Introduction
The progressive elucidation of molecular mechanisms involved in cancer has opened up a new horizon for the development of new antitumoral compounds [
1]. Specifically, for profiling the activity of anticancer drugs, the major indicators of these agents include inhibition of proliferation and induction of apoptosis in cancer cells [
2]. Thus, direct manipulation of the biochemical machinery that regulates apoptosis is an interesting and new therapeutic approach to cancer [
3].
Apoptosis or programmed cell death not only plays a crucial role in tissue development and homeostasis, but is also involved in a range of pathological conditions and now recognized as an important component of multi-step carcinogenesis and therapy resistance [
1,
4]. This physiological phenomenon represents the terminal morphological and biochemical events [
5] and occurs through the activation of a cell-intrinsic suicide program. This program is carried out by internal, as well as external signals, divided in various phases terminating with signals that initiate the process leading to cellular destruction, characterized by DNA fragmentation as well as by loss of mitochondrial membrane integrity and liberation of molecules that initiate intracellular proteases activation [
6]. At the center of the apoptosis machinery is a family of intracellular proteases, known as 'caspases', that are responsible directly or indirectly for the morphological and biochemical events that characterize classical apoptosis [
7].
Apoptotic pathways might be significantly altered in cancer cells with respect to normal cells, and these differences might present a therapeutic window that can be exploited for the development of useful anticancer drugs. Moreover, cancers that possess alterations in proteins involved in cell death signaling are often resistant to chemotherapy and are more difficult to treat using chemotherapeutic agents that primarily work by inducing apoptosis [
8].
Several compounds prepared employing classical organic and combinatorial chemistry have advanced into clinical trials or are already approved. Research in the last decade has revealed a promising future for apoptosis based cancer therapies [
