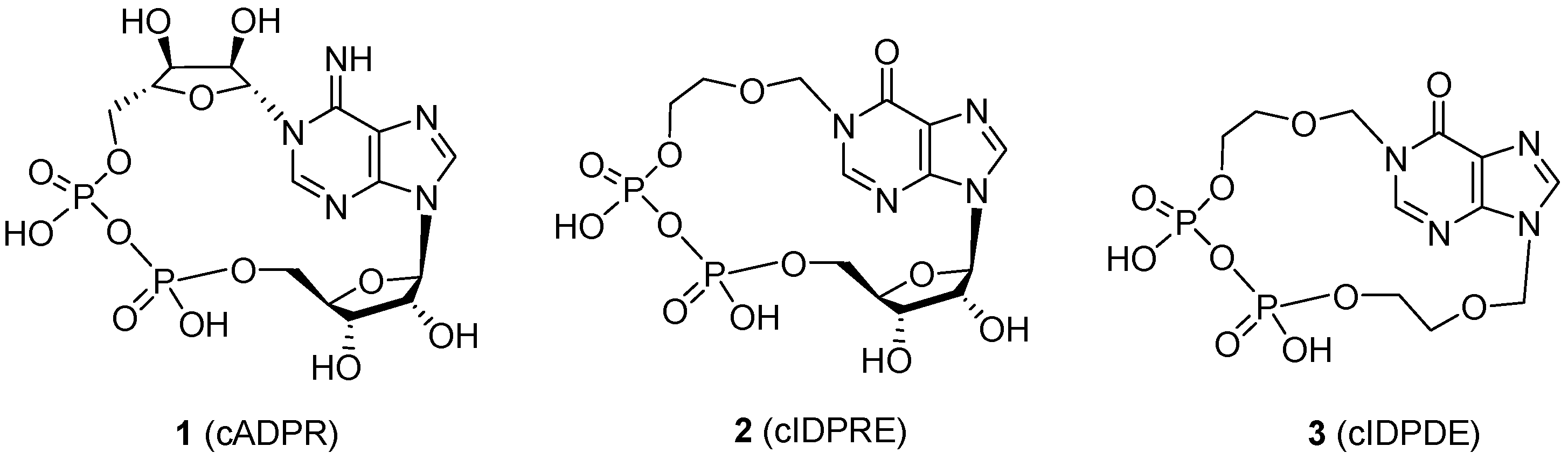
3.2. Synthesis
N9-[(5′
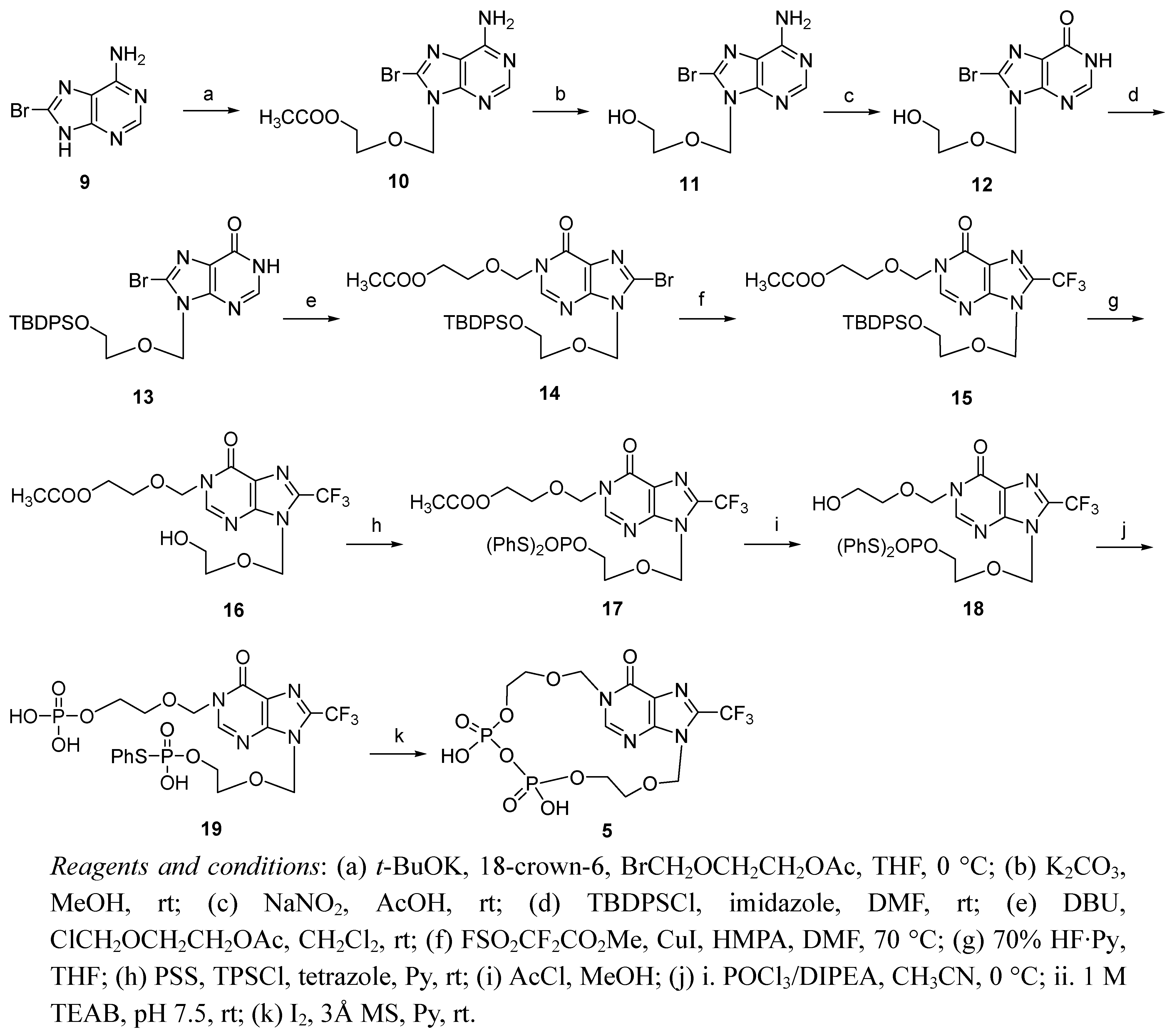
-Acetoxyethoxy)methyl]-8-bromoadenine (
10). To a stirred suspension of 8-bromoadenine (4.5 g, 21.03 mmol) [
14] in anhydrous THF (400 mL) was added potassium
tert-butoxide (2.59 g, 23.13 mmol) and 18-crown-6 (1.11 g, 4.20 mmol). The reaction mixture was stirred at room temperature for 15 min, and then BrCH
2OCH
2CH
2OAc (3.1 mL, 23.13 mmol) [
15] was added dropwise at 0 °C. After being stirred for 30 min at 0 °C, the mixture was filtered and the filtrate is evaporated under reduced pressure. The residue was purified by silica gel column chromatography (PE-EA = 1:2) to afford compound
10 (3.02 g, 44%).
1H-NMR (400 MHz, DMSO-
d6)
δ 1.92 (s, 3H, OAc), 3.69-3.72 (m, 2 H, H
4′), 4.04-4.17 (m, 2 H, H
5′), 5.51 (s, 2H, H
1′), 7.48 (s, 2H, NH
2), 8.16 (s, 1H, H
2).
13C-NMR (100 MHz, DMSO-
d6)
δ 170.1, 154.8, 153.2, 151.2, 126.5, 118.7, 72.3, 67.0, 62.7, 20.5. MS (ESI-TOF
+):
m/z = 330.0 [(M + H)
+].
N9-[(5′-Hydroxylethoxy)methyl]-8-bromohypoxanthine (12). Compound 10 (1.43 g, 4.34 mmol) was dissolved in methanol (120 mL). To the solution was added K2CO3 (73 mg, 0.53 mmol) and stirred for 6 h at room temperature. The mixture was neutralized by addition of 0.1 M HCl solution, and evaporated under reduced pressure. The residue was dissolved in AcOH (70 mL), and a solution of NaNO2 (2.52 g, 36.4 mmol) in H2O (17 mL) was added. The resulting mixture was stirred at room temperature for 24 h. After the mixture was evaporated in vacuo, the