Marine Derived Polysaccharides for Biomedical Applications: Chemical Modification Approaches

Abstract
:Contents
- Introduction
- Polysaccharides as biomaterials
- Alginate: structure and chemical modification
- Alginate-based materials for drug-delivery applications
- Alginate for cell immobilization
- Chitin and chitosan: structure and chemical modification
- Composites and hydrogels based on N-succinylchitosan/alginate blends
- Conclusions
1. Introduction
General principles of chemical modification
- Blending or chemical linkages with synthetic biopolymers;
- Surface coating of micro- or nano-spheres with biocompatible synthetic polymers;
- Crosslinking with different physical or chemical reagents;
- Hydrophobization through alkylation reactions;
- Modulation of guluronic/mannuronic ratio, or of deacetylation degree, respectively.
2. Polysaccharides as biomaterials
Alginate
Chitin and chitosan
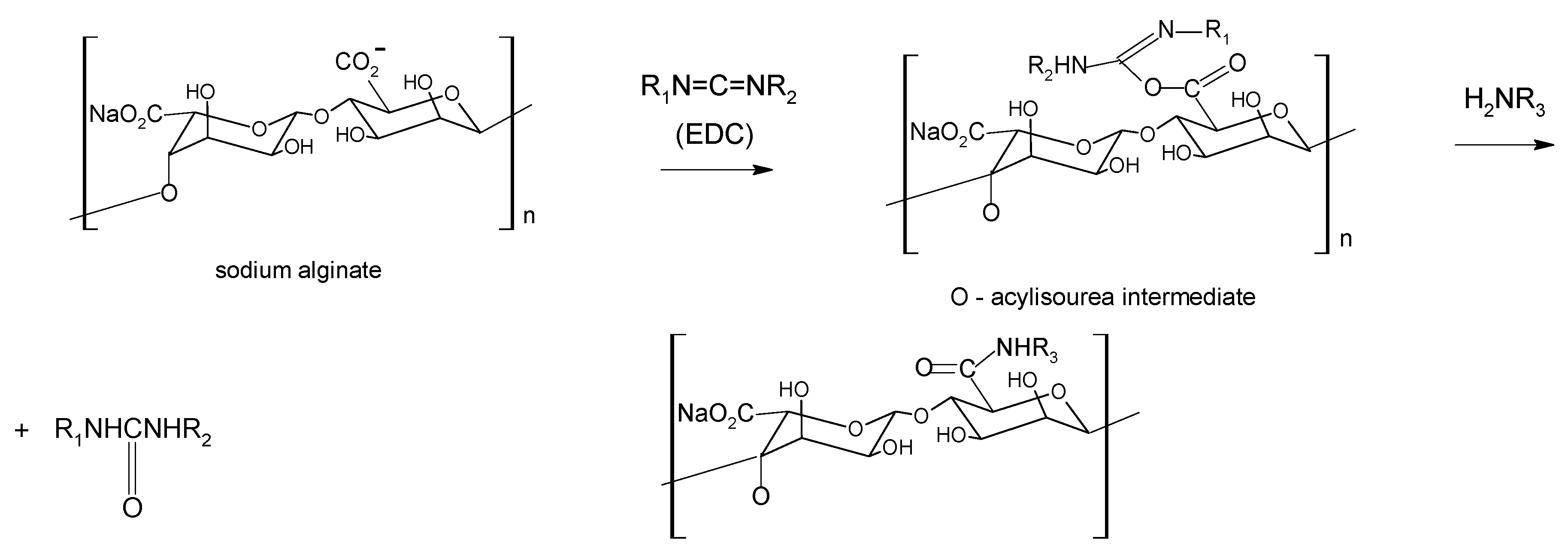
