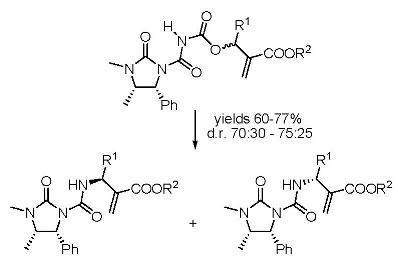
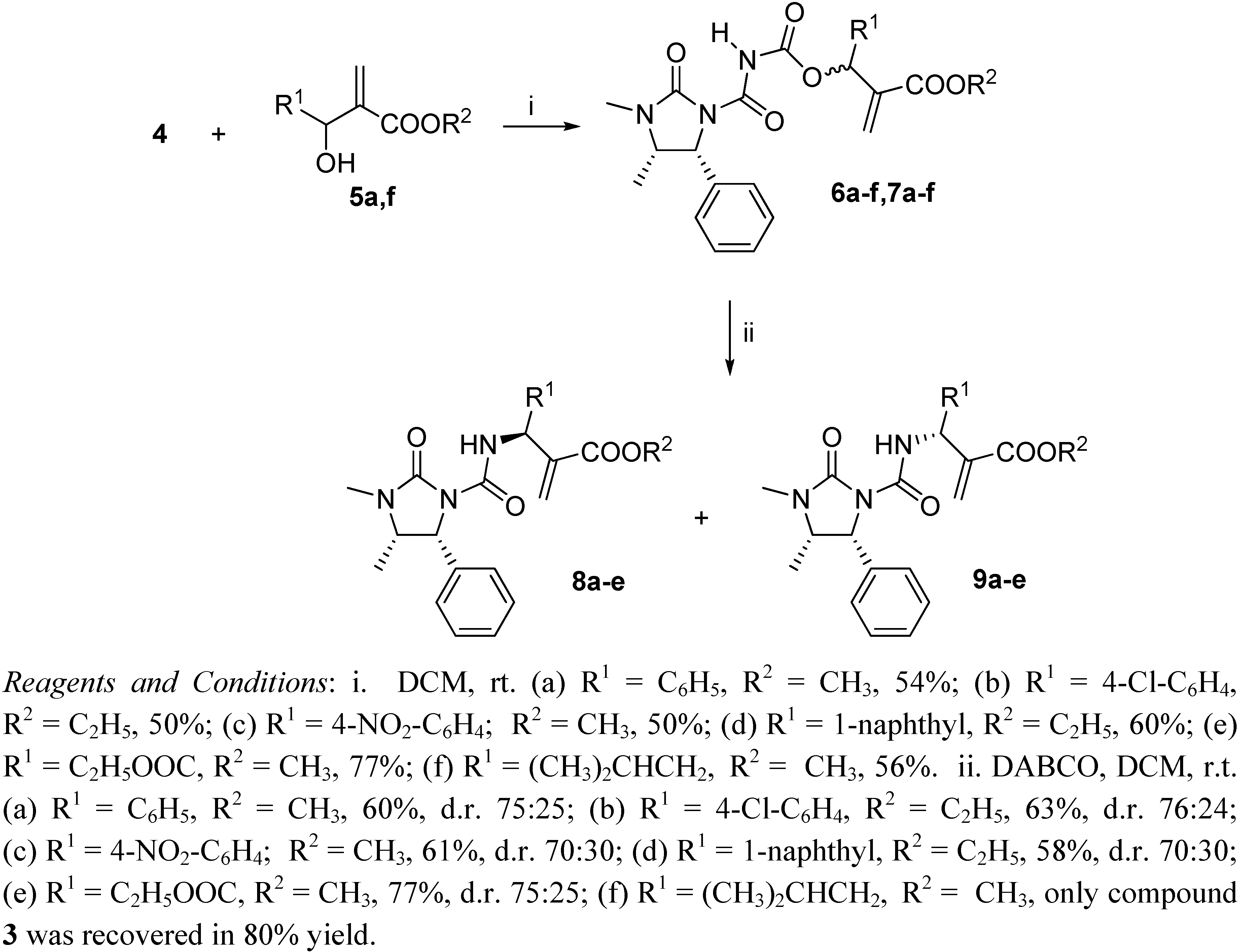
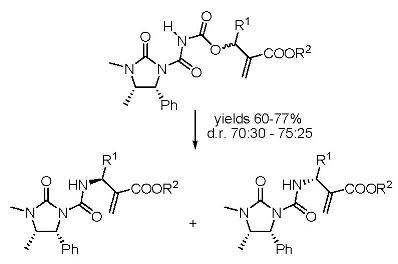
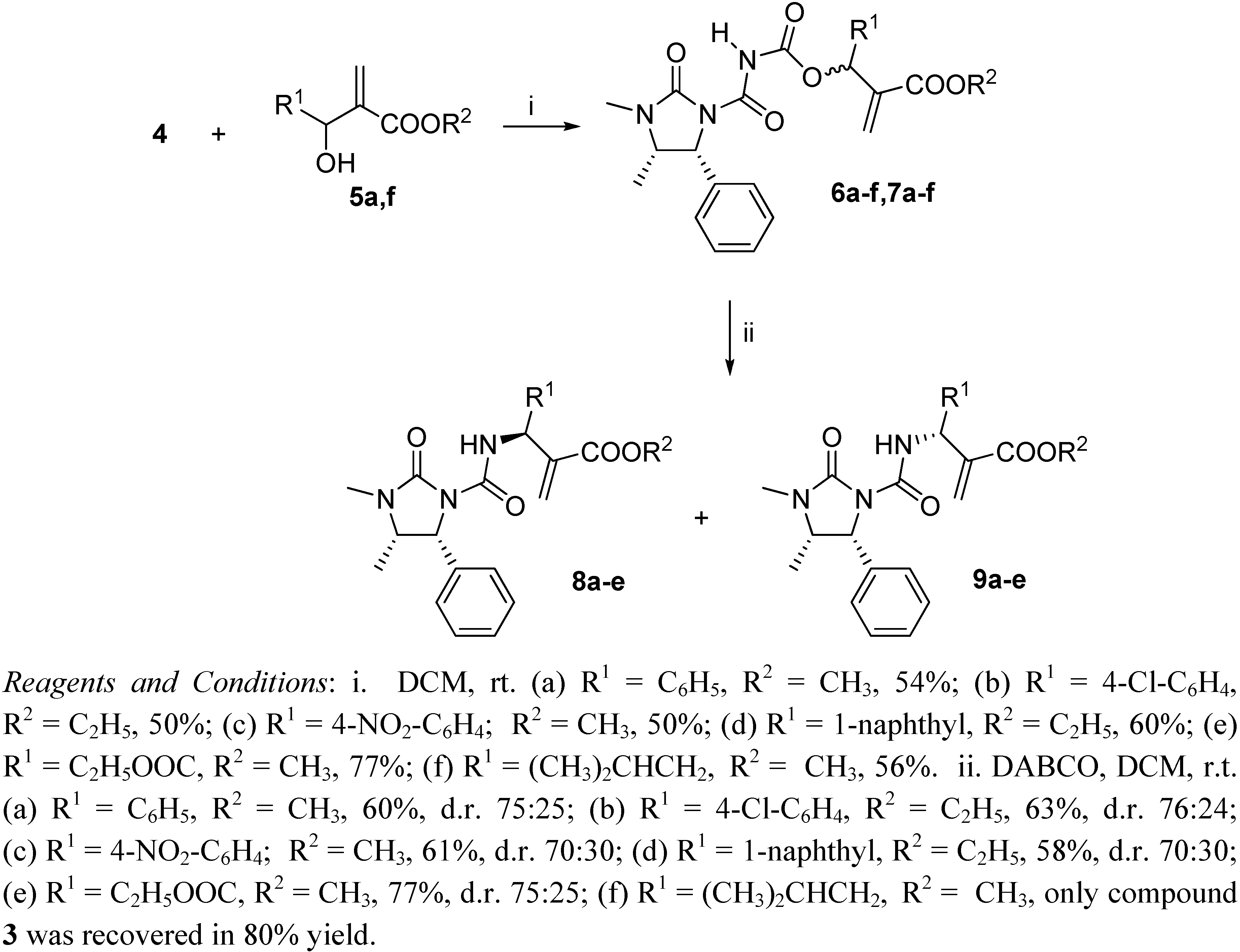
Preparation of derivatives 6 and 7
To a solution containing compound 3 (1.17 g; 5.0 mmol) in dichloroethane (10 mL), oxalyl chloride (0.66 g; 5.2 mmol) was added and the mixture was refluxed for 4 h. The solvent was partially removed and then the Baylis-Hillman adducts 5a-f (3.3 mmol) were added, dissolved in DCM (10 mL). The reaction was stirred for 12 h, volatiles were removed under reduced pressure and the products 6 and 7 were obtained as equimolar mixtures hard to separate by silica gel chromatography (cyclohexane-ethyl acetate 1:1), although small amounts could be obtained pure enough for analytical determinations.
Methyl(1'S,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carbonylcarbamoyloxy)-(phenyl)methyl]acrylate (6a) and its (1'R,4"S,5"R)-isomer 7a: The title compounds were obtained in 54% overall yield as an equimolar diastereomeric mixture. White solid; MS (ESI): m/z 451.2 [M+], 474.2 [M+Na]+; Anal. Calcd. for C24H25N3O6: C, 63.85; H, 5.58; N, 9.31; Found: C, 63.79; H, 5.54; N, 9.35. Isomer 6a: Rf 0.63 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.82 (d, J = 6.6 Hz, 3H, 4"-CH3), 2.82 (s, 3H, 3"-CH3), 3.68 (s, 3H, OCH3), 3.95 (dq, J = 6.6 Hz, J = 8.7 Hz, 1H, H-4"), 5.29 (d, J = 8.7 Hz, 1H, H-5"), 5.97 (bs, 1H, H-1'), 6.41 (s, 1H, =CH2), 6.67 (s, 1H, =CH2), 7.07 - 7.18 (m, 2 ArH), 7.24 - 7.42 (m, 8 ArH), 10.98 (s, 1H, NH); 13C-NMR: δ 14.5, 28.0, 51.9, 54.3, 59.0, 74.4, 126.2, 126.8, 126.9, 127.7, 127.8, 128.3, 128.5, 128.7, 135.6, 137.3, 139.0, 147.2, 149.3, 156.8, 165.2. Isomer 7a: Rf 0.58 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.82 (d, J = 6.6, 3H, 4"-CH3), 2.84 (s, 3H, 3"-CH3), 3.68 (s, 3H, OCH3), 3.96 