residue was partitioned between CHCl3 and H2O. The aqueous phase was extracted again with CHCl3, the organic layer was combined and washed with brine, dried (Na2SO4), filtered and concentrated in vacuo. Flash chromatography (CH2Cl2-MeOH = 40:1) afforded 12 (792 mg, 63% for two steps). 1H-NMR (400 MHz, DMSO-d6) δ 3.46-4.51 (m, 4H, H4′, H5′), 4.63(s, 1H, OH), 5.32 (s, 2H, H1′), 8.14 (s, 1H, H2), 12.56 (s, 1H, NH). MS (ESI-TOF+): m/z = 289.2 [(M + H)+].
N9-[(5′-tert-Butyldiphenylsilyloxyethoxy)methyl]-8-bromohypoxanthine (13). To a solution of 12 (700 mg, 2.42 mmol) in anhydrous DMF (10 mL) was added imidazole (1.86 g, 24.2 mmol) and tert-butyldiphenylsilyl chloride (3.4 mL, 12.1 mmol) under argon, and the mixture was stirred at room temperature for 12 h. And the mixture was evaporated in vacuo, the residue was partitioned between CH2Cl2 and H2O. The aqueous phase was extracted again with CH2Cl2, the organic layer was combined and washed with brine, dried (Na2SO4), filtered and concentrated in vacuo. Flash chromatography (PE-acetone = 5:1) afforded compound 13 (1.21 g, 95%). 1H-NMR (400 MHz, CDCl3) δ 1.06 (s, 9H, (CH3)3C-), 3.71-3.73 (m, 2H, H4′), 3.82-3.84 (m, 2H, H5′), 5.66 (s, 2H, H1′), 7.37-7.69 (m, 10H, ArH), 8.44 (s, 1H, H2),13.19 (s, 1H, NH). 13C-NMR (100 MHz, CDCl3) δ 157.8, 150.8, 146.3, 135.5, 133.3, 129.6, 127.6, 126.4, 124.6, 77.3, 77.0, 76.7, 73.5, 71.1, 62.9, 26.7, 19.0. HRMS (ESI-TOF+): calcd for C24H27BrN4O3Si [(M + H)+] 527.1109, [(M + Na)+] 549.0928, [(M + K)+] 565.0662; found, 527.1109, 549.0931, 565.0667.