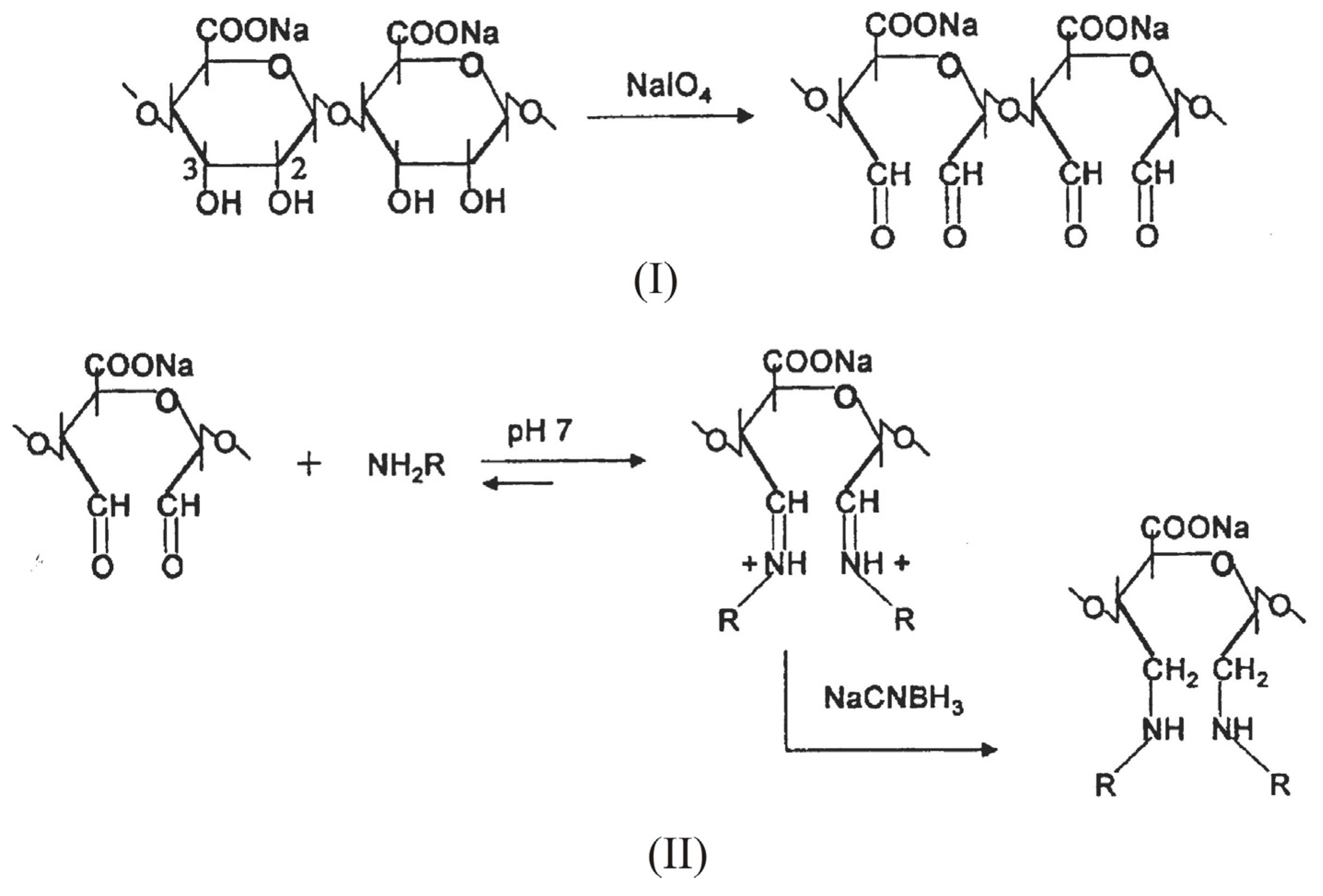
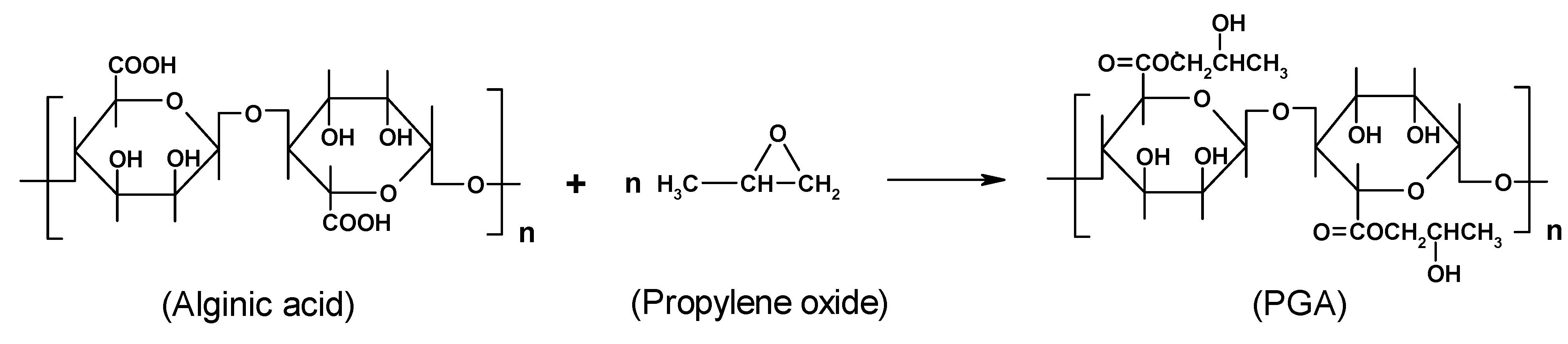
3. Alginate: Structure and chemical modification
Chemical modification
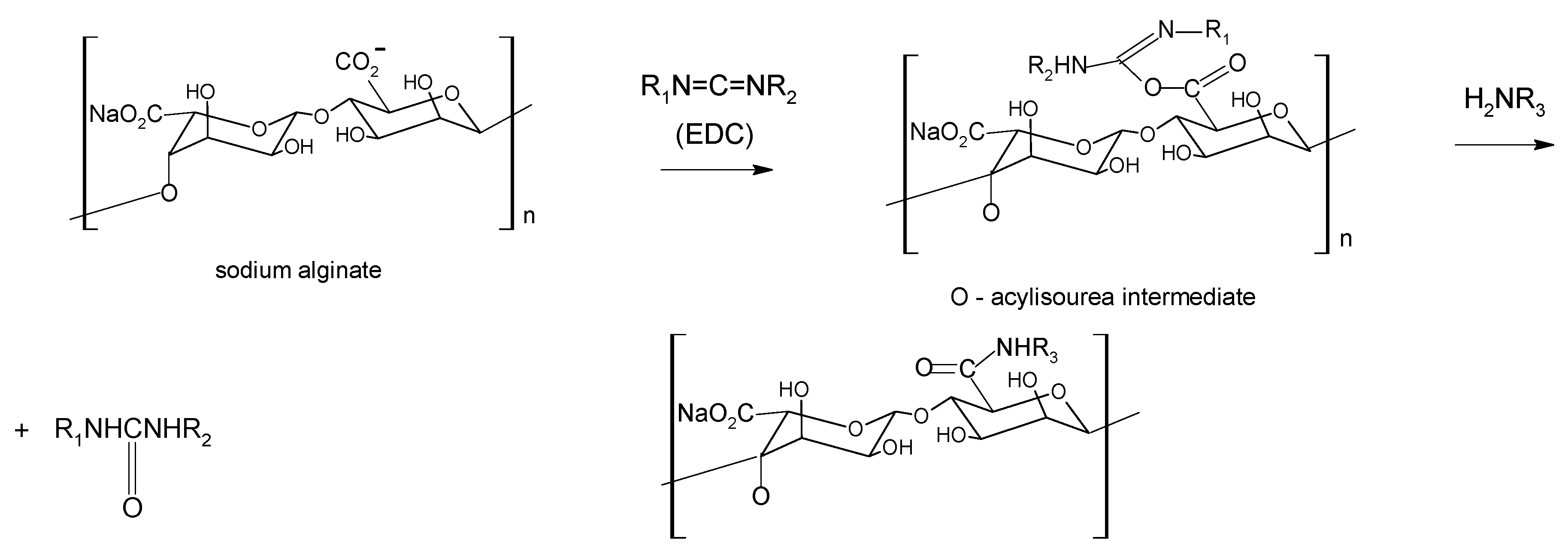

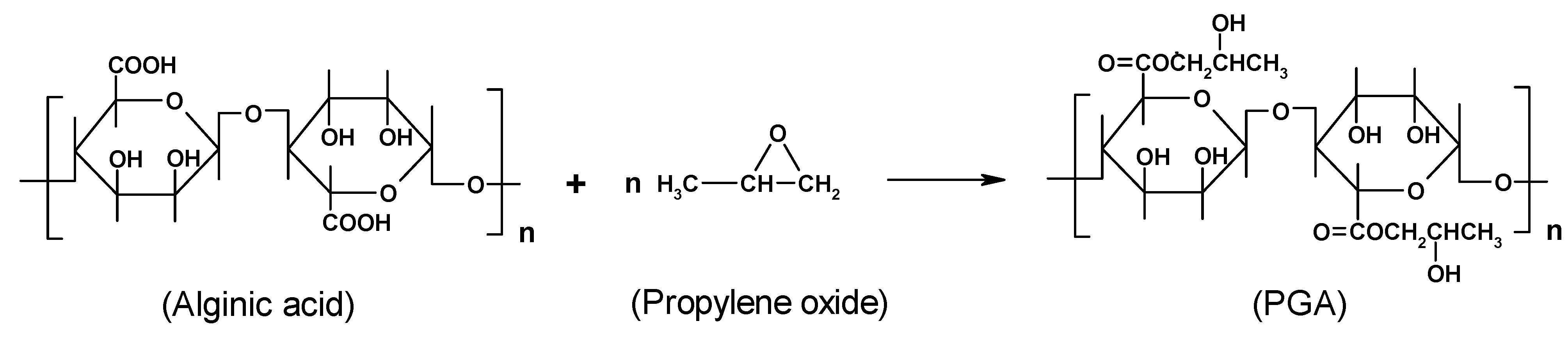
4. Alginate-based materials for drug-delivery applications
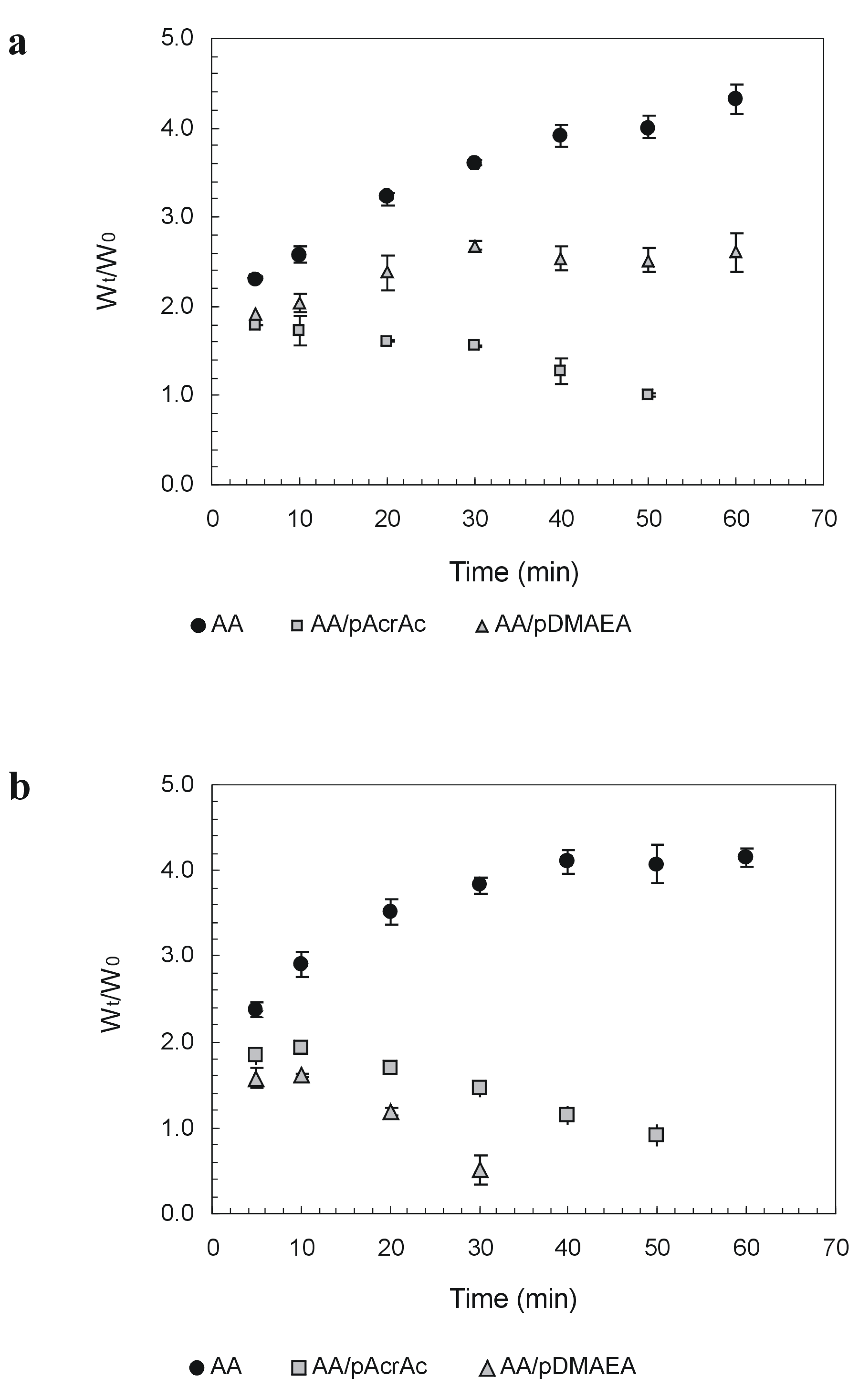