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 5.31 (d, J = 8.8 Hz, 1H, H-5"), 5.97 (bs s, 1H, H-1'), 6.41 (s, 1H, =CH2), 6.68 (s, 1H, =CH2), 7.07 - 7.18 (m, 2 ArH), 7.23 - 7.44 (m, 8 ArH), 10.97 (s, 1H, NH); 13C-NMR: δ 14.6, 28.0, 51.9, 54.3, 59.1, 74.4, 126.0, 126.8, 126.9, 127.8, 128.4, 128.5, 128.6, 135.6, 139.0, 147.3, 149.3, 156.9, 165.2.
Ethyl(1'S,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carbonylcarbamoy-loxy)(4-chlorophenyl)methyl]acrylate (6b) and its (1'R,4"S,5"R)-isomer 7b: The title compounds were obtained in 50% overall yield as an equimolar diastereomeric mixture. White solid; MS (ESI): m/z 499.2 [M]+, 522.2 [M+Na]+; Anal. Calcd. for C25H26ClN3O6: C, 60.06; H, 5.24; N, 8.40; Found: C, 60.02; H, 5.19; N, 8.35. Isomer 6b: Rf 0.67 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.82 (d, J = 6.7 Hz, 3H, 3"-CH3), 1.19 (t, J = 7.2 Hz, 3H, CH3CH2O), 2.83 (s, 3H, 3"-CH3), 3.96 (dq, J = 6.7 Hz, J = 8.8 Hz, 1H, H-4"), 4.13 (q, J = 7.2 Hz, 2H, CH3CH2O), 5.29 (d, J = 8.8 Hz, 1H, H-5"), 5.98 (s, 1H, H-1'), 6.42 (s, 1H, =CH2), 6.62 (s, 1H, =CH2), 7.07 - 7.17 (m, 2 ArH), 7.21 - 7.86 (m, 8 ArH), 11.00 (s, 1H, NH); 13C-NMR: δ 13.9, 14.6, 28.0, 54.4, 59.1, 61.0, 73.9, 126.0, 126.9, 128.4, 128.5, 128.6, 129.3, 134.3, 135.6, 136.1, 136.9, 147.2, 149.2, 156.9, 164.6. Isomer 7b: Rf 0.61 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.83 (d, J = 6.6 Hz, 3H, 3"-CH3), 1.23 (t, J = 7.1 Hz, 3H, CH3CH2O), 2.84 (s, 3H, 3"-CH3), 3.97 (dq, J = 6.6 Hz, J = 8.7 Hz, 1H, H-4"), 4.13 (q, J = 7.1 Hz, 2H, CH3CH2O), 5.30 (d, J = 8.7 Hz, 1H, H-5"), 5.97 (s, 1H, H-1'), 6.41 (s, 1H, =CH2), 6.63 (s, 1H, =CH2), 7.05 - 7.17 (m, 2 ArH), 7.21 - 7.38 (m, 8 ArH), 10.99 (s, 1H, NH); 13C-NMR: δ 14.0, 14.6, 28.1, 54.4, 59.2, 61.0, 73.9, 125.9, 126.9, 128.4, 128.6, 129.4, 134.4, 135.6, 136.0, 139.0, 147.3, 149.3, 156.9, 164.6.