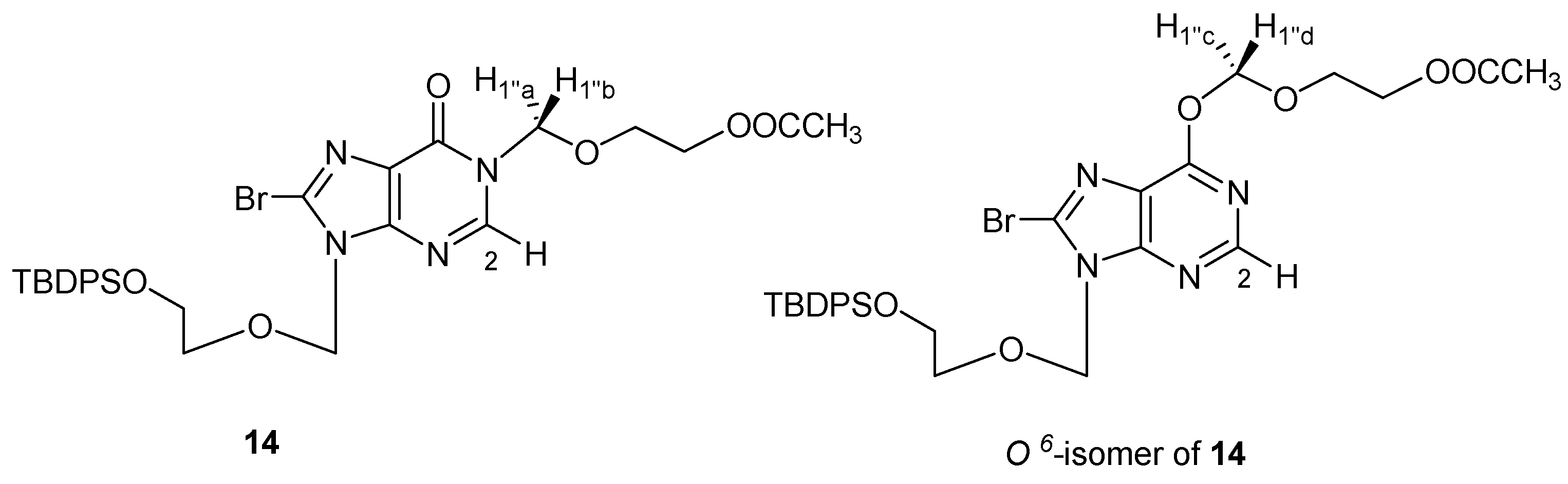
N1-[(5′′
-Acetoxyethoxy)methyl]-N9-[(5′
-tert-butyldiphenylsilyloxyethoxy)methyl]-8-bromohypoxanthine (
14). To the solution of
13 (1.23 g, 2.33 mmol) and DBU (3.5 mL, 23.3 mmol) in anhydrous CH
2Cl
2 (25 mL) was added ClCH
2OCH
2CH
2OAc (1.8 mL, 11.65 mmol) [
15] dropwise at 0 °C. After being stirred for 40 min at room temperature, the solvent was evaporated
in vacuo and the residue was purified by silica gel column chromatography (PE-acetone = 5:1) to afford compound
14 (916 mg, 61%).
1H-NMR (400 MHz, CDCl
3)
δ 1.04 (s, 9H, (CH
3)
3C-), 2.03 (s, 3H, OAc), 3.68-3.70 (m, 2H, H
4′), 3.79-3.82 (m, 2H, H
5′), 3.85-3.87 (m, 2H, H
4′′), 4.18-4.20 (m, 2H, H
5′′), 5.54 (s, 2H, H
1′′), 5.61 (s, 2H, H
1′), 7.35-7.66 (m, 10H, ArH), 8.09 (s, 1H, H
2).
13C-NMR (100 MHz, CDCl
3)
δ 170.7, 155.3, 149.2, 147.8, 135.5, 133.2, 129.7, 127.6, 126.2, 124.3, 75.0, 73.5, 71.0, 68.1, 62.9, 26.7, 20.7, 19.0. HRMS (ESI-TOF
+): calcd for C
29H
35BrN
4O
6Si [(M + H)
+] 643.1582, [(M + Na)
+] 665.1401, [(M + K)
+] 681.1135; found, 643.1563, 665.1377, 681.1117.