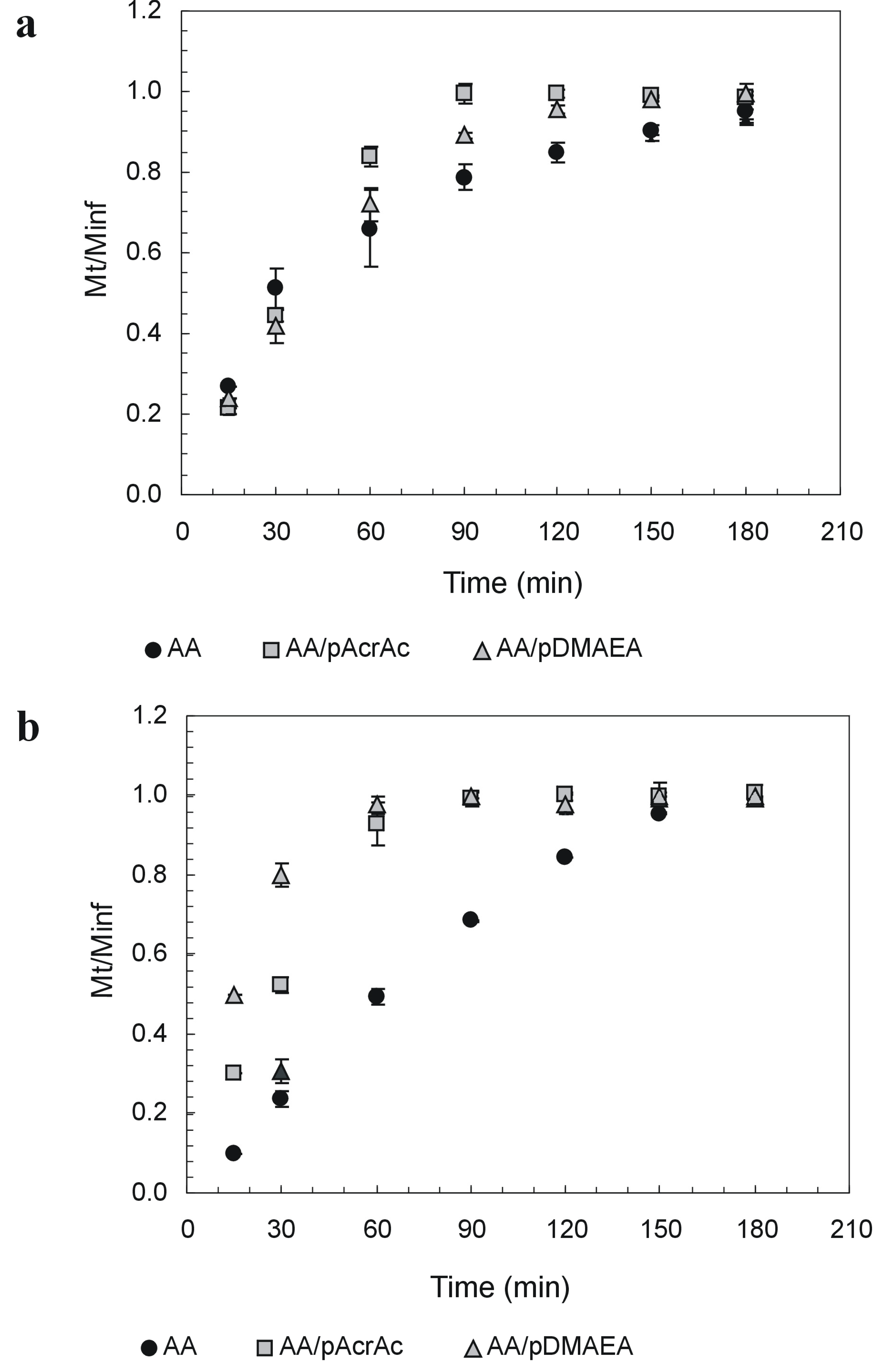
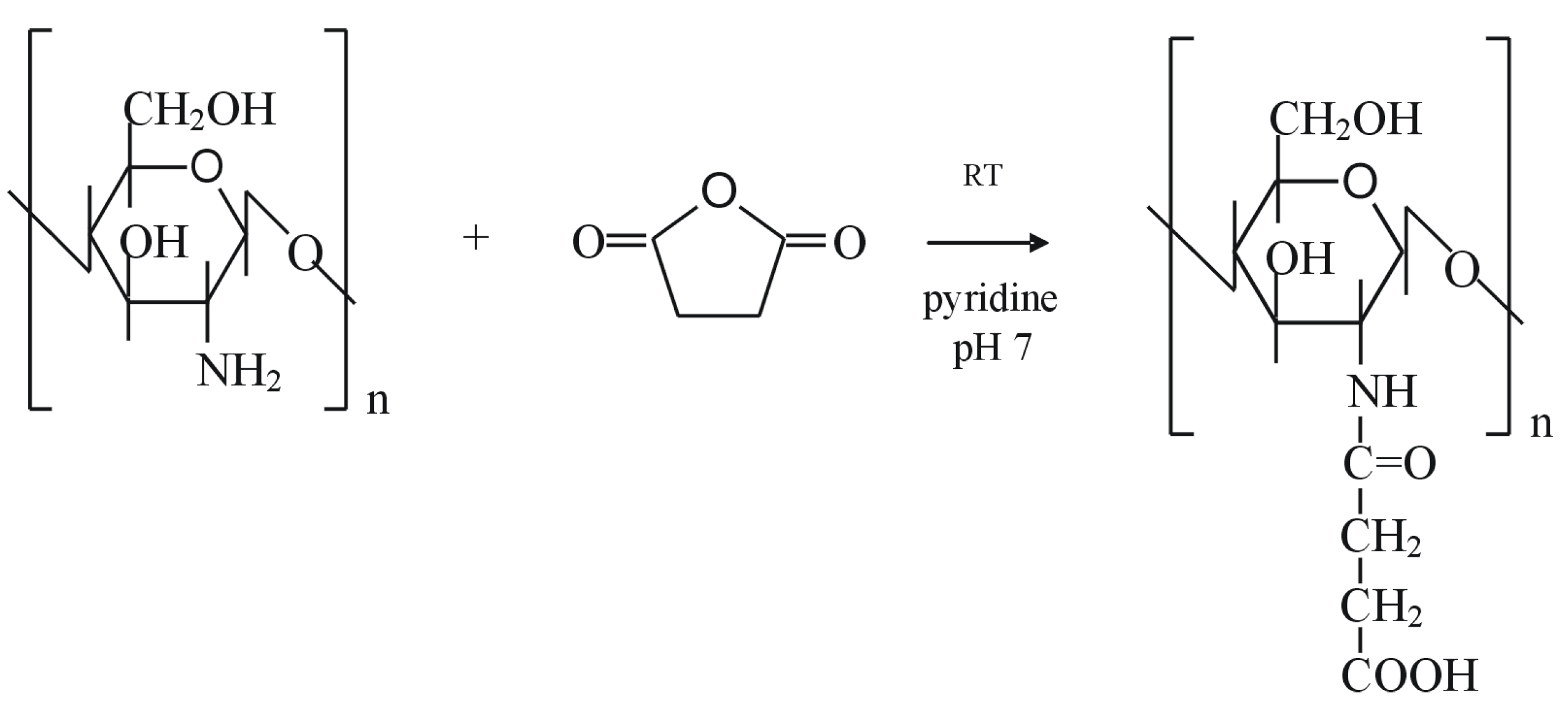
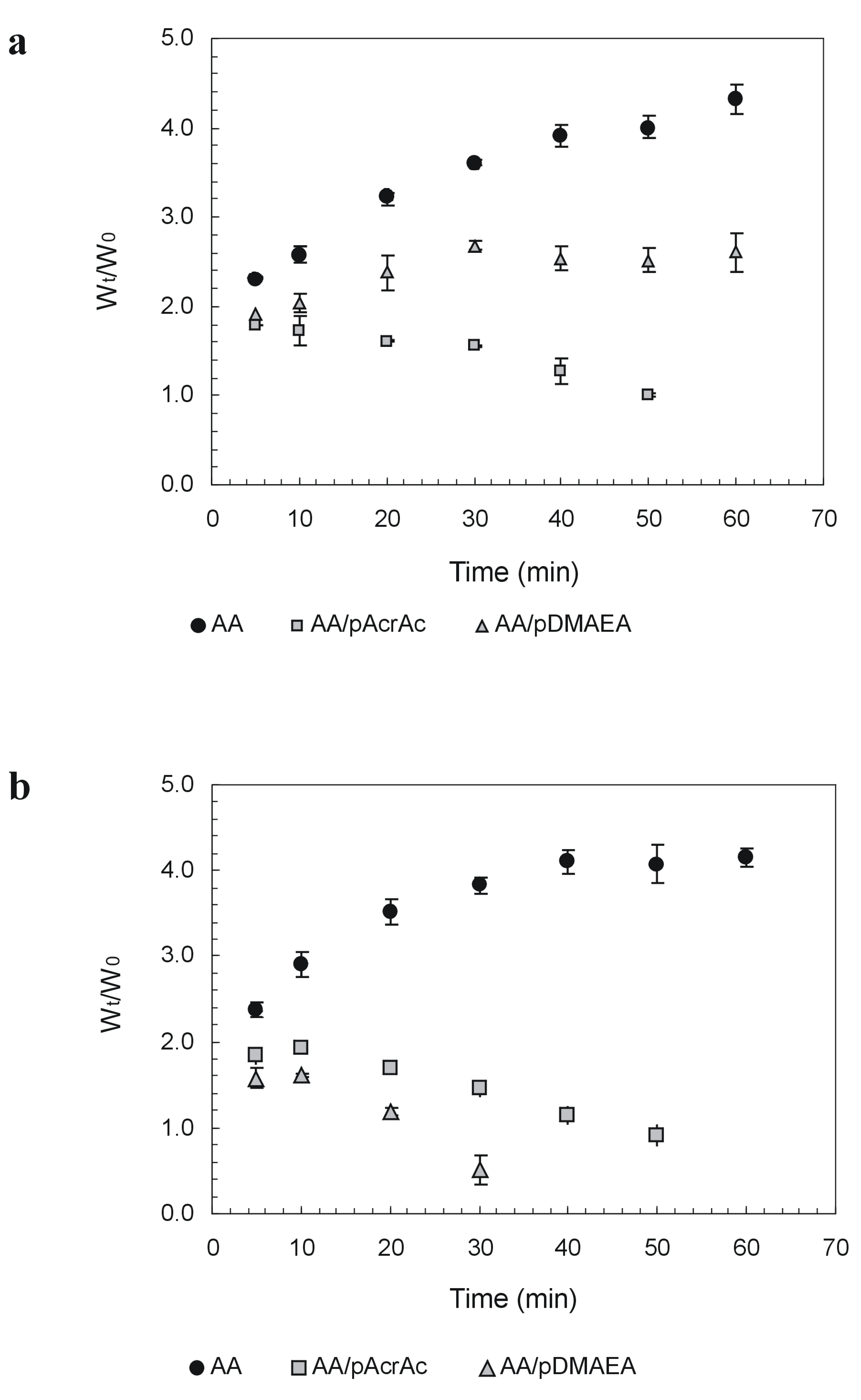
Novel alginate-acrylic polymers as a platform for drug delivery


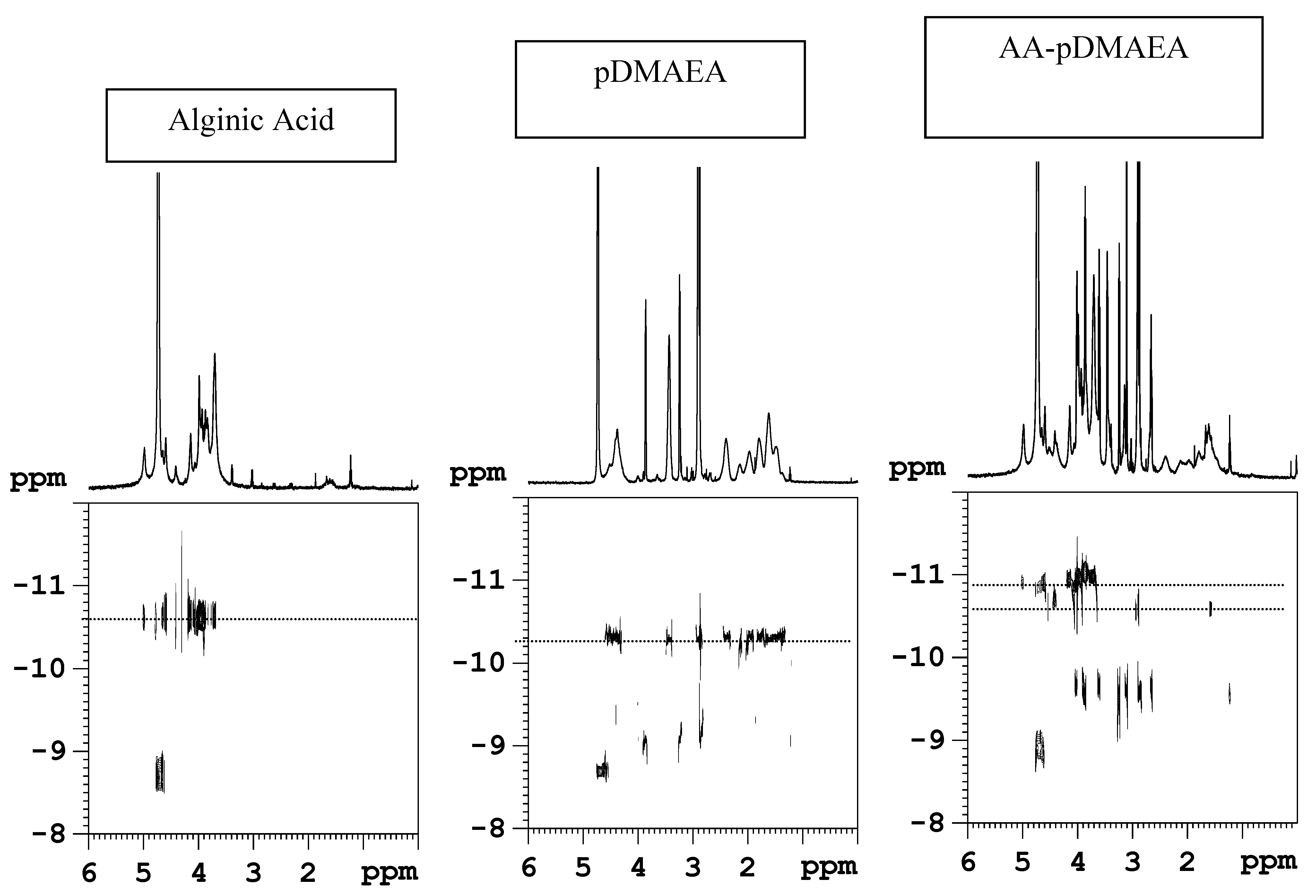{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | pH 1.2 | pH 6.8 | ||
|---|---|---|---|---|
