9 Hz, 2H), 6.53 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 2.9 Hz, 1H), 7.45 (d, J = 8.1 Hz, 2H), 7.57 (d, J = 8.1 Hz, 2H), 7.66 (dd, J = 7.9, 1.9 Hz, 2H); 13C-NMR (300 MHz, CDCl3) δ 47.1 (1C), 55.5 (1C), 55.7 (1C), 98.5 (1C), 98.9 (1C), 104.4 (1C), 111.6 (2C), 122.5 (1C), 125.6 (1C), 125.8 (2C), 127.4 (2C), 128.2 (1C), 129.6 (1C), 132.6 (2C), 142.7 (1C), 151.5 (1C), 158.9 (1C), 162.4 (1C), 194.0 (1C); IR (thin film) cm-1 3345, 3009, 2940, 1636, 1597, 1528, 1505, 1462, 1323, 1161; MS (ESI, positive mode) m/z [M+H]+ 416.14680 (C23H21F3NO3 requires 416.13953).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}