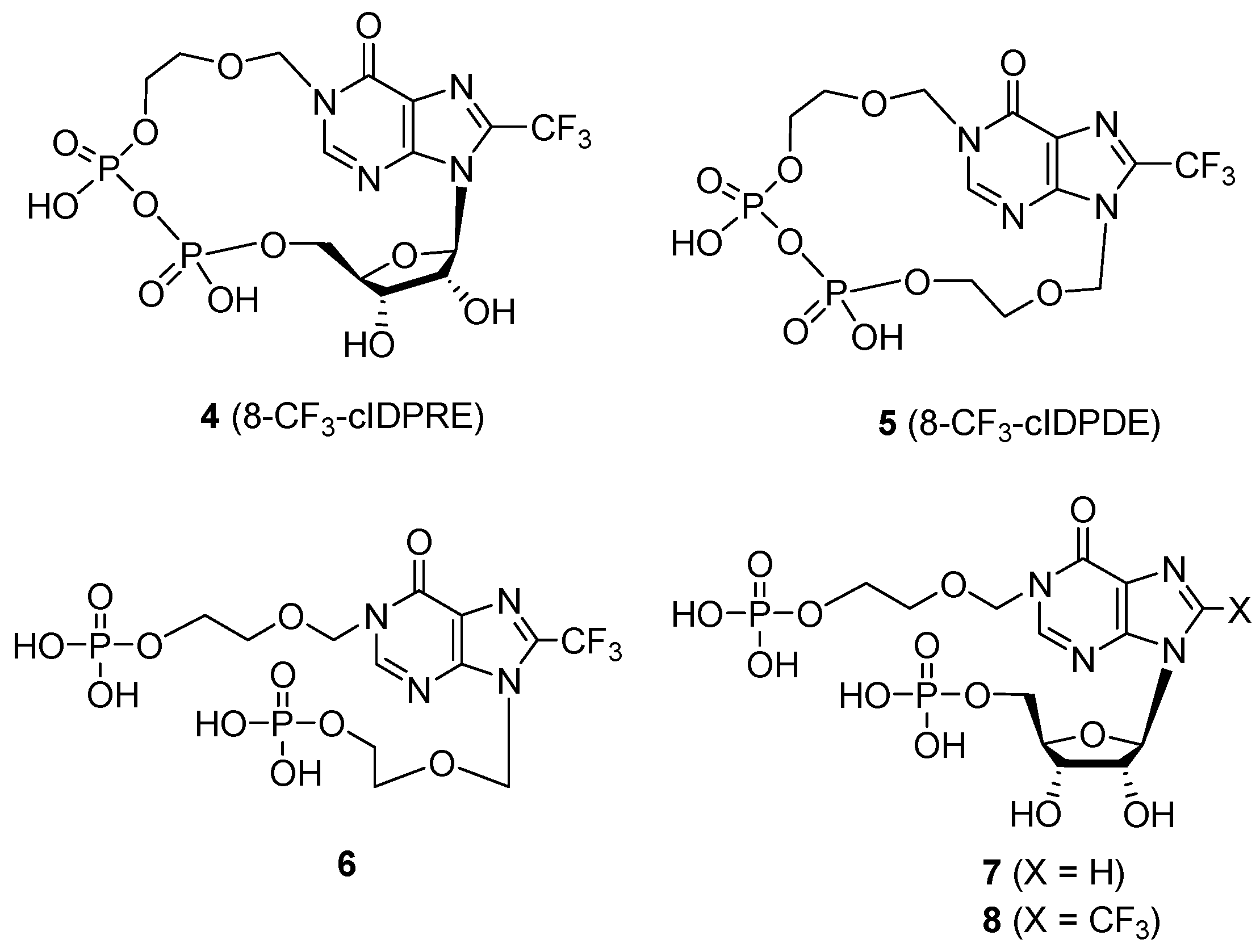
N1-[(5′′-Acetoxyethoxy)methyl]-N9-[(5′-tert-butyldiphenylsilyloxyethoxy)methyl]-8-trifluoromethyl- hypoxanthine (15). To a solution of compound 14 (576 mg, 0.895 mmol) and CuI (206 mg, 1.074 mmol) in anhydrous DMF (33 mL), hexamethylphosphoric triamide (2.39 mL, 13.425 mmol) and FSO2CF2CO2Me (1.71 mL, 13.425 mmol) were added successively. The reaction mixture was stirred for 20 h at 70 °C under argon, then cooled to room temperature, 22 mL of saturated aq. NH4Cl was added and the mixture was extracted with 200 mL of EA-hexanes (7:3). The organic layer was washed successively with sat. aq. NaHCO3, water and brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE-acetone = 9:2) to afford compound 15 (238 mg, 42%) and compound 16 (48 mg, 14%), with compound 14 recovered (96 mg, 17%). 1H-NMR (400 MHz, CDCl3) δ 1.03 (s, 9H, (CH3)3C-), 2.04 (s, 3H, OAc), 3.68-3.70 (m, 2H, H4′), 3.79-3.81 (m, 2H, H5′), 3.87-3.89 (m, 2H, H4′′), 4.20-4.22 (m, 2H, H5′′), 5.56 (s, 2H, H1′′), 5.76 (s, 2H, H1′), 7.35-7.66 (m, 10H, ArH), 8.18 (s, 1H, H2). 13C-NMR (100 MHz, CDCl3) δ 170.7, 156.3, 149.4, 149.3, 138.6 (d, 2JCF = 41 Hz), 135.5, 133.2, 129.7, 127.7, 122.7, 118.2 (q, 1JCF = 270 Hz), 75.1, 73.5, 71.4, 68.3, 63.1, 63.0, 26.7, 20.8, 19.1. 19F-NMR (470 MHz, CDCl3) δ -63.4 (s). HRMS (ESI-TOF+): calcd for C30H35F3N4O6Si [(M + Na)+] 655.2170, [(M + K)+] 671.1904; found, 655.2169, 671.1913.
N1-[(5′′-Acetoxyethoxy)methyl]-N9-[(5′-hydroxylethoxy)methyl]-8-trifluoromethylhypoxanthine (16). A solution of 15 (182 mg, 0.288 mmol) in anhydrous THF (35 mL) was added 70% HF·Py 1.3 mL at −20 °C. The mixture was stirred at 0 °C for 1 h and at room temperature over night. The reaction mixture was quenched with saturated aq. NaHCO3 at 0 °C and diluted with ethyl acetate, then partitioned and the water layer was washed with ethyl acetate again. The organic layer was combined, washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE-EA = 1:5) to afford the compound 16 (91 mg, 82%). 1H-NMR (400 MHz, CDCl3) δ 2.05 (s, 3H, OAc), 3.68-3.74 (m, 4H, H4′, H5′), 3.87-3.89 (m, 2H, H4′′), 4.19-4.22 (m, 2H, H5′′), 5.56 (s, 2H, H1′′), 5.76 (s, 2H, H1′), 8.23 (s, 1H, H2). 13C-NMR (100 MHz, CDCl3) δ 170.8, 156.2, 149.6, 149.3, 138.4 (q, 2JCF = 41 Hz), 122.7, 118.2 (q, 1JCF = 270 Hz), 75.1, 73.2, 71.2, 68.3, 62.9, 61.4, 20.8; 19F-NMR (470 MHz, CDCl3) δ-63.4 (s). HRMS (ESI-TOF+): calcd for C14H17F3N4O6 [(M + Na)+] 417.0992, [(M +K)+], 433.0726; found, 417.0991, 433.0730.