| T1.0 | Weight loss | T1.0 | Weight loss | |
| AA | 180 | 32±2 | 150 | 90±3 |
| AA-pAcrAc | 90 | 100±2 | 60 | 100±5 |
| AA-pDMAEA | 150 | 78±2 | 60 | 100±4 |

| Sample | Work of Detachment |
|---|---|
| (J/m2) | |
| AA | 9.4 |
| AA-pAcrAc | 29.4 |
| AA-pDMAEA | 29.6 |
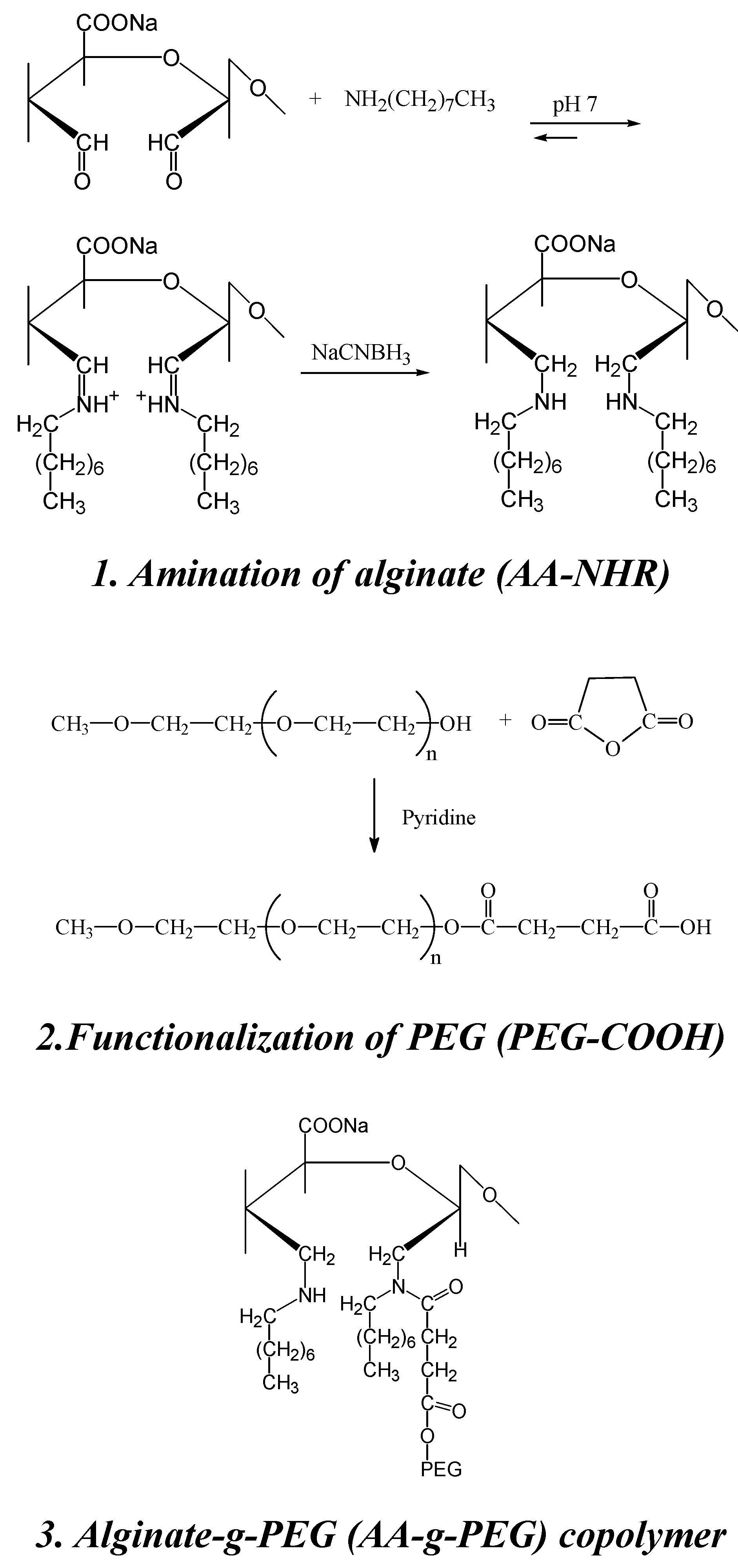