9]. In this scenario of discovery of small molecule modulators of apoptosis, considerable effort is being aimed at improving the prototypic drugs and replacing them by small molecule organic compounds, which could set the stage for future therapeutics [
10].
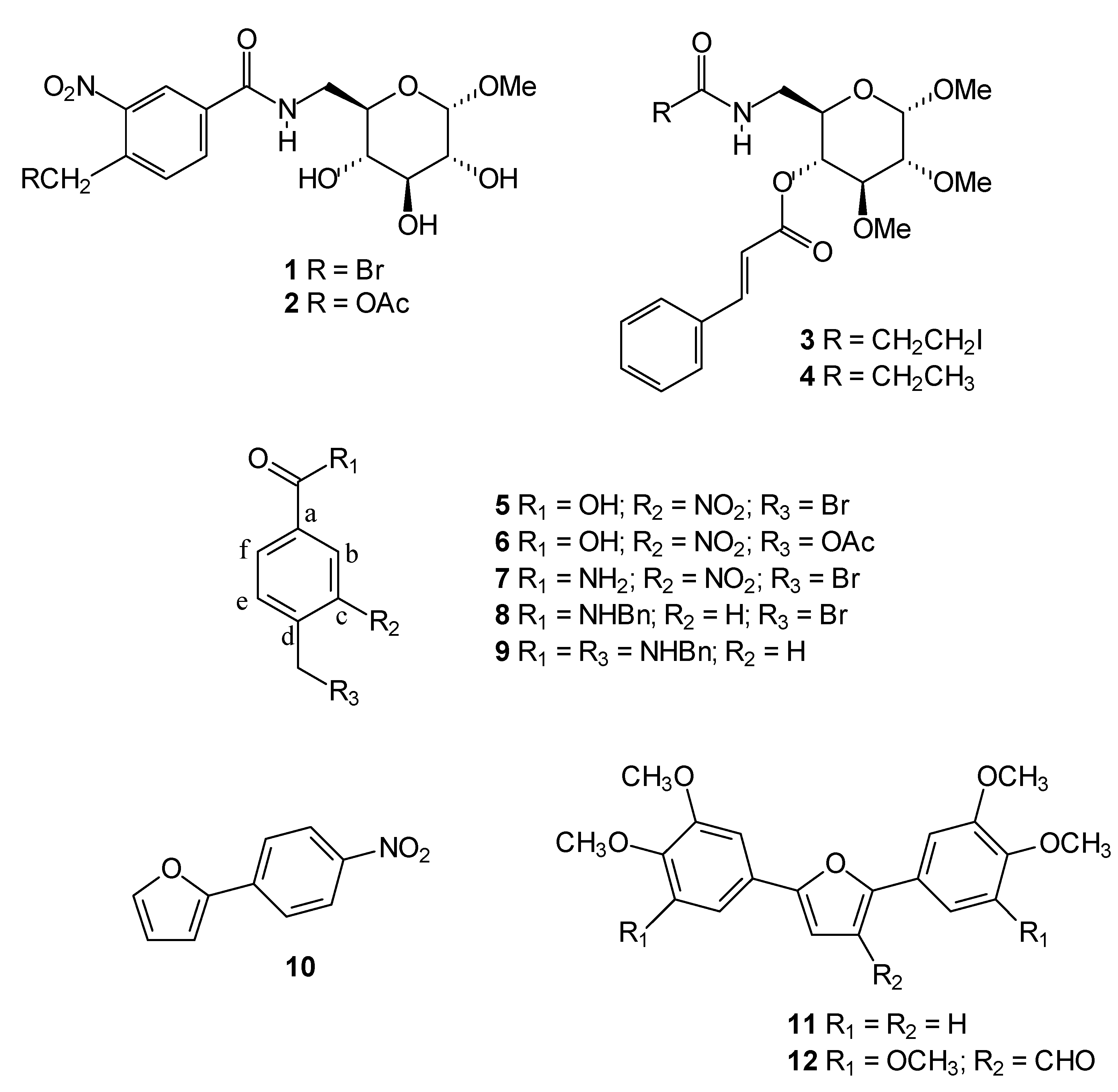
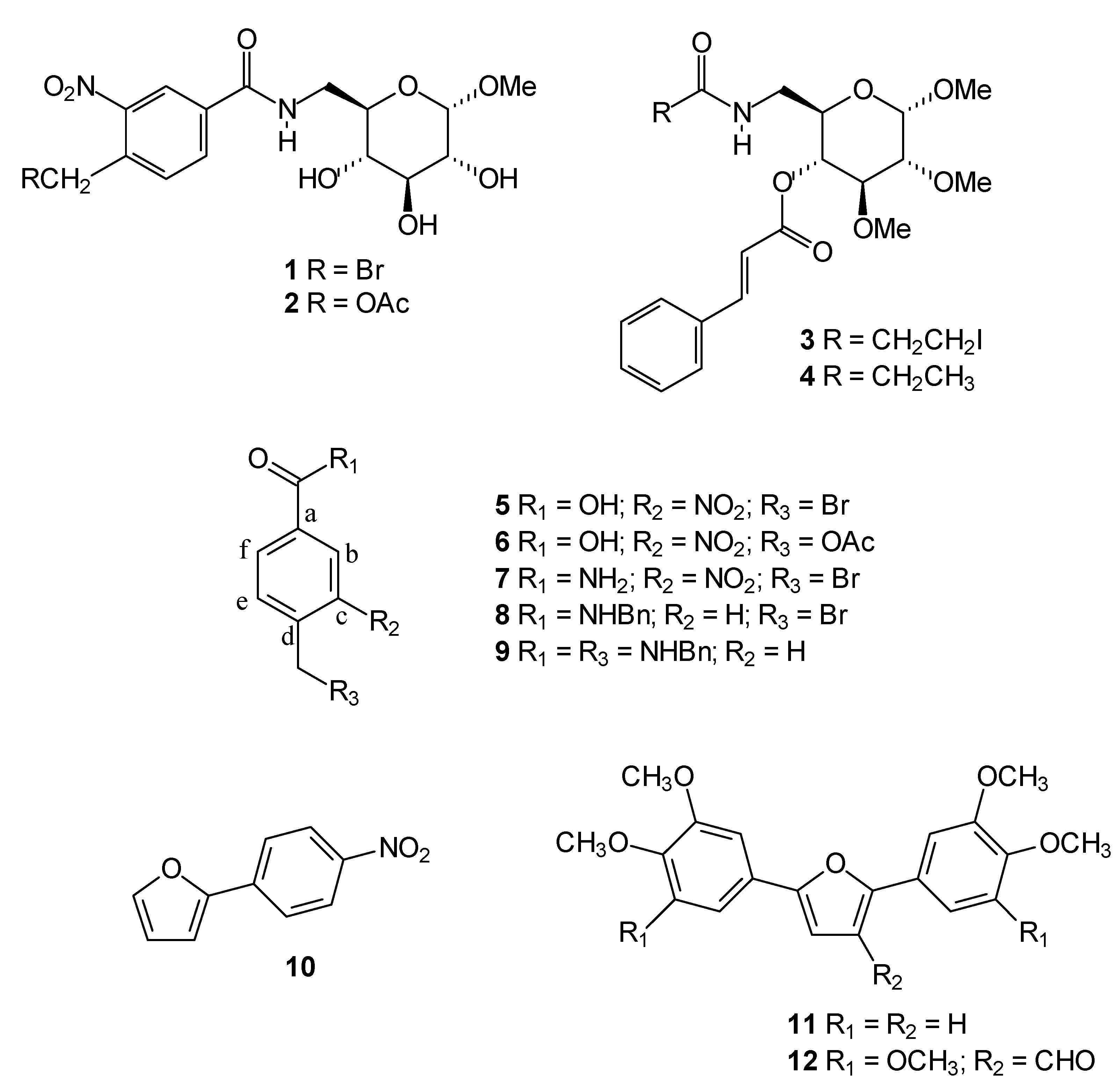
With this in mind, we decided to evaluate the cytotoxic activity of twelve compounds previously synthesized in our laboratory (
Figure 1) to potentially identify novel small-molecule compounds with potential anti-cancer properties. We performed this investigation using an
in vitro bioassay based on their cytotoxic effects against cancer cells, including UACC-62 (human melanoma) and Jurkat (human leukemia T-cell line) measured by the MTT method. The active compounds were investigated to determine if they also act as apoptosis inducers as evaluated by quantification of subdiploid DNA content and caspase 3 activation by flow cytometry.
The carbohydrate derivatives
1-
5 were chosen to evaluate their cytotoxicity because there is evidence that the activity of some compounds can be tuned by changes to the monosaccharide core [
11]. Moreover, carbohydrates play crucial roles in many biological processes and, therefore, their presence can be important to modulate the cytotoxic activity.
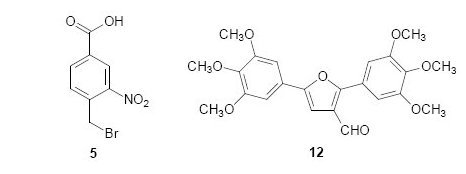
The bromomethyl, acetoxymethyl and iodoethyl groups present in compounds
1-
3, respectively
, are particularly reactive and these compounds can act as alkylating agents. Binding of alkylating agents to cellular DNA is considered to be a lethal event associated with anticancer activity [
12]. Compounds
5-
8, containing the alkylating groups but without the carbohydrate moiety, were included for comparison. Compound
4, presenting the carbohydrate moiety but without the alkylating groups, was also included for comparison.
Figure 1.
Chemical structures of compounds tested as cytotoxic agents.
Figure 1.
Chemical structures of compounds tested as cytotoxic agents.
We have previously reported the trypanocidal activity of compound
9 [