N1-[(5′′
-Acetoxyethoxy)methyl]-N9-[[5′
-bis(phenylthio)phosphoryloxyethoxy]-methyl]-8-trifluoro-methylhypoxanthine (
17). To a solution of
16 (66 mg, 0.167 mmol) in anhydrous pyridine (5 mL) was added TPSCl (302 mg, 1.00 mmol), PSS (571 mg, 1.50 mmol) [
20], and tetrazole (105 mg, 1.50 mmol), and the mixture was stirred at room temperature for 12 h. The mixture was evaporated, and the residue was purified by silica gel column chromatography (PE-EA = 1:2) to give compound
17 (86 mg, 79%).
1H NMR (400 MHz, CDCl
3)
δ 2.05 (s, 3H, OAc), 3.82-3.84 (m, 2H, H
4′), 3.86-3.88 (m, 2H, H
5′), 4.18-4.21 (m, 2H, H
4′′), 4.31-4.35 (m, 2H, H
5′′), 5.56 (s, 2H, H
1′′), 5.68 (s, 2H, H
1′), 7.33-7.52 (m, 10H, ArH), 8.22 (s, 1H, H
2).
13C-NMR (100 MHz, CDCl
3)
δ 170.8, 156.3, 149.7, 149.3, 138.5 (q,
2JCF = 41 Hz), 135.3, 129.7, 129.4, 125.9, 122.7, 118.2 (q,
1JCF = 269 Hz), 75.2, 73.0, 69.0, 68.4, 66.2, 62.9, 20.8.
19F NMR (470 MHz, CDCl
3)
δ-63.4 (s).
31P-NMR (D
2O, 243 MHz, decoupled with
1H)
δ50.41 (s). HRMS (ESI-TOF
+): calcd for C
30H
35F
3N
4O
6Si [(M + H)
+], 659.1005; found, 659.1006.
N1-[(5′′-Phosphonoxyethoxy)methyl]-N9-[[5′-(phenylthio)phosphoryloxyethoxy]methyl]-8-trifluoro- methylhypoxanthine (19). Compound 17 (54 mg, 0.082 mmol) was dissolved in MeOH (4 mL), and a solution of acetyl chloride (7 μL, 0.098 mmol) in anhydrous CH2Cl2 (1 mL) was added at −20 °C. The mixture was stirred at 0 °C for 30 min and raised to room temperature for 24 h, then neutralized by sat. aq. NaHCO3 solution. The mixture was evaporated, and the residue was partitioned between CH2Cl2 and H2O. The aqueous phase was extracted again with CH2Cl2, the organic layers were combined and washed with brine, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE-EA =1:10) to give compound 18 (31 mg). The deacetylated product 18 (31 mg, 0.050 mmol) was dissolved in anhydrous CH3CN (8 mL). DIPEA (65 μL, 0.375 mmol) and POCl3 (28 μL, 0.300 mmol) were added successively to the solution at −20 °C, and the mixture was stirred at 0 °C for 14 h, and then added 5mL of TEAB (1 M, pH 7.5) at 0 °C and stirred at room temperature for 6 h. After evaporation under reduced pressure, the residue was partitioned between H2O and CHCl3, and the aqueous layer was washed with CHCl3 and evaporated in vacuo. The residue was dissolved in 5 mL of TEAB buffer (0.05 M, pH 7.5), then applied to a C18 reversed-phase column (2.2 × 25 cm). The column was developed using a linear gradient of 0-40% CH3CN in TEAB buffer (0.05 M, pH 7.5) within 30 min to afford 19 (27 mg, 41% for two steps) as its triethylammonium salt. 1H-NMR (400 MHz, D2O) δ 3.70-3.73 (m, 4H, H4′, H5′), 3.85-3.89 (m, 2H, H4′′), 3.94-3.98 (m, 2H, H5′′), 5.45 (s, 2H, H1′′), 5.64 (s, 2H, H1′), 7.09-7.29 (m, 5H, ArH), 8.41 (s, 1H, H2). 13C-NMR (100 MHz, D2O) δ 157.6, 150.9, 149.5, 138.8 (q, 2JCF = 41 Hz), 132.7, 129.6, 128.9, 127.7, 122.1, 117.8 (q, 1JCF = 270 Hz), 76.4, 73.3, 69.3, 64.9, 64.3, 46.6, 8.2. 19F-NMR (470 MHz, D2O) δ-63.0 (s). 31P-NMR (D2O, 243 MHz, decoupled with 1H) δ 1.10 (s), 17.80 (s). HRMS (ESI-TOF−) calcd for C18H21N4O10P2SF3 [(M − H)−], 603.0333; found, 603.0331.