Methyl (1'S,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carbonylcarbamoyloxy)-(4-nitrophenylmethyl)]acrylate (6c) and its (1'R,4"S,5"R)-isomer (7c): The title compounds were obtained in 50% overall yield as an equimolar diastereomeric mixture. White solid; MS (ESI): m/z 496.2 [M]+, 519.2 [M+Na]+; Anal. Calcd. for C24H24N4O8: C, 58.06; H, 4.87; N, 11.28; Found: C, 58.00; H, 4.84; N, 11.33. Isomer 6c: Rf 0.56 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.83 (d, J = 6.6 Hz, 3H, 4"-CH3), 2.84 (s, 3H, 3"-CH3), 3.69 (s, 3H, OCH3), 3.97 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 5.30 (d, J = 8.8 Hz, 1H, H-5"), 6.08 (d, J = 1.3 Hz, 1H, H-1'), 6.47 (s, 1H, =CH2), 6.70 (s, 1H, =CH2), 7.08 - 7.18 (m, 2 ArH), 7.25 - 7.35 (m, 3 ArH), 7.58 (d, J = 8.9 Hz, 2 ArH), 8.16 (d, J = 8.9 Hz, 2 ArH), 11.09 (s, 1H, NH); 13C-NMR: δ 14.6, 26.9, 28.1, 52.1, 54.4, 59.2, 73.5, 123.7, 126.9, 127.3, 128.5, 128.7, 135.6, 138.1, 144.7, 147.2, 147.9, 149.2, 156.9, 164.8. Isomer 7c: Rf 0.49 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.83 (d, J = 6.6 Hz, 3H, 4"-CH3), 2.84 (s, 3H, 3"-CH3), 3.69 (s, 3H, OCH3), 3.98 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 5.30 (d, J = 8.8 Hz, 1H, H-5"), 6.06 (d, J = 1.1 Hz, 1H, H-1'), 6.46 (s, 1H, =CH2), 6.71 (s, 1H, =CH2), 7.08 - 7.18 (m, 2 ArH), 7.21 - 7.35 (m, 3 ArH), 7.58 (d, J = 8.8 Hz, 2 ArH), 8.17 (d, J = 8.8 Hz, 2 ArH), 11.09 (s, 1H, NH); 13C-NMR: δ 14.6, 26.9, 28.1, 52.1, 54.4, 59.2, 73.5, 123.7, 126.8, 126.9, 127.2, 127.8, 128.1, 128.5, 128.7, 135.5, 138.1, 144.6, 147.2, 147.8, 149.2, 156.9, 164.8.
Ethyl (1'S,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carbonylcarbamoyloxy)-(naphthalen-1-ylmethyl)]acrylate (6d) and its (1'R,4"S,5"R)-isomer (7d): The title compounds were obtained in 60% overall yield as an equimolar diastereomeric mixture. White solid; MS (ESI): m/z 515.2 [M]+, 538.2 [M+Na]+; Anal. Calcd. for C29H29N3O6: C, 67.56; H, 5.67; N, 8.15; Found: C, 67.51; H, 5.62; N, 8.19. Isomer 6d: Rf 0.62 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.80 (d, J = 6.6 Hz, 3H, 4"-CH3), 1.17 (t, J = 7.1 Hz, 3H, CH3CH2O), 2.81 (s, 3H, 3"-CH3), 3.92 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 4.12 (q, J = 7.1 Hz, 2H, CH3CH2O), 5.28 (d, J = 8.8 Hz, H-5"), 6.02 (d, J = 1.3 Hz, 1H, H-1'), 6.46 (s, 1H, =CH2), 6.85 (s, 1H, =CH2), 7.09 - 7.15 (m, 2 ArH), 7.21 - 7.38 (m, 3ArH), 7.40 - 7.51 (m, 3ArH), 7.74 - 7.87 (m, 4ArH), 11.02 (s, 1H, NH); 13C-NMR: δ 14.0, 14.6, 26.9, 28.0, 54.4, 59.1, 60.9, 74.7, 125.3, 126.1, 126.3, 127.0, 127.4, 127.6, 128.1, 128.2, 128.4, 128.6, 133.0, 133.3, 134.8, 135.7, 139.3, 147.3, 149.4, 156.9, 164.8. Isomer 7d: Rf 0.56 (cyclohexane-AcOEt 1:1); 1H- NMR: δ 0.76 (d, J = 6.6 Hz, 3H, 4"-CH3), 1.18 (t, J = 7.1 Hz, 3H, CH3CH2O), 2.79 (s, 3H, 3"-CH3), 3.90 (dq, J= 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 4.12 (q, J = 7.1 Hz, 2H, CH3CH2O), 5.28 (d, J = 8.8 Hz, 1H, H-5"), 6.05 (s, 1H, H-1'), 6.47 (s, 1H, =CH2), 6.88 (s, 1H, =CH2), 7.05 -7.16 (m, 2 ArH), 7.20 - 7.32 (m, 3 ArH), 7.40 - 7.55 (m, 3 ArH), 7.75 - 7.91 (m, 4 ArH), 11.07 (s, 1H, NH); 13C-NMR: δ 13.8, 14.4, 26.8, 27.9, 54.2, 59.0, 60.8, 74.6, 125.2, 125.8, 126.0, 126.2, 126.8, 127.3, 127.5, 128.1, 128.2, 128.4, 132.9, 133.2, 134.6, 135.6, 139.2, 147.2, 149.3, 156.7, 164.6.