13]. Some studies have demonstrated a correlation between trypanocidal and antitumor activities [
14]. Thus, we decided to investigate the effects of compound
9 on the growth of cancer cells. Recently, we also reported the evaluation of the cytotoxicity activity of arylfurans
10 and
11 against the human cancer cells lines MCF-7 (breast), TK-10 (renal) and UACC-62 (melanoma) [
15], which motived us to perform a more detailed investigation of the ability of these compounds to induce apoptosis. Diarylfuran
12 was also selected based on its structural similarity to the diarylfuran
11.
Chemistry
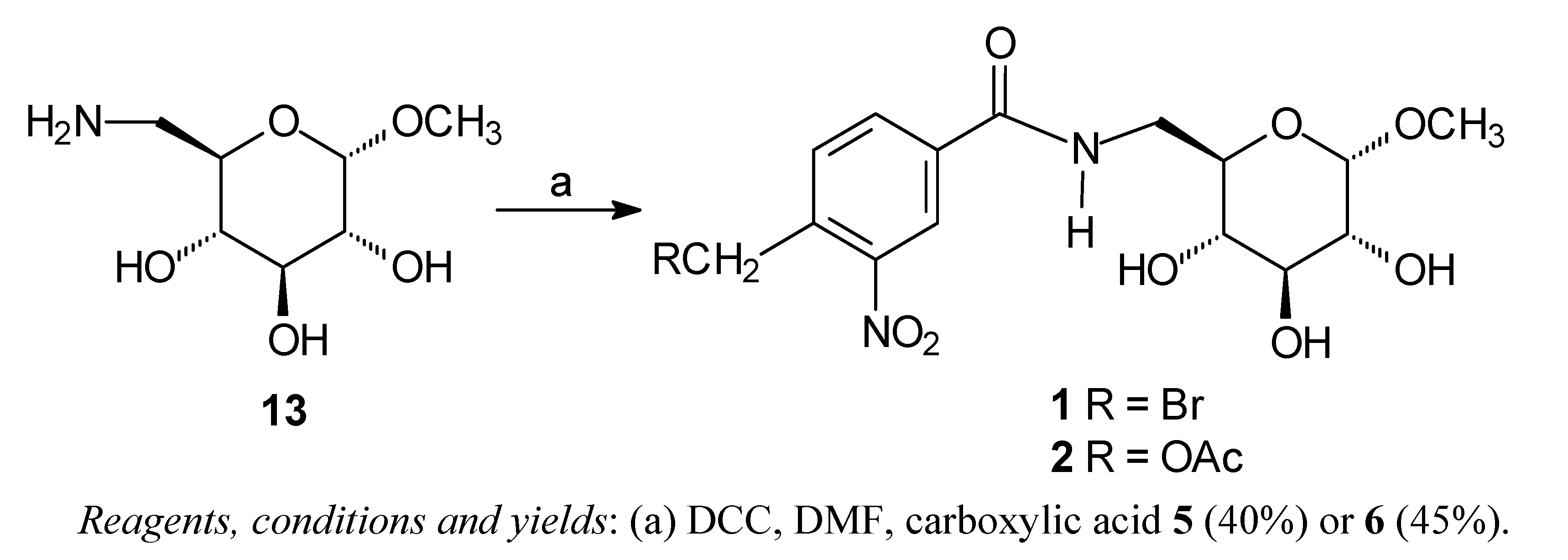
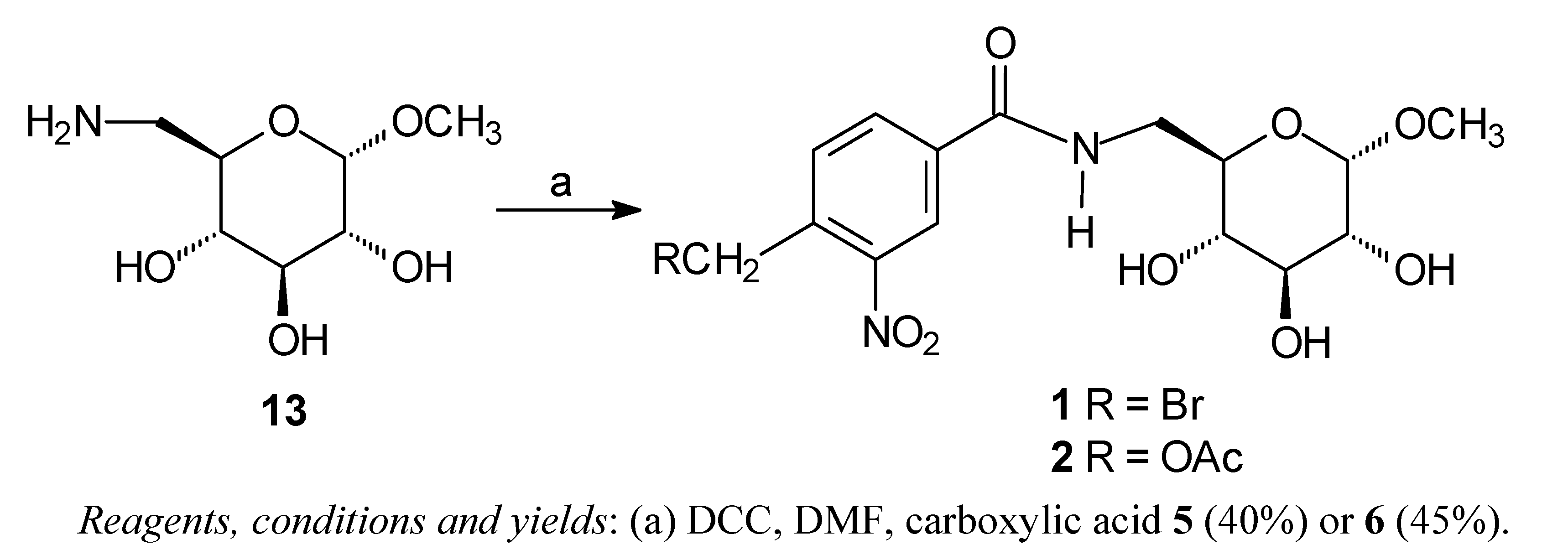
The carbohydrate derivatives
1 and
2 were prepared by treatment of methyl 6-amino-6-deoxy-α-D-glucopyranoside
13 [
16] with carboxylic acid
5 or
6 and DCC (
Scheme 1). The amido-esters
3 and
4 were synthesized from readily available methyl α-D-glucopyanoside in seven steps using methods previously reported by us [
17].
The compounds
5 and
6 were prepared in two and three steps, respectively, from
p-toluic acid, according to literature procedures [
18,
19,
20]. The benzamide
7 was obtained from the direct reaction of carboxylic acid
5 and NH
4Cl/SiO
2, triethylamine (TEA) and tosyl chloride under solvent-free conditions [
21]. Synthesis of the amides
8 and
9, was carried out by reaction of the acyl chloride, obtained from 4-(bromomethyl)benzoic acid [
19] by means of reflux with thionyl chloride, with benzylamine. Benzylamine was added either with an equimolar amount to obtain the amide
8 or in an excess to obtain the amide
9.
Scheme 1.
Synthesis of the amides 1 and 2.
Scheme 1.
Synthesis of the amides 1 and 2.
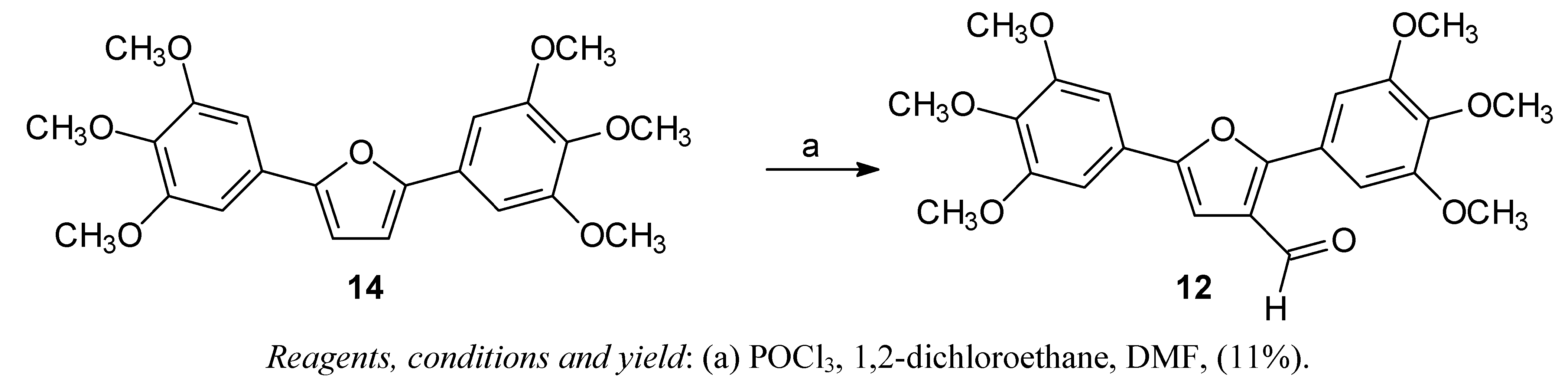
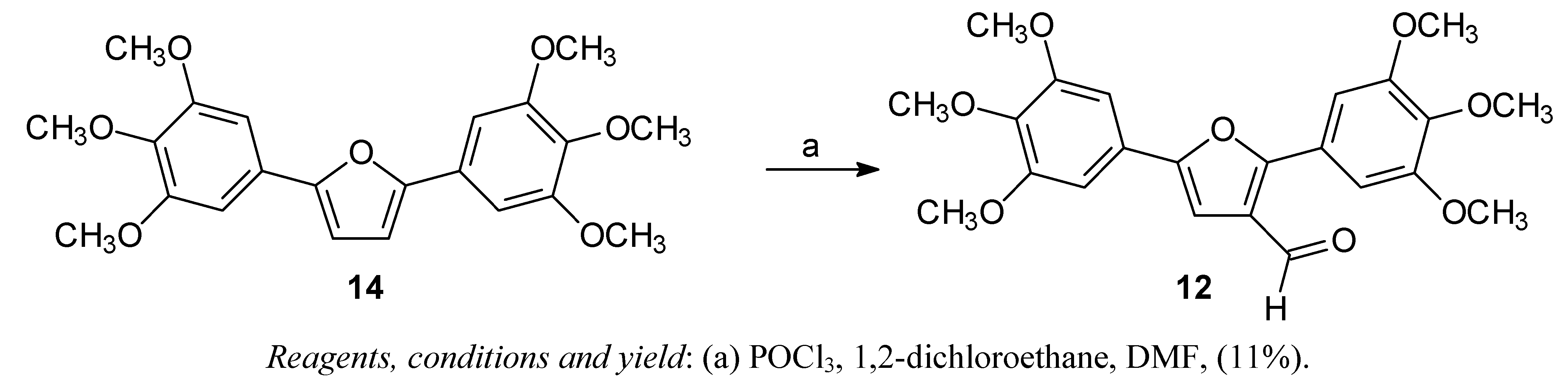
The arylfuran
10 was synthesized in one step using the classical Meerwein arylation (treatment of furan with diazonium salts in presence of cupric salts), as described in previous work [
22]. The diarylfuran
11 and 2,5-bis-(3,4,5-trimethoxy)furan
14 were prepared in two steps via a Stille coupling reaction [
22]. The diarylfuran
12 was obtained from
14 by Vilsmeier-Haack formylation (
Scheme 2).