N1-[(5′′-O-Phosphorylethoxy)methyl]-N9-[(5′-O-phosphorylethoxy)methyl]-8-trifluoromethylhypo- xanthine-cyclic pyrophosphate (5). A solution of 19 (5 mg, 6.1 μmol) in anhydrous pyridine (4.5 mL) was added slowly over 20 h, utilizing a syringe pump, to a mixture of I2 (36 mg, 142 μmol) and 3 Å molecular sieves (0.36 g), in pyridine (40 mL) at room temperature in the dark. The molecular sieves were filtered off with Celite and washed with H2O. The combined filtrate was evaporated, and the residue was partitioned between CHCl3 and H2O. The aqueous layer was evaporated, and the residue was dissolved in 0.05 M TEAB buffer, which was applied to C18 reversed-phase column (2.2 × 25 cm). The column was developed using a linear gradient of 0-20% CH3CN in TEAB buffer (0.05 M, pH 7.5) within 30 min to give 5 as its triethylammonium salt (3.0 mg, 71%). 1H-NMR (400 MHz, D2O) δ 3.70-3.78 (m, 4H, H4′, H5′), 3.81-3.83 (m, 2H, H4′′), 3.88-3.90 (m, 2H, H5′′), 5.54 (s, 2H, H1′′), 5.75 (s, 2H, H1′), 8.49 (s, 1H, H2). 19F-NMR (470 MHz, D2O) δ-62.5 (s). 31P-NMR (D2O 121.5 MHz, decoupled with 1H) δ −10.07 (d, JP,P = 18.2 Hz), −10.42 (d, JP,P = 18.2 Hz). HRMS (ESI-TOF−) calcd for C12H15N4O10P2F3 [(M − H)−], 493.0143; found, 493.0146.
N1-[(5′′-Phosphonoxyethoxy)methyl]-N9-[(5′-Phosphonoxyethoxy)methyl]-8- trifluoromethylinosine (6). Compound 16 (20 mg, 0.051 mmol) was dissolved in methanol (2 mL). To the solution was added K2CO3 (1 mg, 7.24 μmol) at room temperature and stirred for 6h. The mixture was neutralized by addition of 0.01 M HCl solution, and removed of the solvent in vacuo. The residue was partitioned between CHCl3 and H2O, and the organic layer was washed with brine, dried (Na2SO4), and evaporated, affording compound 20 (16 mg). Compound 20 (16 mg, 0.045 mmol) was dissolved in anhydrous CH3CN (5 mL). DIPEA (94 μL, 0.54 mmol) and POCl3 (42 μL, 0.45 mmol) were added successively to the solution at −20 °C. The mixture was stirred at 0 °C for 16 h, and then added 5 mL of TEAB (1 M, pH 7.5) at 0 °C and stirred for 6 h at room temperature. After evaporation under reduced pressure, the residue was partitioned between H2O and CHCl3, and the aqueous layer was washed with CHCl3 and evaporated in vacuo. The residue was dissolved in 5 mL of TEAB buffer (0.05 M, pH 7.5), and applied to a C18 reversed-phase column (2.2 × 25 cm). The column was developed using a linear gradient of 0-40% CH3CN in TEAB buffer (0.05 M, pH 7.5) within 30 min to give 6 (22.3 mg, 62% for two steps) as its triethylammonium salt. 1H-NMR (400 MHz, D2O) δ 3.73-3.79 (m, 4H, H4′, H5′), 3.85-3.92 (m, 4H, H4′′, H5′′), 5.45 (s, 2H, H1′′), 5.77 (s, 2H, H1′), 8.51 (s, 1H, H2). 13C-NMR (100 MHz, D2O) δ 157.8, 151.0, 149.7, 138.6 (q, 2JCF = 41 Hz), 122.2, 117.9 (q, 1JCF = 270 Hz), 76.4, 73.3, 69.5, 69.2, 64.1, 63.8. 19F-NMR (470 MHz, D2O) δ -62.9 (s). 31P-NMR (D2O, 243 MHz, decoupled with 1H) δ 0.19 (s), 0.22 (s). HRMS (ESI-TOF−) calcd for C12H17N4O11P2F3 [(M − H)−], 511.0248; found, 511.0246.
N1-[(5′′
-Phosphonoxyethoxy)methyl]-5′
-O-phosphoryl-2′
,3′
-O-isopropylidene-inosine(
23). Compound
21 (49 mg, 0.116 mmol) [
6] was dissolved in 24 mL of methanol. To the solution was added K
2CO
3 (2 mg, 14.5 μmol) and stirred at room temperature for 6h. The mixture was neutralized by addition of 0.1 M HCl solution, and removed of the solvent
in vacuo. The residue was partitioned between CHCl
3 and H
2O, and the organic layer was washed with brine, dried (Na
2SO
4), and evaporated, affording compound
22 (38mg)
. Compound
22 (38 mg, 0.099 mmol) was dissolved in anhydrous CH
3CN (5 mL). DIPEA (0.21 mL, 1.19 mmol) and POCl
3 (91 μL, 0.99 mmol) were added successively to the solution at −20 °C. The mixture was stirred at 0 °C for 16 h, and then was neutralized by addition of 1 M NaOH solution. And the resulting mixture was stirred at room temperature for 2 h. After evaporated under reduced pressure, the residue was partitioned between H
2O and CHCl
3, and the aqueous layer was washed with CHCl
3 and evaporated
in vacuo. The residue was dissolved in 5 mL of TEAB buffer (0.05 M, pH 7.5), and applied to a C
18 reversed-phase column (2.2 × 25 cm)
. The column was developed using a linear gradient of 0-40% CH
3CN in TEAB buffer (0.05 M, pH 7.5) within 30 min to give
23 (61 mg, 71% for two steps) as its triethylammonium salt.