Methyl (1'R,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carbonylcarbamoyloxy)-(ethoxycarbonylmethyl]acrylate (6e) and its (1'S,4"S,5"R)-isomer (7e): The title compounds were obtained in 77% overall yield as an equimolar diastereomeric mixture. White solid; MS (ESI): m/z 447.2 [M]+, 470.2 [M+Na]+; Anal. Calcd. for C21H25N3O8: C, 56.37; H, 5.63; N, 9.39; Found: C, 56.33; H, 5.57; N, 9.43. Isomer 6e: Rf 0.57 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.80 (d, J = 6.6, 3H, 4"-CH3), 1.21 (t, J = 7.0 Hz, 3H, CH3CH2O), 2.80 (s, 3H, 3"-CH3), 3.77 (s, 3H, OCH3), 3.93 (dq, J = 6.6, J = 8.7, 1 H, H-4"), 4.17 (q, J = 7.0 Hz, 2H, CH3CH2O), 5.29 (d, J = 8.7, 1H, H-5"), 5.94 (s, 1H, H-1'), 6.00 (s, 1H, =CH2), 6.47 (s, 1H, =CH2), 7.06 – 7.21 (m, 3 ArH), 7.22 – 7.93 (m, 7 ArH + NH); + NH); 13C-NMR:: δ 14.5, 28.0, 51.9, 54.3, 59.0, 74.4, 126.2, 126.9, 127.7, 127.8, 128.3, 128.5, 128.7, 135.6, 137.3, 139.0, 147.2, 149.3, 156.8, 165.2. Isomer 7e: Rf 0.51 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.83 (d, J = 6.6, 3H, 4"-CH3), 1.22 (t, J = 7.0 Hz, 3H, CH3CH2O), 2.81 (s, 3H, 3"-CH3), 3.76 (s, 3H, OCH3), 3.95 (dq, J = 6.6, J = 8.8, 1H, H-4"), 4.19 (q, J = 7.0 Hz, 2H, CH3CH2O), 5.31 (d, J = 8.8, 1H, H-5"), 5.95 (s, 1H, H-1'), 5.99 (s, 1H, =CH2), 6.48 (s, 1H, =CH2), 7.07 – 7.20 (m, 3 ArH), 7.26 – 7.47 (m, 7 ArH + NH); 13C-NMR: δ 14.6, 28.0, 51.9, 54.3, 59.1, 74.4, 126.0, 126.8, 126.9, 127.8, 128.4, 128.5, 128.6, 135.6, 139.0, 147.3, 149.3, 156.8, 165.2.