Scheme 2.
Synthesis of the diarylfuran 12.
Scheme 2.
Synthesis of the diarylfuran 12.
Antiproliferative activity
Screening of synthesized substances was carried out using two human cancer cell lines: UACC-62 (human melanoma) and Jurkat (derived from human T-cell leukaemia). Proliferation percentage was determined by the modified 3-(4,5-dmethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [
23], based on the ability of a mitochondrial dehydrogenase enzyme from viable cells to cleave the tetrazolium rings of the pale yellow MTT and form dark blue formazan crystals. Both lineages were incubated with the test substances at 100 μM for 48 hours and the cell proliferation/viability determination using the survival percentage obtained with the cell treated only with the vehicle (0.1% aqueous DMSO) as reference. In all experiments, etoposide (20 μM) was the positive control of a reference chemotherapeutic used in clinic. The results are expressed as the average of triplicate assays.
Analysis of DNA Fragmentation
In order to study the relationship between cell proliferation inhibition and the induction of apoptosis, we decided to study the subdiploid DNA contends as indicative of DNA fragmentation by apoptosis. We selected active cytotoxic substances to one or both cell lines and evaluated their pro-apoptotic potential. We used the method to detect apoptotic nuclei, as described by Nicoletti and colleagues [
24]. This flow cytometric method is useful for measuring the percentage of apoptotic nuclei after propidium iodide staining in hypotonic buffer for assessing apoptosis. The cells were treated with 100 μM of compounds for 18 h and incubated for 4 h at 4 °C, and PI fluorescence of individual nuclei was measured by flow cytometry. The percentage of hypodiploid nuclei correlates with the extent of apoptosis in the samples. The results represent the average ± SD in triplicate samples. Every experiment was repeated at least three times. With regard to apoptosis induction, a result is considered positive when the obtained level of DNA fragmentation at least doubles the values obtained for the control cultures that were treated only with the solvent.
Caspase 3 Activation
We evaluated if the DNA fragmentation was connected with caspase-activation dependence since various anticancer drugs have been reported to induce caspase-3 activation leading to apoptosis. Therefore, we examined involvement of the principal executing caspases - caspase-3, which clearly emerged as the single most important cysteine protease during the execution phase of apoptosis. The percentage of caspase-3 dimerized (actived) was determined by a single staining with a Caspase 3-FITC Antibody from BD Biosciences to detect the quantity of the apoptotic cells. This assay permits the confirmation of involvement of this enzyme in the cell death process; that is considered to be preliminary data for the determination of the mechanism of action.
Results and Discussion
The effects of compounds
1-
12 on the growth and viability of UACC-62 and Jurkat cells were investigated and the results are summarized in
Table 1. The results are given in percent of cell growth compared to the untreated control cells (DMSO 0.1%).
Among the twelve compounds tested, four (5, 8, 11 and 12) displayed anti-proliferation effects (proliferation less than 60%) against one or both of the cancer cells lines. Compound 5 was found to be more toxic to the Jurkat cell line than to UACC-62. The results revealed the interesting effect presented by compound 11, which exhibited cytotoxicity only against UACC-62 cells. In contrast, compounds 8 and 12 presented cytotoxic activity against both cells lines. The cytotoxicity of compounds 5 and 8 might be associated to their intrinsic alkylating properties. However, compound 1 and 7, bearing benzylic bromine substituent as 5 and 8, were inactive. The low cytotoxic activity of the compounds 1 and 7 may be related to their inadequate physicochemical properties and inability to cross cells membranes. The mechanism of action of diarylfurans 11 and 12 has not yet been proposed.
Table 1.
Effects of compounds synthesized on the growth of human cancer cells lines UACC-62 and Jurkat.
Table 1.
Effects of compounds synthesized on the growth of human cancer cells lines UACC-62 and Jurkat.