1H-NMR (400 MHz, D
2O)
δ 1.31, 1.53 (each s, each 3H, 2 × CH
3), 3.72-3.74 (m, 2H, H
5′), 3.85-3.89 (m, 2H, CH
2O), 3.91-3.94 (m, 2H, CH
2OP), 4.52-4.56 (m, 1H, H
4′), 5.06 (dd, 1H,
JH3′,H4′ = 1.6 Hz,
JH3′, H2′ = 6.0 Hz, H
3′), 5.29 (dd, 1H,
JH2′,H1′= 2.8 Hz,
JH2′,H3′ = 6.0 Hz, H
2′), 5.47 (d, 1H,
JH1″b, H1″a = 10.8 Hz, H
1″b), 5.51 (d, 1H,
JH1″a, H1″b = 10.8 Hz, H
1″a), 6.17 (d, 1H,
JH1′,H2′ = 2.8 Hz, H
1′), 8.23, 8.34 (each s, each 1H, H
8, H
2).
31P-NMR (D
2O, 121.5 MHz, decoupled with
1H)
δ 1.79 (s), 1.91 (s). HRMS(ESI-TOF
−): calcd for C
14H
24N
4O
13P
2 [(M − H)
−], 541.0742; found, 541.0733.
N1-[(5′′-Phosphonoxyethoxy)methyl]-5′-O-phosphorylinosine (7). A solution of 23 (25 mg, 33.6 μmol) in 60% HCOOH (6 mL) was stirred for 8 h, and then 14 mL of TEAB (1M, pH 7.5) was added. The solution was evaporated under reduced pressure. The residue was dissolved in 0.05 M TEAB buffer (4.0 mL), which was applied to C18 reversed-phase column (2.2 × 25 cm). The column was developed using a linear gradient of 0-40% CH3CN in TEAB buffer (0.05 M, pH 7.5) within 30 min to afford 7 as its triethylammonium salt (20.2 mg. 85%). 1H-NMR (400 MHz, D2O) δ 3.72-3.74 (m, 2H, H5′), 3.85-3.88 (m, 2H, CH2O), 3.98-4.01 (m, 2H, CH2OP), 4.23-4.26 (m, 1H, H4′), 4.34-4.37 (m, 1H, H3′), 4.59-4.61 (m, 1H, H2′), 5.46-5.52 (m, 2H, H1″), 6.01 (d, 1H, JH1′,H2′ = 5.6 Hz, H1′), 8.31, 8.36 (each s, each 1H, H8, H2). 31P-NMR (D2O, 243 MHz, decoupled with 1H) δ 0.81 (s), 0.92 (s). HRMS (ESI-TOF−): calcd for C14H24N4O13P2 [(M − H)−], 501.0429; found, 501.0426.
N1-[(5′′-Phosphonoxyethoxy)methyl]-5′-O-phosphoryl-2′
,3′
-O-isopropylidene-8-trifluoromethylinosine (
26). By a similar procedure that described for
6,
26 was synthesized from
24 [
9], as its triethylammonium salt, in 57% yield for two steps.
1H-NMR (400 MHz, D
2O)
δ 1.29, 1.50 (each s, each 3H, 2×CH
3), 3.71-3.73 (m, 2H, H
5′), 3.82-3.96 (m, 4H, CH
2O, CH
2OP), 4.35-4.39 (m, 1H, H
4′), 5.19 (dd, 1H,
JH3′,H4′ = 4.0 Hz,
JH3′,H2′ = 6.8 Hz, H
3′), 5.40 (d, 1H,
JH1″a,H1″b=10.8 Hz, H
1″a), 5.55-5.60 (m, 2H, H
1″b, H
2′), 6.23 (d, 1H,
JH1′,H2′ = 2.0 Hz, H
1′), 8.44 (s, 1H, H
2).
13C-NMR (100 MHz, D
2O)
δ 157.8, 150.6, 148.9, 138.3 (q,
2JCF = 40Hz), 122.9, 117.8 (q,
1JCF = 270 Hz), 115.4, 90.0, 86.7, 86.6, 83.7, 81.1, 76.4, 69.2, 64.2, 63.8, 25.9, 24.2.
19F-NMR (470 MHz, D
2O)
δ -61.9 (s).
31P-NMR (D
2O, 243 MHz, decoupled with
1H)
δ 0.54 (s), 0.70 (s). HRMS(ESI-TOF
−): calcd for C
17H
23F
3N
4O
13P
2 [(M − H)
−], 609.0612; found, 609.0615.