Methyl (3S,4'S,5'R)-3-(3',4'-dimethyl-2'-oxo-5'-phenylimidazolidine-1'-carbonylcarbamoyloxy)-5-methyl-2-methylenehexanoate (6f) and its (3R,4'S,5'R)-isomer (7f): The title compounds were obtained in 56% overall yield as an equimolar diastereomeric mixture. White solid; MS (ESI): m/z 431.2 [M]+, 454.2 [M+Na]+; Anal. Calcd. for C22H29N3O6: C, 61.24; H, 6.77; N, 9.74. Found: C, 61.18; H, 6.73; N, 9.69. Isomer 6f: Rf 0.53 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.81 (d, J = 6.6, 3H, CH3), 0.90 (d, J = 6.6, 3H, CH3), 0.93 (d, J = 6.6, 3H, 4'-CH3), 1.45 – 1.73 (m, 3H, CH-CH2), 2.83 (s, 3H, 3'-CH3), 3.73 (s, 3H, OCH3), 3.96 (dq, J = 6.6, J = 8.8, 1H, H-4'), 5.30 (d, J = 8.8, 1H, H-5'), 5.66 (dd, J = 4.0, J = 8.8, 1H, H-3), 5.82 (s, 1H, =CH2), 6.25 (s, 1H, =CH2), 7.09 – 7.18 (m, 3 ArH), 7.26 – 7.35 (m, 2 ArH + NH); 13C NMR (50 MHz, CDCl3) δ 14.5, 21.6, 23.1, 24.7, 26.8, 29.1, 43.7, 51.8, 54.3, 59.1, 72.0, 125.3, 126.9, 128.3, 128.5, 135.7, 140.3, 147.3, 149.7, 156.9, 165.5. Isomer 7f: Rf 0.44 (cyclohexane-AcOEt 1:1); 1H-NMR: δ 0.79 (d, J = 6.6, 3H, CH3), 0.88 (d, J = 5.3, 3H, CH3), 0.91 (d, J = 5.3, 3H, 4'-CH3), 1.41 - 1.71 (m, 3H, CH-CH2), 2.81 (s, 3H, 3'-CH3), 3.73 (s, 3H, OCH3), 3.95 (dq, J = 5.3, J = 8.8, 1H, H-4'), 5.28 (d, J = 8.8, 1H, ), 5.65 (dd, J = 3.3, J = 8.9, 1H, H-3), 5.79 (s, 1H, =CH2), 6.24 (s, 1H, =CH2), 7.07 – 7.15 (m, 3 ArH), 7.23 – 7.35 (m, 2 ArH + NH); 13C-NMR: δ 14.6, 21.5, 23.1, 24.6, 26.8, 27.9, 43.7, 51.7, 54.3, 59.0, 71.8, 125.0, 126.9, 128.2, 128.5, 135.7, 140.4, 147.3, 149.6, 158.9, 165.5.
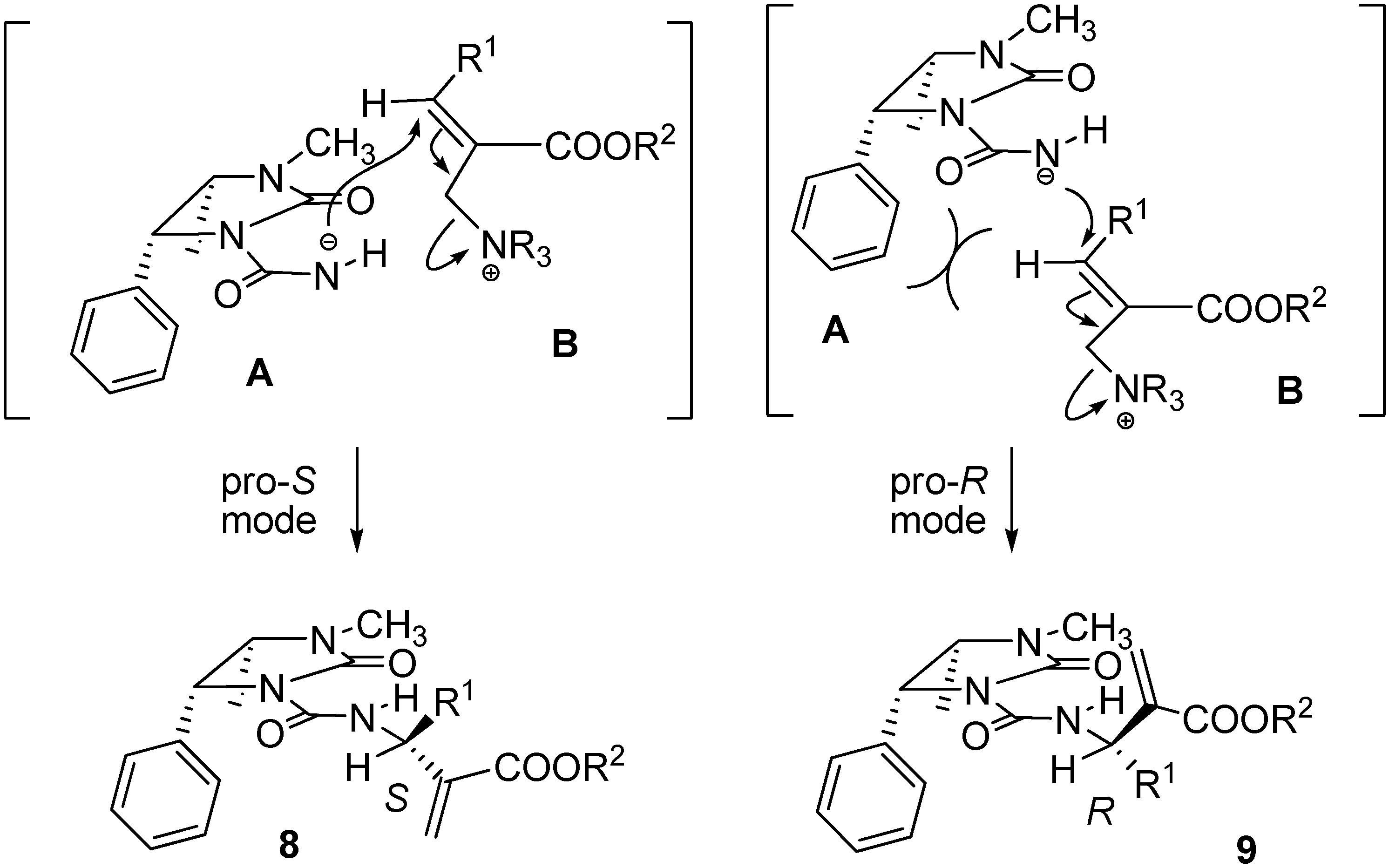
Preparation of compounds 8 and 9
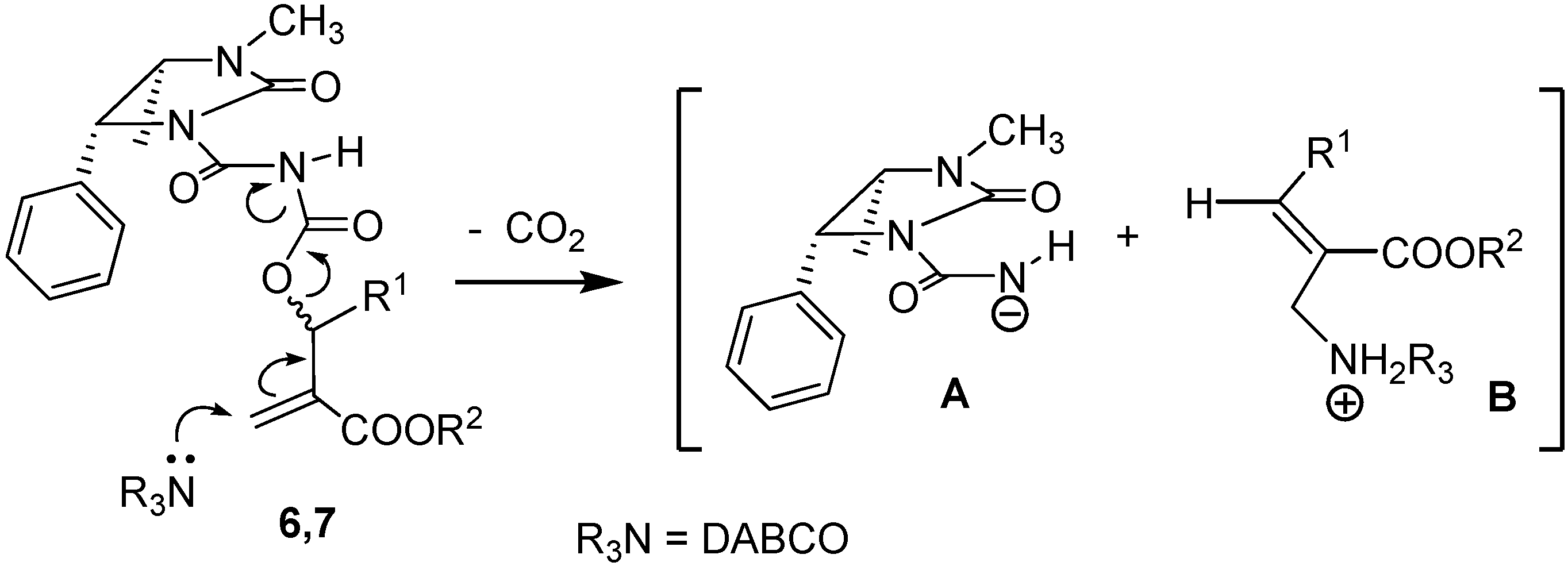
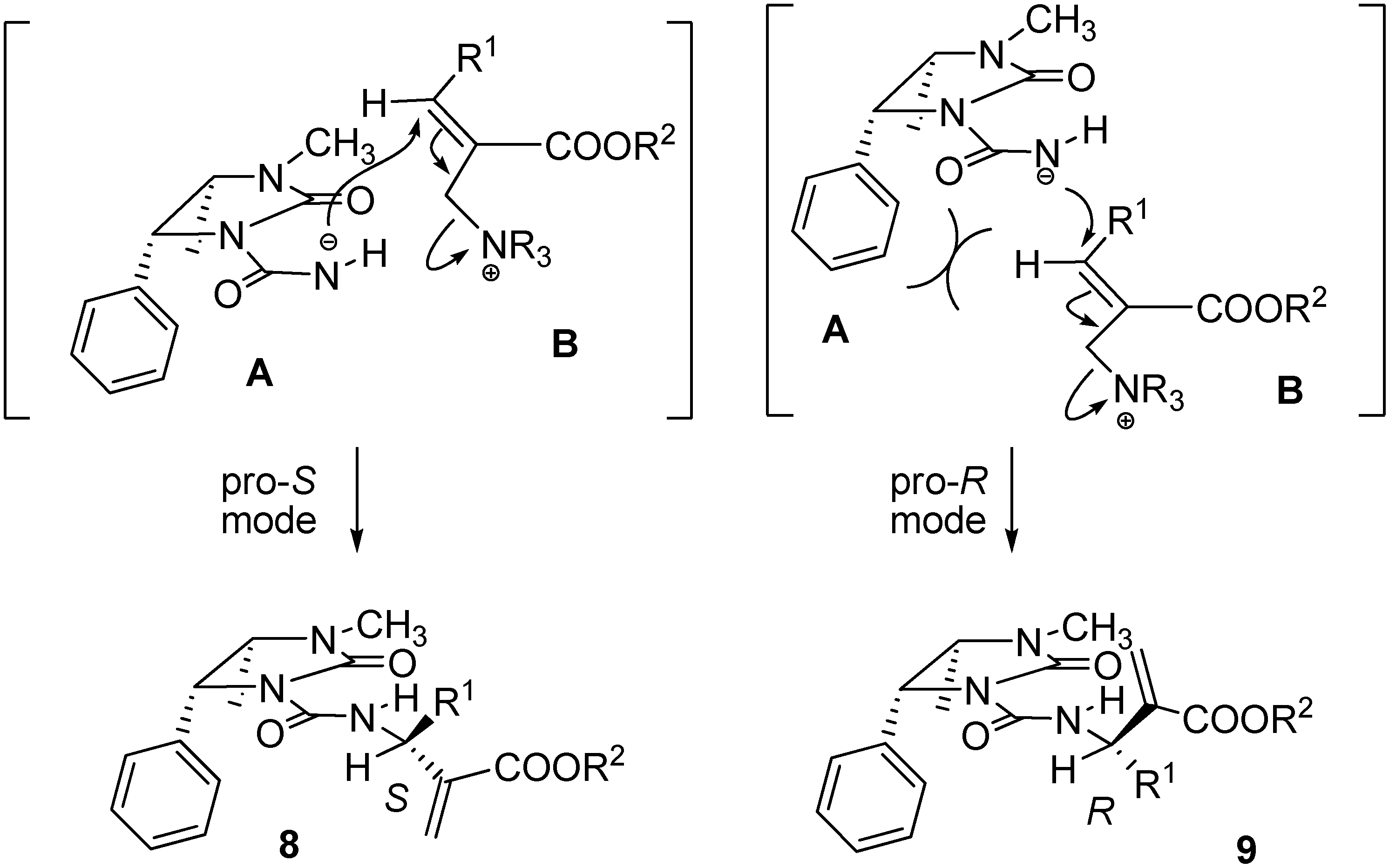
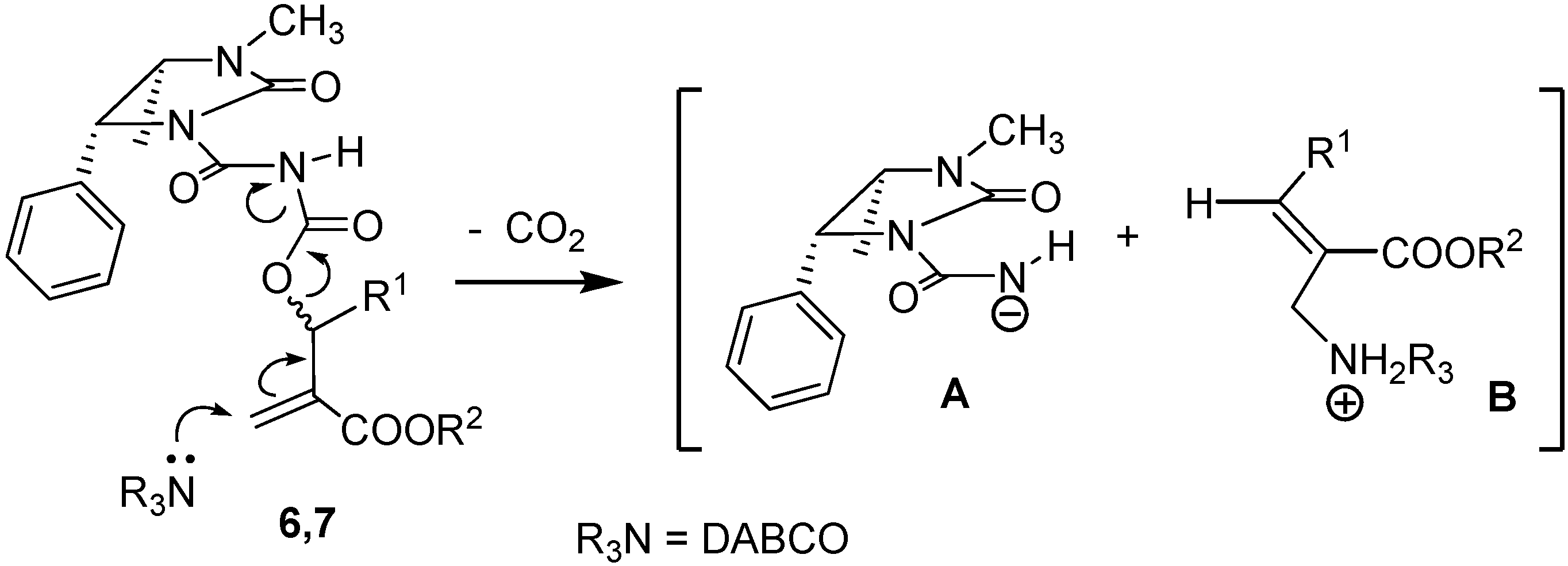