| Compound
a | % Proliferation versus control |
|---|
| UACC-62 (melanoma) | Jurkat (lymphoma) |
|---|
| 1 | 105 ± 16 | 88 ± 9 |
| 2 | 117 ± 28 | 107 ± 9 |
| 3 | 104 ± 32 | 92 ± 9 |
| 4 | 99 ± 14 | 128 ± 22 |
| 5 | 84 ± 17 | 41 ± 18 |
| 6 | 106 ± 16 | 110 ± 10 |
| 7 | 106 ± 25 | 93 ± 5 |
| 8 | 39 ± 14 | 32 ± 5 |
| 9 | 110 ± 15 | 99 ± 6 |
| 10 | 87 ± 13 | 113 ± 19 |
| 11 | 25 ± 6 | 120 ± 14 |
| 12 | 60 ± 4 | 54 ± 12 |
| Cell control | 100 | 100 |
| Etoposide | 70 ± 3 | 60 ± 15 |
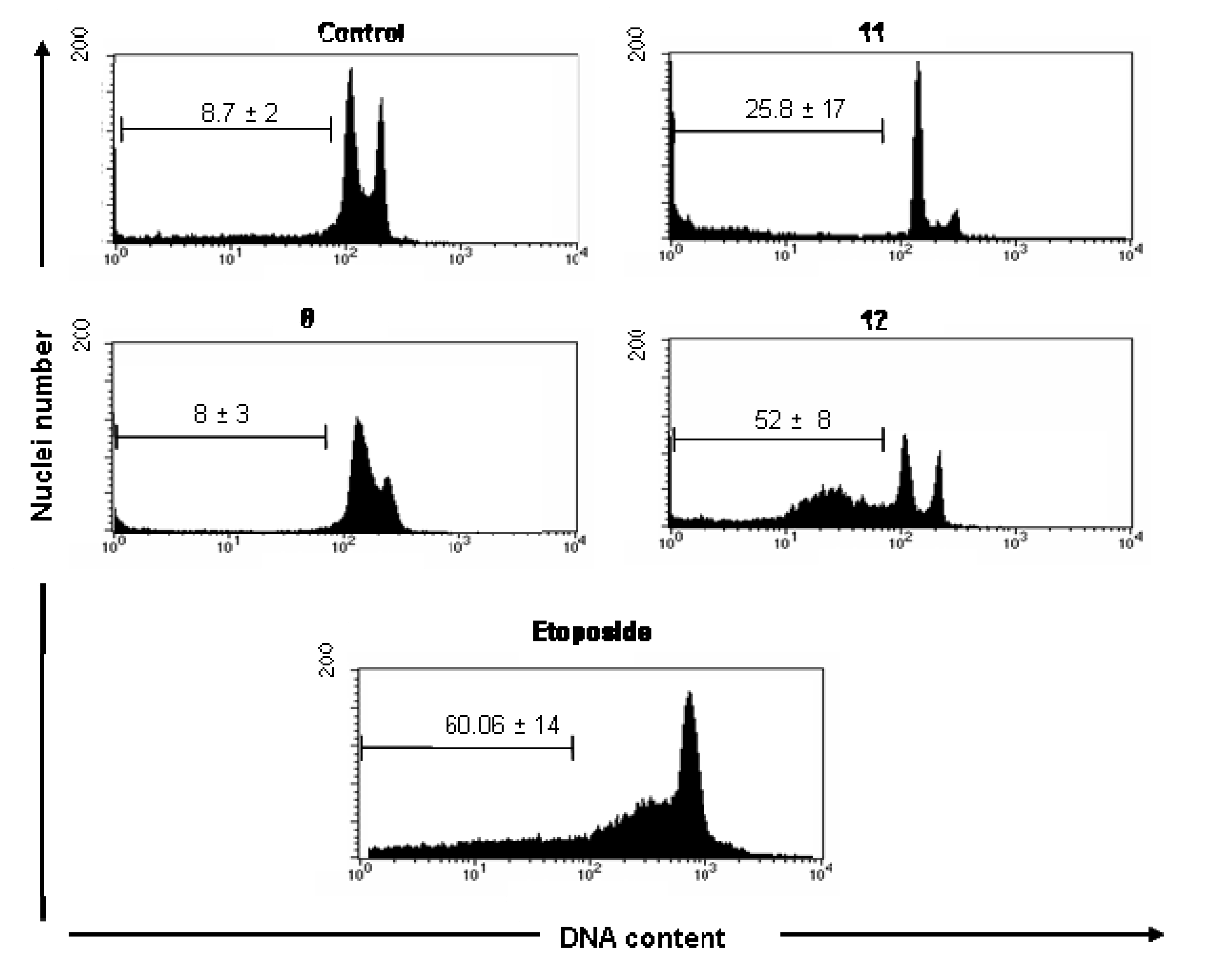
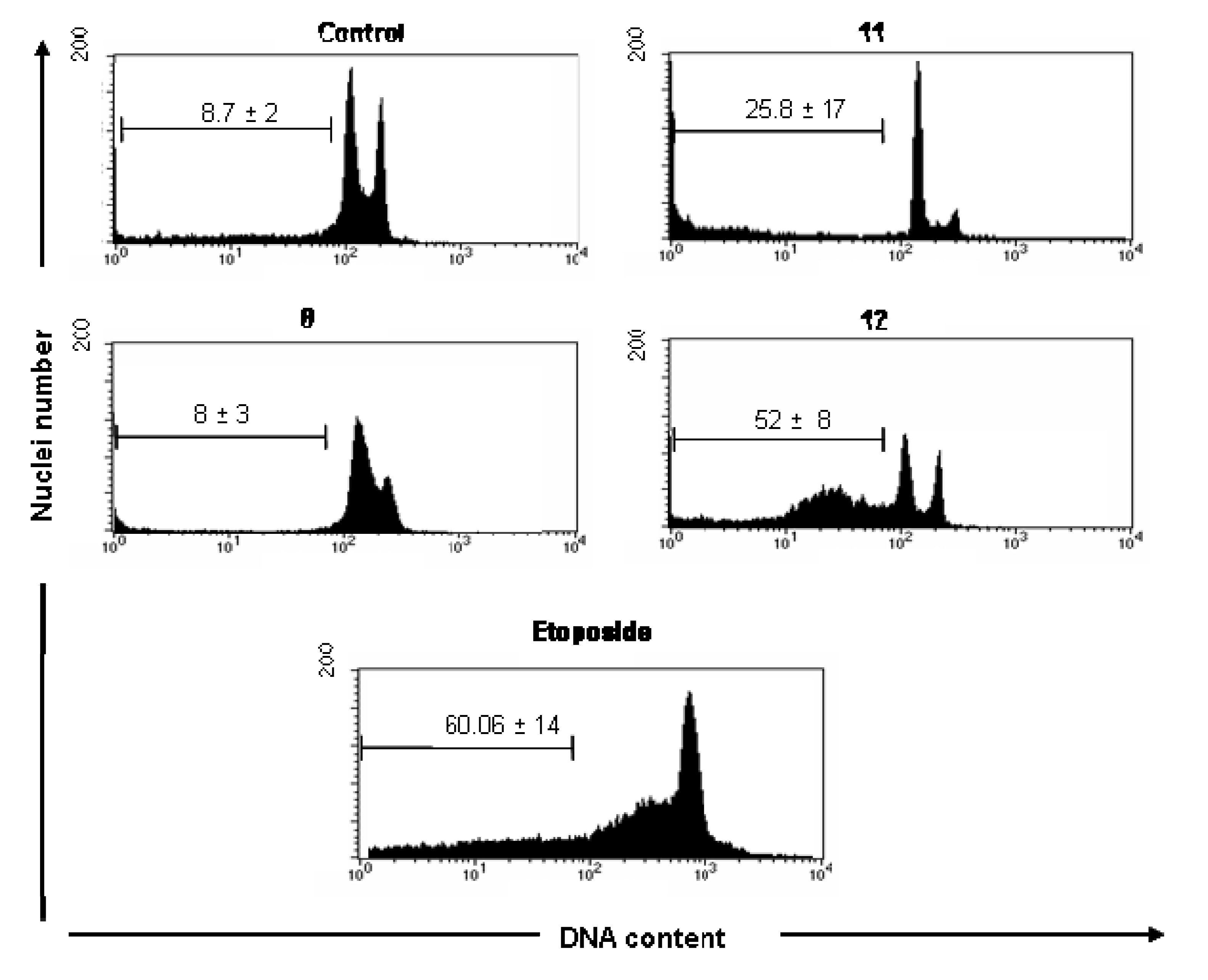
A significant increase of subdiploid DNA content in UACC-62 cells was observed following treatment with compounds
11 (25.8 ± 17) and
12 (52 ± 8), assayed by increase of sub-G1 peak (
Figure 2) when compared with cell control (8.7 ± 2% of cells). Although the compound
8 significantly inhibited the UACC-62 cell proliferation (
Table 1), it showed no significantly effect on induction of DNA fragmentation measuring the DNA subdiploid content (8 ± 4), suggesting that the cytotoxic effect of this compound probable involves other mechanism different of apoptosis. In these cells, as expected, etoposide significantly induced apoptosis (49 ± 14) after 18 h of culture.