N1-[(5′′-Phosphonoxyethoxy)methyl]-5′-O-phosphoryl-8-trifluoromethylinosine (8). A solution of 26 (15 mg, 18.47 μmol) in 10% HCOOH (7.5 mL) was stirred at room for 60 h, and then 11 mL of TEAB (1 M, pH 7.5) was added. The solution was evaporated in vacuo. The residue was dissolved in 0.05 M TEAB buffer (2.0 mL), which was applied to C18 reversed-phase column (2.2 cm × 25 cm). The column was developed using a linear gradient of 0-40% CH3CN in TEAB buffer (0.05 M, pH 7.5) within 30 min to afford compound 8 (9.7 mg, 68%) as its triethylammonium salt, with the compound 26 (2.2 mg, 15%) recovered. 1H-NMR (400 MHz, D2O) δ 3.76-3.78 (m, 2H, H5′), 3.87-3.91 (m, 2H, CH2O), 4.02-4.13 (m, 2H, CH2OP), 4.21-4.25 (m, 1H, H4′), 4.55-4.57 (m, 1H, H3′), 5.20-5.23 (m, 1H, H2′), 5.46 (d, 1H, JH1″b, H1″a = 10.6 Hz,H1′b), 5.59 (d, 1H, JH1″a, H1″b = 10.6 Hz, H1″a), 5.99 (d, 1H, JH1″,H2′ = 5.6 Hz, H1′), 8.47 (s, 1H, H2). 13C-NMR (100 MHz, D2O) δ 157.9, 150.3, 149.4, 138.9 (q, 2JCF = 40Hz), 123.2, 117.8 (q, 1JCF = 269 Hz), 89.8, 84.4, 76.3, 72.0, 70.0, 69.3, 64.4, 64.1. 19F-NMR (470 MHz, D2O) δ -61.7 (s). 31P-NMR (D2O, 121.5 MHz, decoupled with 1H) δ 6.25(s), 6.27 (s). HRMS(ESI-TOF−): calcd for C14H19F3N4O13P2 [(M − H)−], 569.0303; found, 569.0315.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
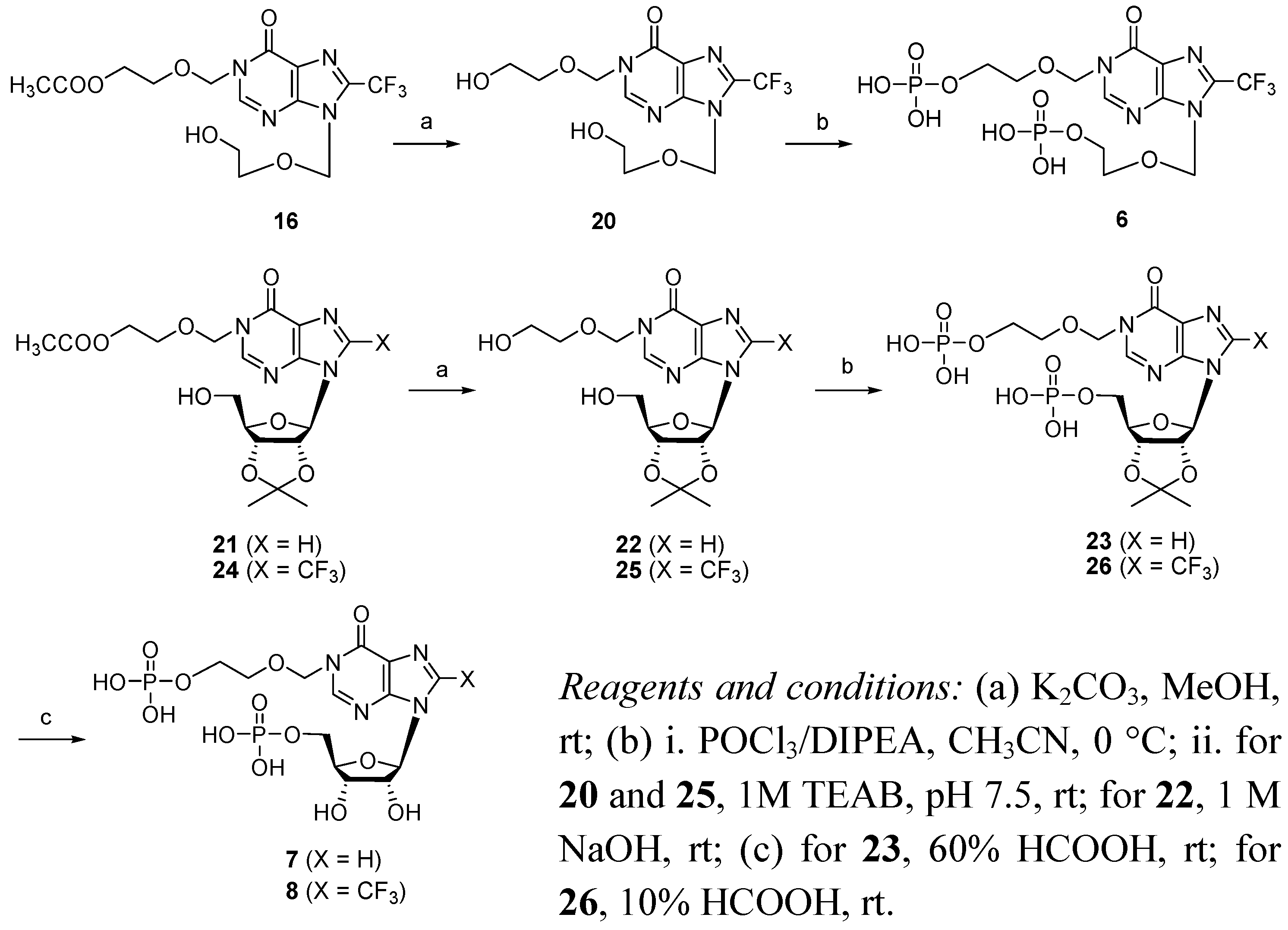{kind=link}