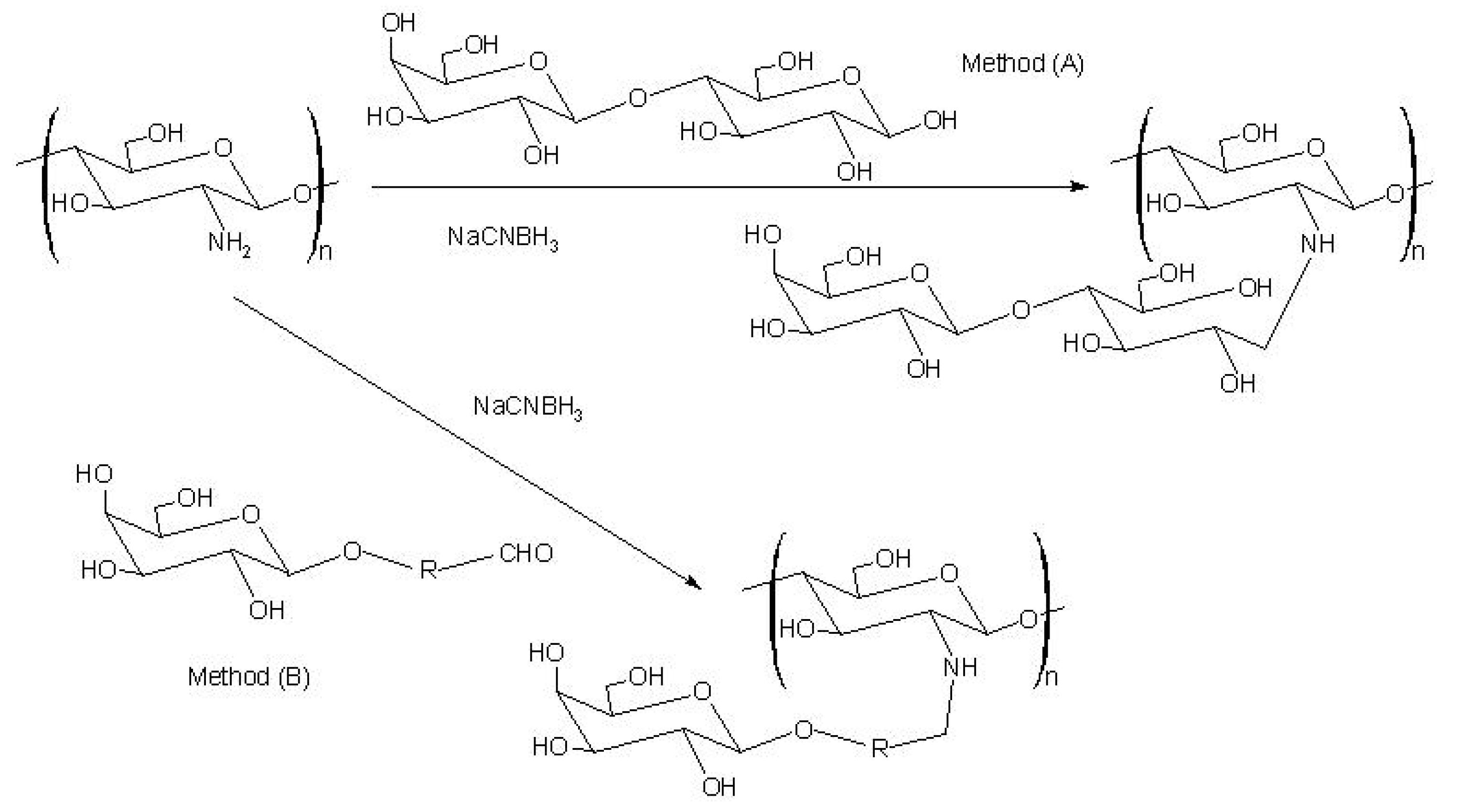
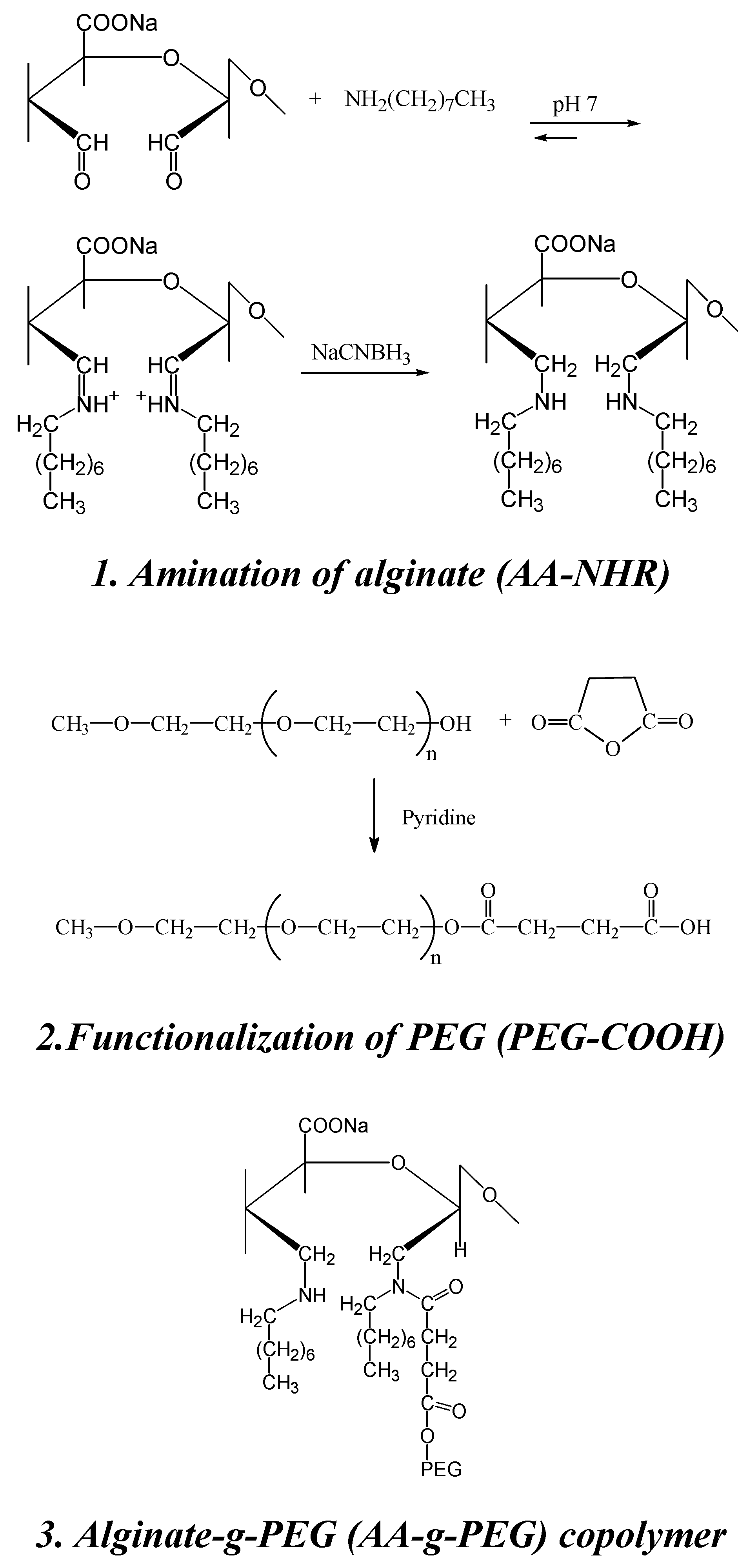
5. Alginate for cell immobilization
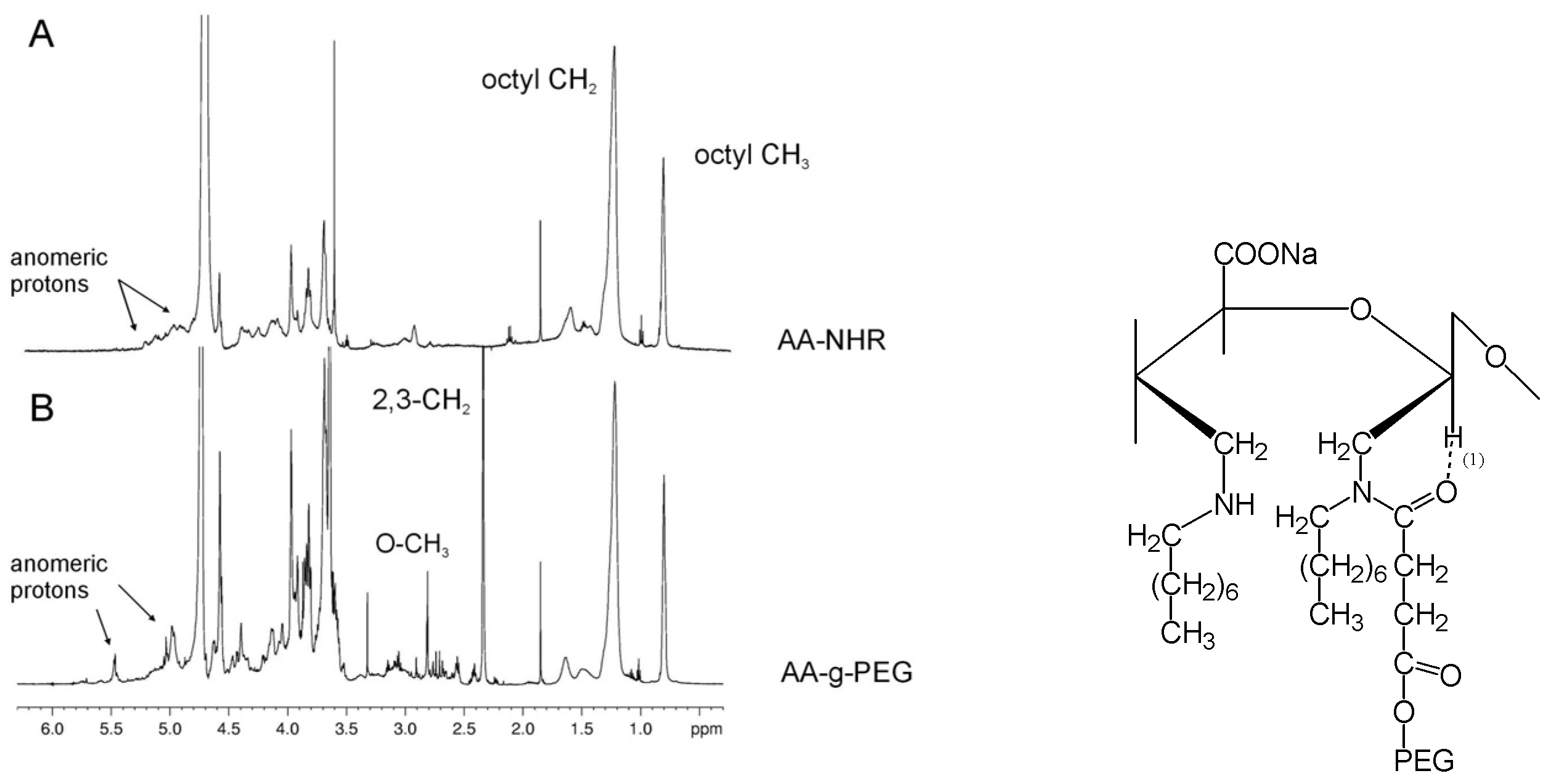
Synthesis of a novel alginate-poly(ethylene glycol) graft copolymer
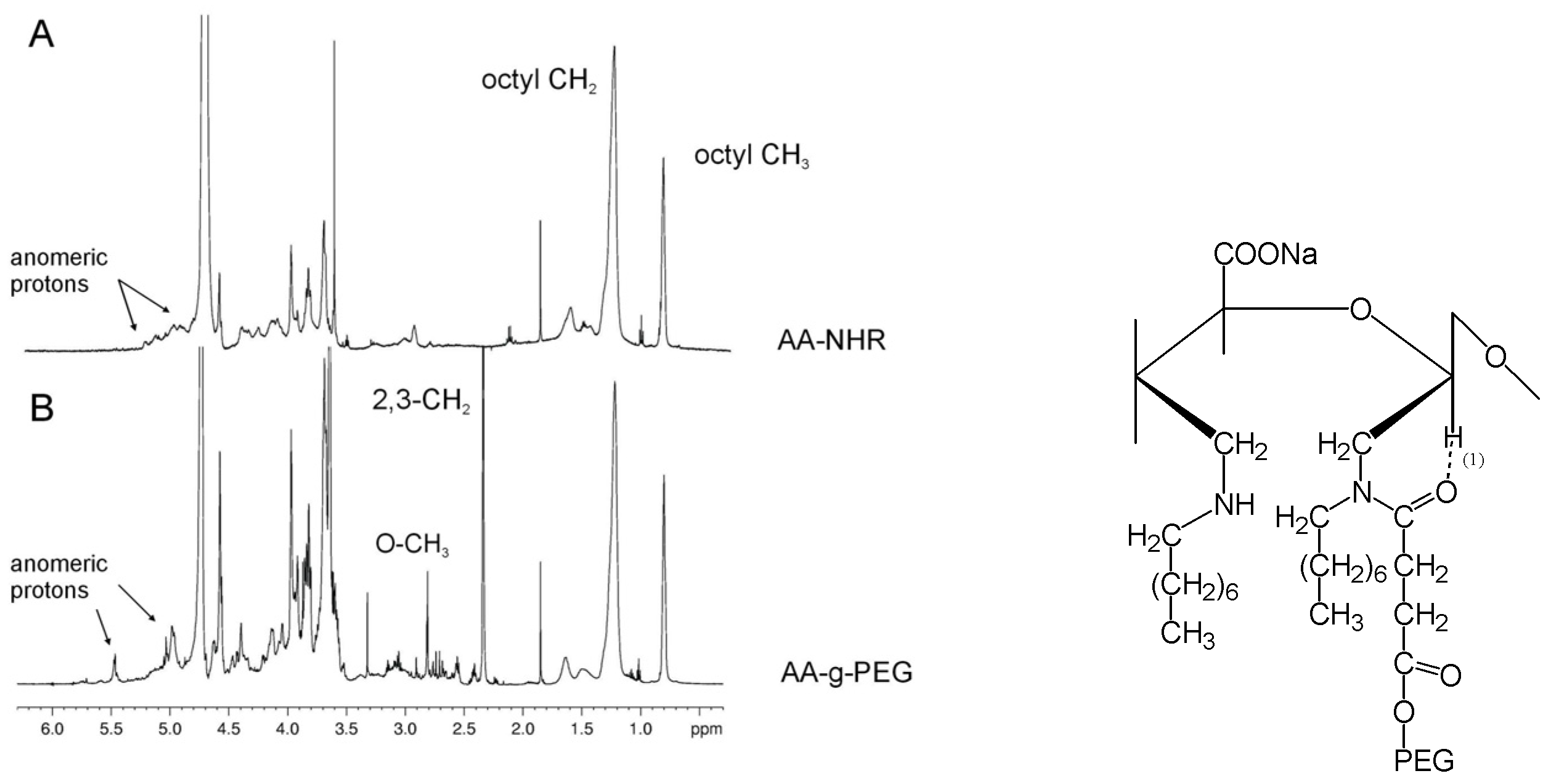
6. Chitin and chitosan: structure and chemical modification