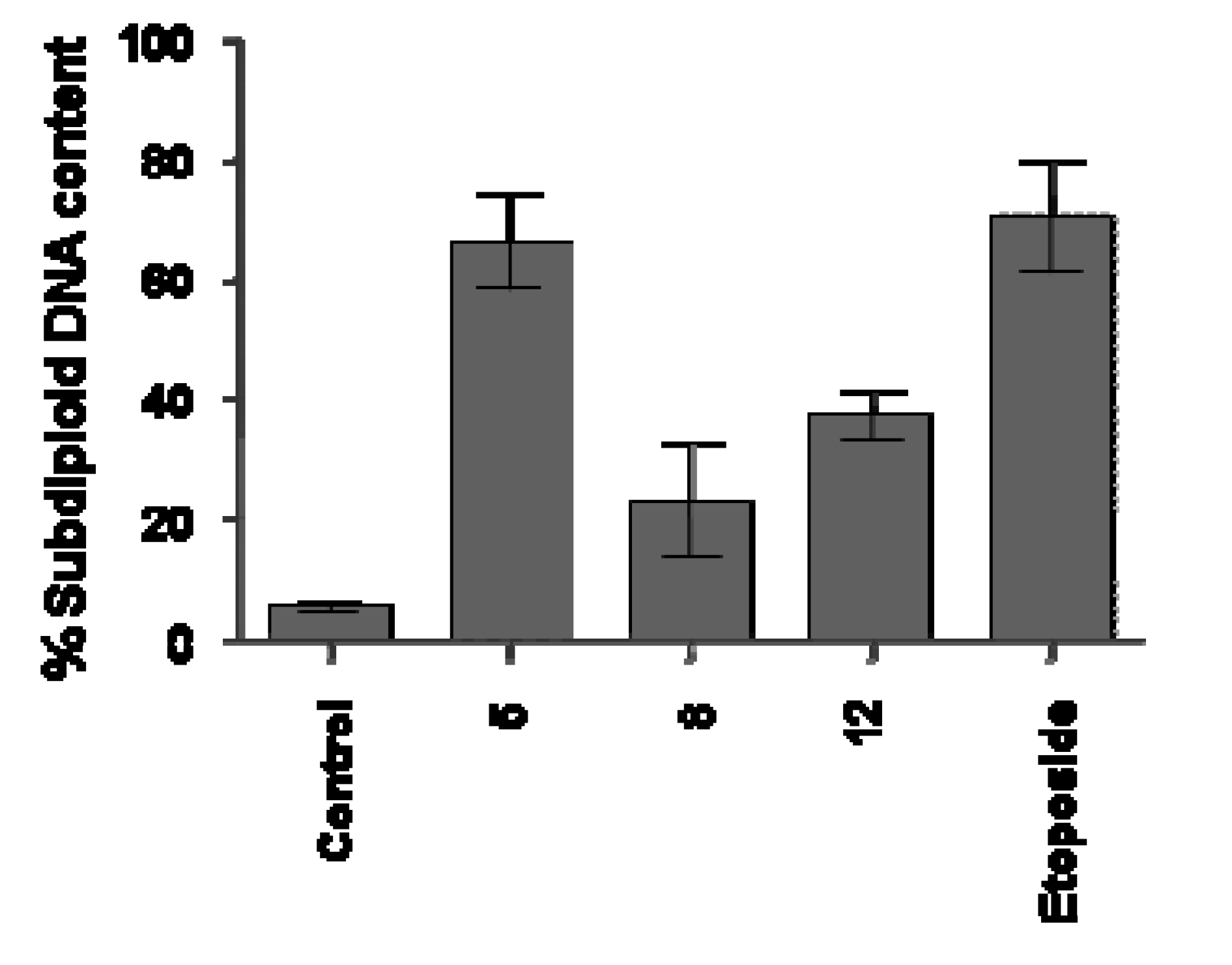
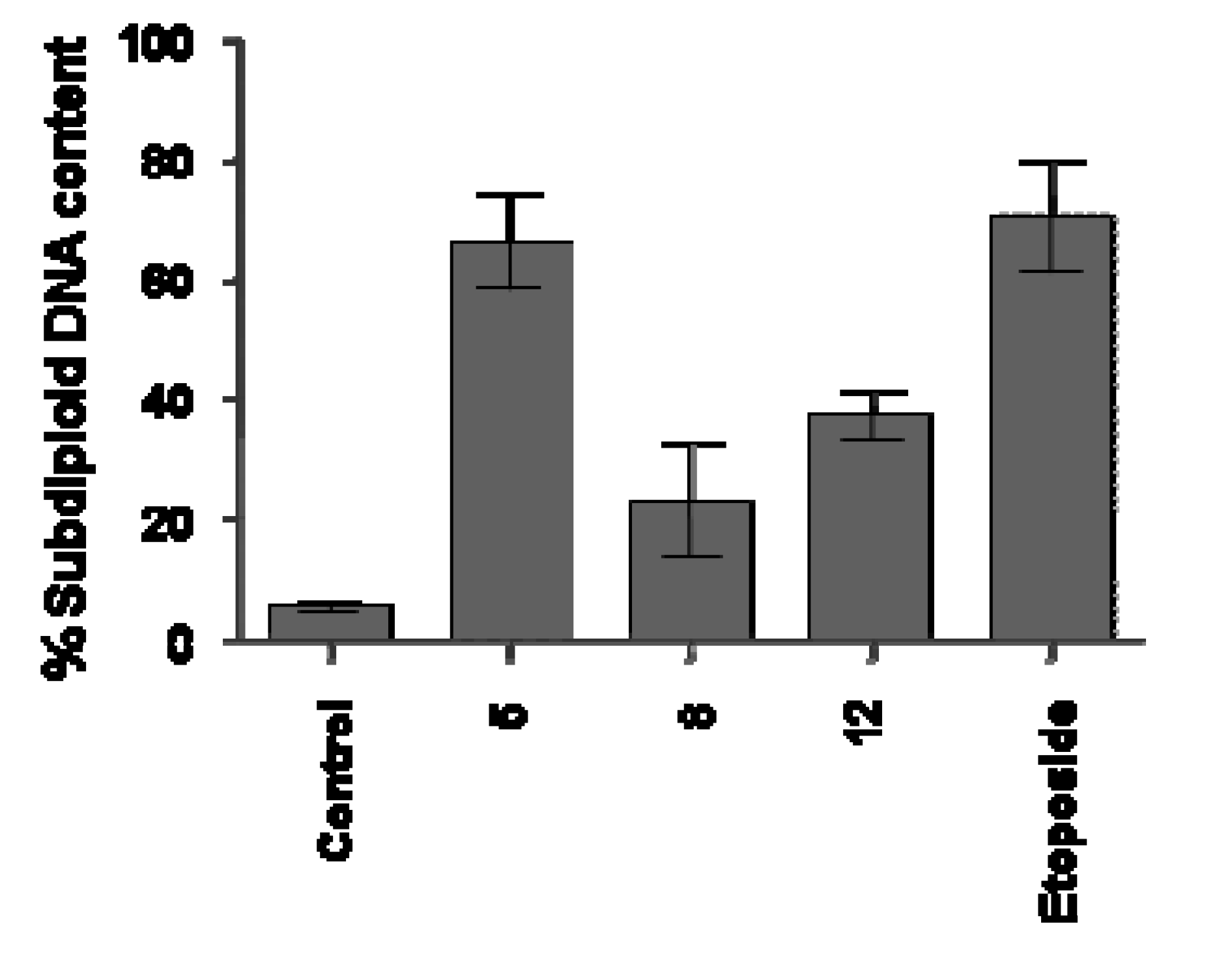
Different results were observed with Jurkat cells. Compounds
5, 8 and
12 that significantly reduced the cell proliferation (
Table 1) demonstrated different impact on DNA fragmentation induction on this line. As shown in
Figure 3, the data provided strong evidence that the reduction of proliferation in Jurkat cells after treatment with
8, as previously verified to UACC-62 cells, is not connected with apoptosis induction, as demonstrated by reduced sub diploid DNA content, after 18 h of treatment with this compound. However, the data clearly showed that compounds
5 and
12, demonstrated a significant induction of DNA fragmentation (66 ± 17.4% and 37.4 ± 5.9%, respectively) compared with the cell control (5.5 ± 1.9%). In these experiments, etoposide (73 ± 7%) used as the reference substance and known for its proapoptotic behavior, induced DNA fragmentation in a significantly way, as previously described for this chemotherapeutic agent used in clinic.