To a solution containing an equimolar mixture of acyl carbamates 6 and 7 (3.0 mmol) in dry DCM (8.0 mL), DABCO (0.07 g; 0.6 mmol) was slowly added at r.t. After 20 min, 2 M HCl (10 mL) was added and the mixture was extracted with DCM (2 × 35 mL). After drying (Na2SO4) and removal of the volatiles, the residue was purified by silica gel chromatography (cyclohexane:ethyl acetate 60:40), to give pure compounds 8 and 9.
Methyl (1'S,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carboxamido)(phenyl)-methyl]acrylate (8a) and its (1'R,4"S,5"R)-isomer (9a): The title compounds were obtained in 60% overall yield as 75:25 diastereomeric mixture. MS (ESI): m/z 407.2 [M]+, 430.2 [M+Na]+; Anal. Calcd. for C23H25N3O4: C, 67.80; H, 6.18; N, 10.31; Found: C, 67.84; H, 6.13; N, 10.27. Isomer 8a: Rf 0.47 (cyclohexane-AcOEt 1:1); White crystals; Mp 50-52 °C; 1H-NMR: δ 0.80 (d, J = 6.6 Hz, 3H, 4"-CH3), 2.80 (s, 3H, 3"-CH3), 3.64 (s, 3H, OCH3), 3.91 (dq, J = 6.6 Hz, J= 8.8 Hz, 1H, H-4"), 5.26 (d, J = 8.7 Hz, 1H, H-1'), 5.82 (s, 1H, =CH2), 5.89 (d, J = 8.8 Hz, 1H, H-5"), 6.29 (s, 1H, =CH2), 7.11 - 7.19 (m, 2 ArH), 7.20 - 7.41 (m, 8 ArH), 9.21 (d, J = 8.7, 1H, NH); 13C-NMR: δ 14.7, 26.9, 28.0, 51.8, 54.5, 54.8, 59.3, 125.7, 126.8, 126.9, 127.4, 128.0, 128.5, 136.8, 140.1, 140.3, 151.6, 157.9, 165.9; [α]D -122.0 (c 0.5, CHCl3). Isomer 9a: Rf 0.27 (cyclohexane-AcOEt 1:1); White crystals; Mp 123-125 °C; 1H-NMR: δ 0.81 (d, J = 6.6 Hz, 3H, 4"-CH3), 2.81 (s, 3H, 3"-CH3), 3.66 (s, 3H, OCH3), 3.94 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 5.28 (d, J = 8.5 Hz, 1H, H-1'), 5.89 (d, J = 8.8 Hz, 1H, H-5"), 5.91 (s, 1H, =CH2), 6.36 (s, 1H, =CH2), 7.08 - 7.17 (m, 2 ArH), 7.20 - 7.39 (m, 8 ArH), 9.15 (d, J = 8.5 Hz, 1H, NH); 13C- NMR: δ 14.8, 26.9, 28.0, 51.8, 54.6, 54.9, 59.4, 126.1, 126.7, 126.8, 127.0, 127.3, 127.8, 128.1, 128.3, 128.5, 128.6, 136.8, 139.6, 140.6, 151.8, 157.9, 165.9; [α]D 93.0 (c 0.5, CHCl3).