Figure 2.
Cell cycle analysis of UACC-62 cells in the absence (control, DMSO 0.5%) and presence of 100 μM of 8, 11 and 12 compounds. Logarithmic representations of fluorescence intensity obtained by PI-staining to distinguish live from apoptotic cells. Sub-G1 peaks are clearly evident after 11 and 12 treatments. Pro-apoptotic drugs, etoposide and camptothencin were used as positive control. Representative data (mean ± SD) of three experiments performed in triplicate. * Statistically different of untreated cell control (p < 0.05).
Figure 2.
Cell cycle analysis of UACC-62 cells in the absence (control, DMSO 0.5%) and presence of 100 μM of 8, 11 and 12 compounds. Logarithmic representations of fluorescence intensity obtained by PI-staining to distinguish live from apoptotic cells. Sub-G1 peaks are clearly evident after 11 and 12 treatments. Pro-apoptotic drugs, etoposide and camptothencin were used as positive control. Representative data (mean ± SD) of three experiments performed in triplicate. * Statistically different of untreated cell control (p < 0.05).
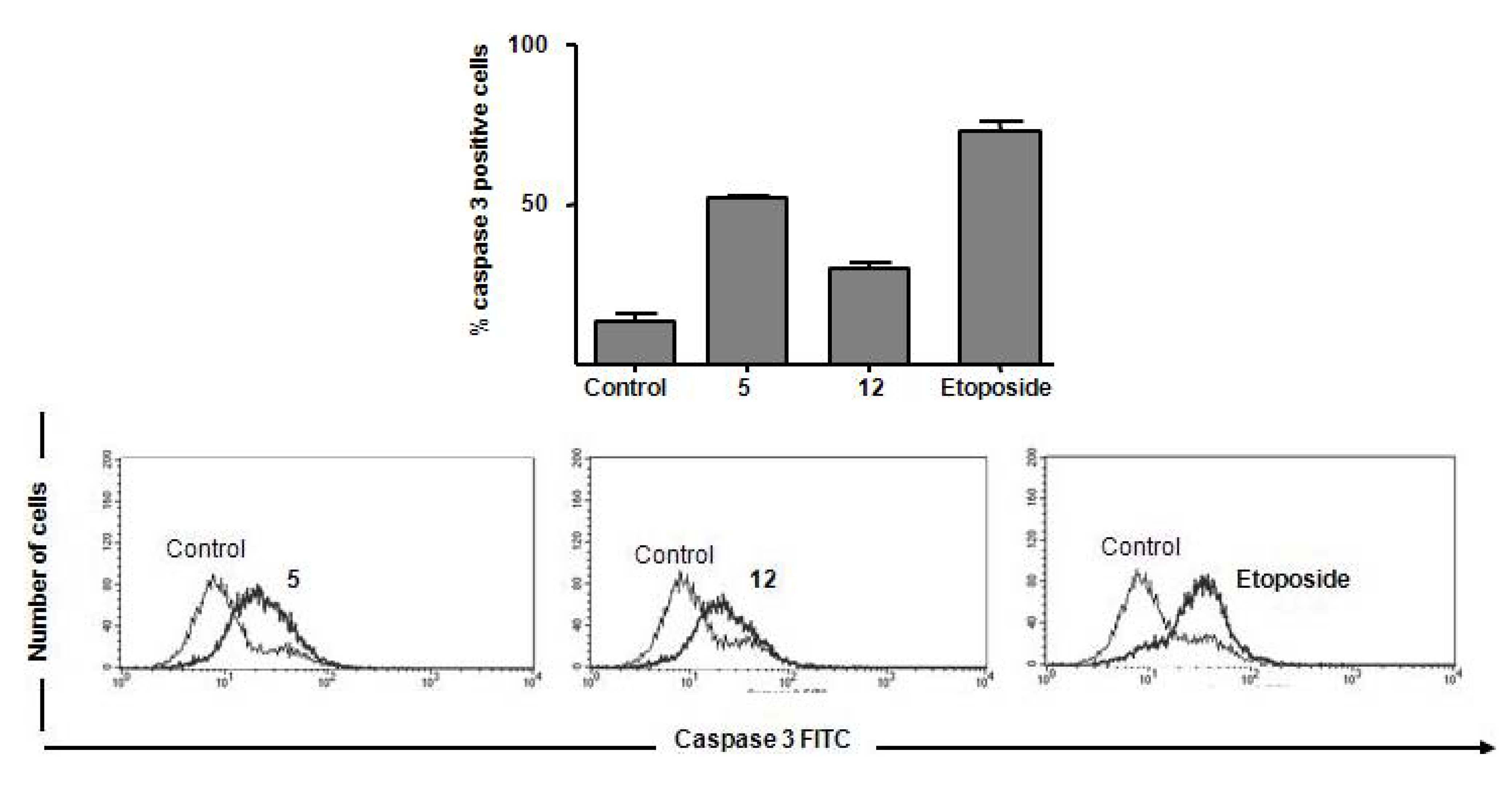
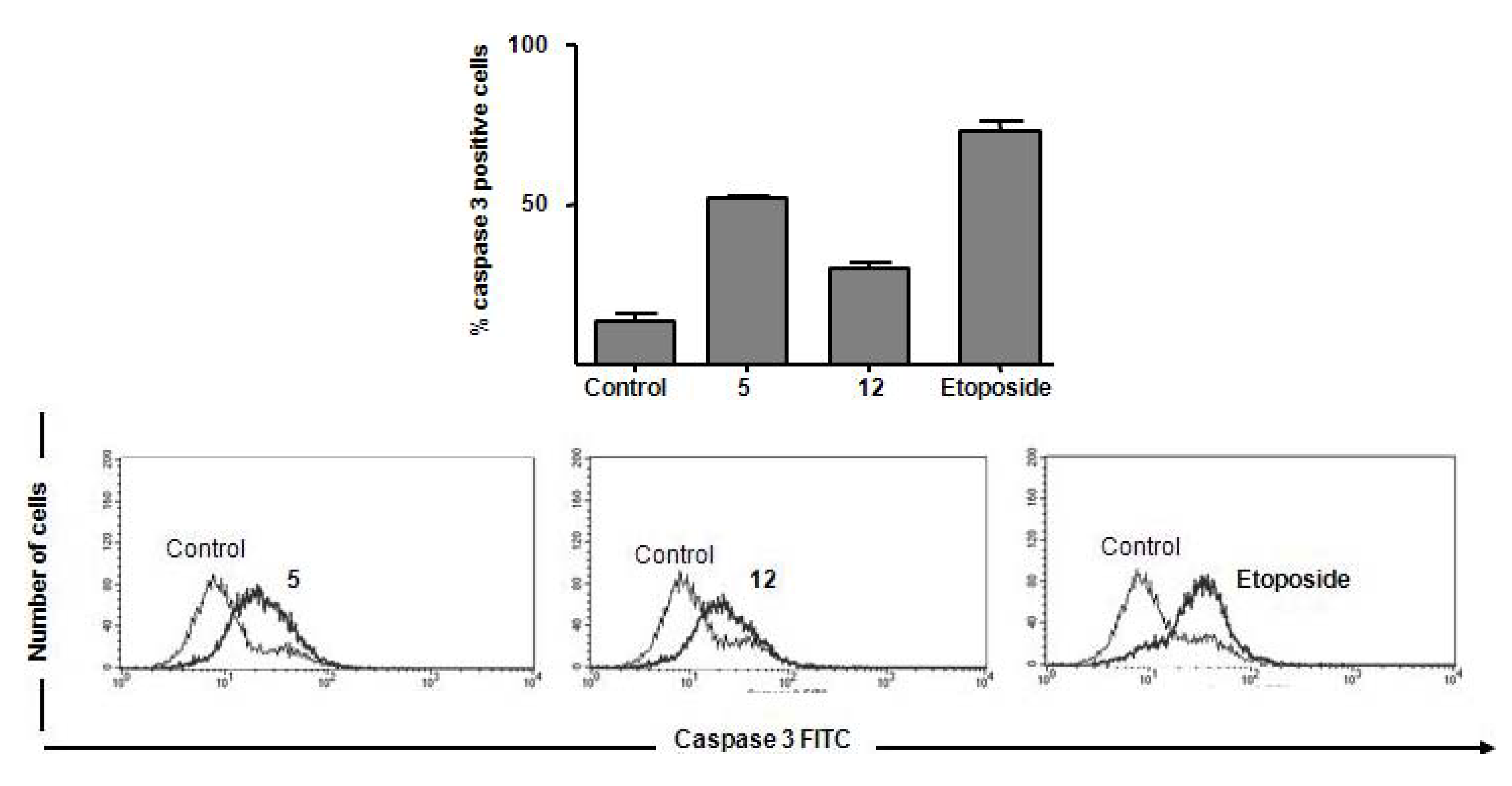
To gain insights into the mechanism by which the cytotoxic compounds induce apoptosis, we also investigated their effects on caspases. We focused on caspase-3, which is activated by a number of apoptotic signals. This enzyme is a main executor of apoptosis playing a central role in its biological processing and has been reported that activation of caspase-3 is an essential event for the induction of oligonucleosomal DNA fragmentation [
25]. Compounds
5,
8 and
12 induced an increase in the amount of subdiploid DNA, indicating internucleosomal DNA breakdown, as previously shown (
Figure 3). Using the 50th percentile greater values of DNA fragmentation induction by actives substances as a cut-off point, we investigated the substances
5 and
12 on caspase-3 activation in Jurkat cells. Flow cytometry measurements were corroborated by activation of caspase-3 observed in
5 and
12-treated Jurkat cells (
Figure 4). Substance
5 showed additional pro-apoptotic potential when compared with
12 (52 ± 1.3% of caspase-3 positive cells versus 30 ± 2.5%, respectively). Etoposide induced 76 ± 5.8% of caspase activation in Jurkat cells. Cell control (12 ± 1.5%) is represented. Etoposide exerts its antineoplastic activity by inhibiting topoisomerase II which leads to DNA strand breaks, inhibition of DNA replication, and apoptotic cell death.
Figure 3.
Flow cytometry analyses of DNA content of Jurkat cells treated with synthetic compounds 5, 8 and 12 for 18h. Cells were analyzed with the FACScan flow cytometer as described in the Experimental section.
Figure 3.
Flow cytometry analyses of DNA content of Jurkat cells treated with synthetic compounds 5, 8 and 12 for 18h. Cells were analyzed with the FACScan flow cytometer as described in the Experimental section.
One of mechanisms involved on apoptotic mechanism induced by etoposide involves the release of mitochondrial cytochrome c leading to the activation of caspase-9. Caspase-9 triggers the activation of caspase-3 [
25,
26]. Caspase 3 is the major death executioner that orchestrate the dismantling of diverse cell structures through cleavage of specific substrates including the cleavage of ICAD (inhibitor of caspase-activated DNase) releases CAD (caspase-activated DNase), which can then catalyze inter-nucleosomal DNA cleavage [
27]. Since oligonucleosomal DNA fragmentation requires activation of caspase-3 [
28], it seems reasonable to consider that the oligonucleosomal DNA fragmentation observed on Jurkat cells after treatment with substances
5 and
12 is connected with caspase-3 activation consequently with their pro-apoptotic potential.
Figure 4.
Impact of compounds 5 and 12 on caspase-3 activation. Jurkat cells were treated with compounds for 18 hours and labeled with anti-caspase 3 FITC, following flow cytometric analysis.
Figure 4.
Impact of compounds 5 and 12 on caspase-3 activation. Jurkat cells were treated with compounds for 18 hours and labeled with anti-caspase 3 FITC, following flow cytometric analysis.
Our results demonstrated that
5 and
12 have a pro-apoptotic profile at higher concentrations (100 µM). Langer and co-workers [
29] confirmed that owing to the expectation of weak potency of the primary hits, it is necessary to perform biochemical assays of the low molecular weight compounds at relatively high concentrations. Moreover, an actual sample of the presumed hit structure must demonstrate activity in a primary biological assay and an important aspect of selecting chemical series for follow-up is their binding mechanism. In this context, the substances related in the present work can be useful as hit compound to the development of leads to de development of new anticancer drugs. Experiments to determine the IC50 values, as well evaluation of the pro-apoptotic mechanism induced and by compounds and their impact on normal cell lines are currently under investigation in our laboratory.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}