Ethyl (1'S,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carboxamido)(4-chloro-phenyl)methyl]acrylate (8b) and its (1'R,4"S,5"R)-isomer (9b): The title compounds were obtained in 63% overall yield as 76:24 diastereomeric mixture. MS (ESI): m/z 455.2 [M] +, 478.2 [M+Na]+; Anal. Calcd. for C24H26ClN3O4: C, 63.22; H, 5.75; N, 9.22; Found: C, 63.17; H, 5.71; N, 9.26. Isomer 8b: Rf 0.47 (cyclohexane-AcOEt 1:1); White crystals; Mp 46-48 °C; 1H-NMR: δ 0.80 (d, J = 6.6 Hz, 3H, 4"-CH3), 1.16 (t, J = 7.2 Hz, 3H, CH3CH2O), 2.80 (s, 3H, 3"-CH3), 3.94 (dq, J = 6.6 Hz, J= 8.8 Hz, 1H, H-4"), 4.09 (50% q, J = 7.2 Hz, 2H, CH3CH2O), 4.10 (50% q, J = 7.2 Hz, 2H, CH3CH2O), 5.26 (d, J = 8.3 Hz, 1H, H-1'), 5.81 (s, 1H, =CH2), 5.86 (d, J = 8.8 Hz, 1H, H-5"), 6.31 (s, 1H, =CH2), 7.10 - 7.18 (m, 2 ArH), 7.22 - 7.38 (m, 7 ArH), 9.22 (d, J = 8.3 Hz, 1H, NH); 13C-NMR: δ 13.9, 14.8, 26.9, 28.1, 54.4 (50%), 54.6 (50%), 59.3, 61.0, 125.9, 126.8, 128.1, 128.3, 128.4, 128.5, 128.6, 133.2, 136.8, 139.0, 140.2, 151.7, 157.9, 165.3; [α]D -125.0 (c 0.5, CHCl3). Isomer 9b: Rf 0.29 (cyclohexane-AcOEt 1:1); Low melting white solid; 1H-NMR: δ 0.80 (d, J = 6.6 Hz, 3H, 4"-CH3), 1.17 (t, J = 7.2 Hz, 3H, CH3CH2O), 2.80 (s, 3H, 3"-CH3), 3.93 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 4.10 (q, J = 7.2 Hz, 3H, CH3CH2O), 5.26 (d, J = 8.8 Hz, 1H, H-1'), 5.86 (d, 1H, J = 8.8 Hz, 1H, H-5"), 5.89 (s, 1H, =CH2), 6.37 (s, 1H, =CH2), 7.09 - 7.18 (m, 2 ArH), 7.19 - 7.38 (m, 7 ArH), 9.17 (d, J = 8.8, 1H, NH); 13C-NMR: δ 13.9, 14.8, 28.0, 29.7, 54.4 (50%), 54.6 (50%), 59.4, 60.9, 126.4, 126.8, 128.1, 128.2, 128.5, 133.1, 136.8, 138.5, 140.3, 151.8, 157.9, 165.3; [α]D 155.0 (c 0.5, CHCl3).
Methyl (1'S,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carboxamido)(4-nitro-phenyl)methyl]acrylate (8c) and its (1'R,4"S,5"R)-isomer (9c): The title compounds were obtained in 61% overall yield as 70:30 diastereomeric mixture. MS (ESI): m/z 452.2 [M]+, 475.2 [M+Na]+; Anal. Calcd. for C23H24N4O6: C, 61.05; H, 5.35; N, 12.38; Found: C, 61.00; H, 5.31; N, 12.35. Isomer 8c: Rf 0.50 (cyclohexane-AcOEt 1:1); White crystals; Mp. 55-57 °C; 1H-NMR: δ 0.82 (d, J = 6.6 Hz, 3H, 4"-CH3), 2.83 (s, 3H, 3"-CH3), 3.68 (s, 3H, OCH3), 3.95 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 5.26 (d, J = 8.7 Hz, 1H, H-1'), 5.92 (s, 1H, =CH2), 5.95 (d, J = 8.8 Hz, 1H, H-5"), 6.38 (s, 1H, =CH2), 7.10 - 7.22 (m, 2 ArH), 7.25 - 7.41 (m, 3 ArH), 7.51 (d, J= 8.5 Hz, 2 ArH), 8.16 (d, J = 8.5 Hz, 2 ArH), 9.44 (d, J = 8.7 Hz, 1H, NH); 13C-NMR: δ 14.8, 28.1, 52.1 (50%), 52.2 (50%), 54.6 (50%), 54.8 (50%), 59.4, 76.4, 123.8, 126.8, 127.5, 127.8, 128.2, 128.6, 136.7, 139.1, 147.8, 151.9, 157.9, 165.5; [α]D -107.0 (c 0.5, CHCl3). Isomer 9c: Rf 0.20 (cyclohexane-AcOEt 1:1); White crystals; Mp. 62-64 °C; 1H-NMR: δ 0.83 (d, J = 6.6 Hz, 3H, 4"-CH3), 2.83 (s, 3H, 3"-CH3), 3.70 (s, 3H, OCH3), 3.96 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 5.28 (d, J = 8.8 Hz, 1H, H-1'), 5.97 (s, 1H, =CH2), 5.97 (d, J = 8.5 Hz, 1H, H-5"), 6.43 (s, 1H, =CH2), 7.11 - 7.18 (m, 2 ArH), 7.25 - 7.38 (m, 3 ArH), 7.44 (d, J = 8.5 Hz, 2 ArH), 8.13 (d, J = 8.5 Hz, 2 ArH), 9.37 (d, J = 8.8 Hz, 1H, NH); 13C-NMR: δ 14.8, 28.1, 52.1, 54.6 (50%), 54.8 (50%), 59.5, 76.4, 123.7, 126.8, 127.4, 128.0, 128.3, 128.6, 136.7, 139.3, 147.4, 152.0, 156.2 (50%), 157.8 (50%), 165.4; [α]D 78.9 (c 0.5, CHCl3).
Ethyl (1'S,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carboxamido) (naphthalen-1-yl)methyl]acrylate (8d) and its (1'R,4"S,5"R)-isomer (9d): The title compounds were obtained in 58% overall yield as 70:30 diastereomeric mixture. MS (ESI): m/z 471.2 [M]+, 494.2 [M+Na]+; Anal. Calcd. for C28H29N3O4: C, 71.32; H, 6.20; N, 8.91. Found: C, 71.36; H, 6.15; N, 8.87. Isomer 8d: Rf 0.50 (cyclohexane-AcOEt 1:1); White crystals; Mp. 53-55 °C; 1H-NMR: δ 0.79 (d, J = 6.6 Hz, 3H, 4"-CH3), 1.13 (t, J = 7.1 Hz, 3H, CH3CH2O), 2.79 (s, 3H, 3"-CH3), 3.88 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 4.08 (q, J = 7.1 Hz, 2H, CH3CH2O), 5.28 (d, J = 8.5 Hz, 1H, H-1'), 5.90 (s, 1H, =CH2), 6.10 (d, J = 8.8 Hz, 1H, H-5"), 6.38 (s, 1H, =CH2), 7.15 - 7.21 (m, 2 ArH), 7.25 - 7.52 (m, 6 ArH), 7.74 - 7.86 (m, 4 ArH), 9.31 (d, J = 8.5, 1H, NH); 13C-NMR: δ 13.9, 14.7, 28.0, 54.5, 55.0, 59.3, 60.8, 125.3, 125.6, 125.7, 125.8, 126.0, 126.8, 127.4, 127.6, 127.7, 128.0, 128.3, 128.5, 132.8, 133.4, 137.0, 137.8, 140.6, 151.7, 157.9, 165.5; [α]D -106.0 (c 0.5, CHCl3). Isomer 9d: Rf 0.35 (cyclohexane-AcOEt 1:1); White crystals; Mp. 56-57 °C; 1H-NMR: δ 0.79 (d, J = 6.6 Hz, 3H, 4"-CH3), 1.16 (t, J = 7.1 Hz, 3H, CH3CH2O), 2.80 (s, 3H, 3"-CH3), 3.91 (dq, J = 6.6 Hz, J = 8.8 Hz, 1H, H-4"), 5.32 (d, J = 8.8 Hz, 1H, H-1'), 6.00 (s, 1H, =CH2), 6.11 (d, J = 8.7 Hz, 1H, H-5"), 6.46 (s, 1H, =CH2), 7.14 - 7.20 (m, 2 ArH), 7.24 - 7.33 (m, 3 ArH), 7.39 - 7.49 (m, 3 ArH), 7.73 - 7.84 (m, 4 ArH), 9.29 (d, J = 8.7 Hz, 1H, NH); 13C-NMR: δ 13.8, 4.6, 27.9, 54.4, 54.8, 59.3, 60.7, 124.9, 125.4, 125.7, 125.9, 126.0, 126.7, 127.4, 127.9, 128.1, 128.4, 128.6, 132.6, 133.2, 136.8, 137.2, 140.7, 151.8 157.8, 165.4; [α]D 54.0 (c 0.5, CHCl3).
Methyl (1'R,4"S,5"R)-2-[(3",4"-dimethyl-2"-oxo-5"-phenylimidazolidine-1"-carboxamido) (ethoxy-carbonyl)methyl]acrylate (8e) and its (1'S,4"S,5"R)-isomer (9e): The title compounds were obtained in 77% overall yield as 75:25 diastereomeric mixture. MS (ESI): m/z 403.2 [M]+, 426.2 [M+Na]+; Anal. Calcd. for C20H25N3O6: C, 59.54; H, 6.25; N, 10.42; Found: C, 59.50; H, 6.21; N, 10.46. Isomer 8e: Rf 0.35 (cyclohexane-AcOEt 1:1); White crystals; Mp. 78-80 °C; 1H-NMR: δ 0.78 (d, J = 6.6 Hz, 3H, 4"-CH3), 1.21 (t, J = 7.1 Hz, 3H, CH3CH2O), 2.80 (s, 3H, 3"-CH3), 3.78 (s, 3H, OCH3), 3.88 (dq, J = 6.6 Hz, J = 8.4 Hz, 1H, H-4"), 4.15 (q, J = 7.1 Hz, 2H, CH3CH2O), 5.26 (d, J = 8.7 Hz, 1H, H-1'), 5.33 (d, J = 8.4 Hz, 1H, H-5"), 5.85 (s, 1H, =CH2), 6.31 (s, 1H, =CH2), 7.09 - 7.18 (m, 2 ArH), 7.22 - 7.35 (m, 3 ArH), 9.22 (d, J = 8.7 Hz, 1H, NH); 13C-NMR: δ 14.0, 14.7, 28.1, 52.1, 54.5, 54.6, 59.2, 61.7, 126.8, 128.0, 128.4, 129.3, 136.5, 136.8, 152.2, 157.6, 165.5, 169.7; [α]D 17.8 (c 0.5, CHCl3). Isomer 9e: Rf 0.25 (cyclohexane-AcOEt 1:1); Low melting white solid; 1H-NMR: δ 0.80 (d, J = 6.6 Hz, 3H, 4"-CH3), 1.21 (t, J = 7.1 Hz, 3H, CH3CH2O), 2.81 (s, 3H, 3"-CH3), 3.79 (s, 3H, OCH3), 3.91 (dq, J = 6.6 Hz, J= 8.8 Hz, 1H, H-4"), 4.18 (q, J = 7.1 Hz, 2H, CH3CH2O), 5.23 (d, J = 8.8 Hz, 1H, H-5"), 5.28 (d, J = 8.2 Hz, 1H, H-1'), 5.92 (s, 1H, =CH2), 6.37 (s, 1H, =CH2), 7.10 - 7.21 (m, 2 ArH), 7.25 - 7.38 (m, 3 ArH), 9.25 (d, J = 8.2 Hz, 1H, NH); 13C-NMR: δ 14.0, 14.8, 28.1, 52.1, 54.5, 54.8, 59.4, 61.8, 126.9, 128.1, 128.5, 129.6, 136.8, 152.1, 157.6, 165.5, 169.5; [α]D - 41.7 (c 0.5, CHCl3).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}