Definition and Composition of Chitosan: Degree of Deacetylation (DD)
Characteristics of Chitosan
Chitin chemical modification
Chitosan chemical modification
O-/N- carboxyalkylation
Sulfonation
Acylation
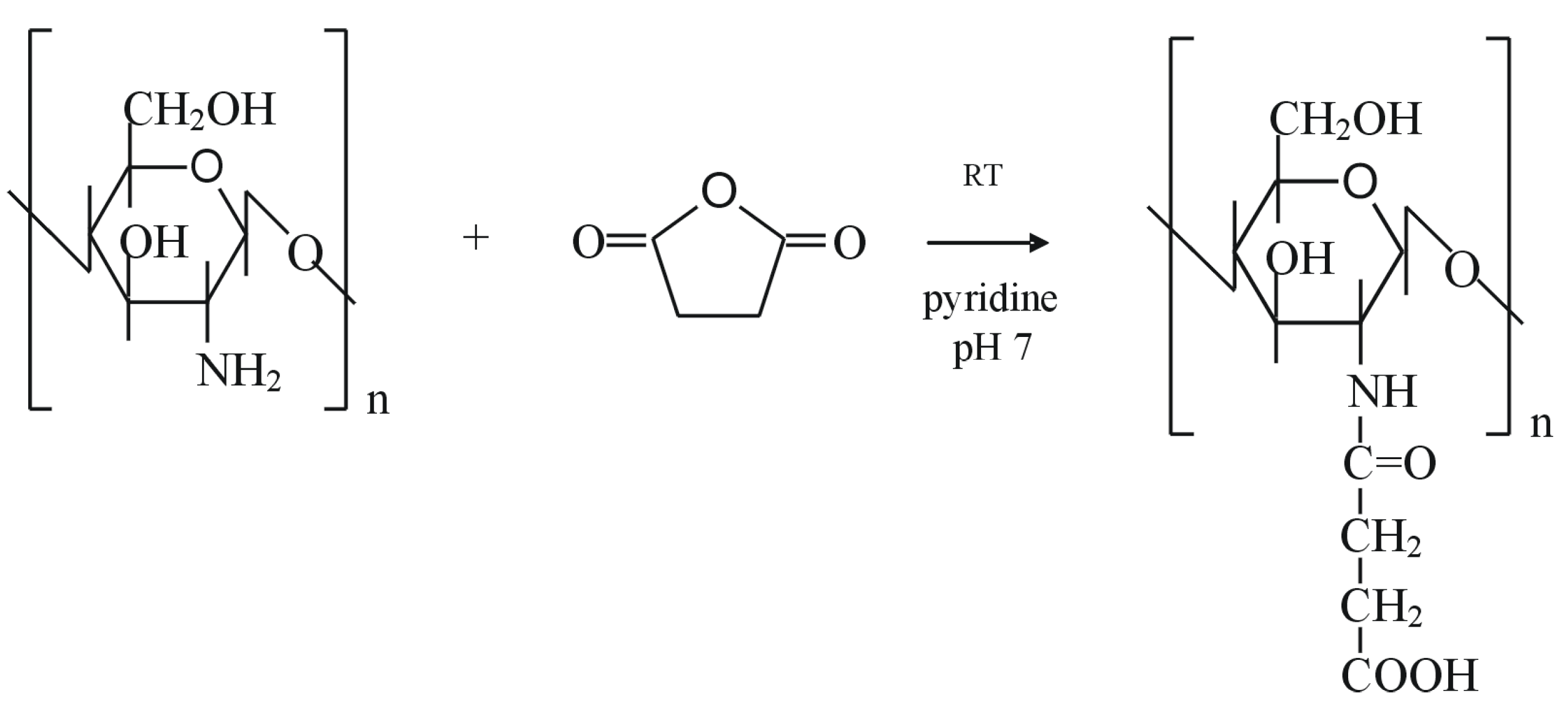
Sugar-modified chitosan

Graft copolymerization
Chitosan crosslinking
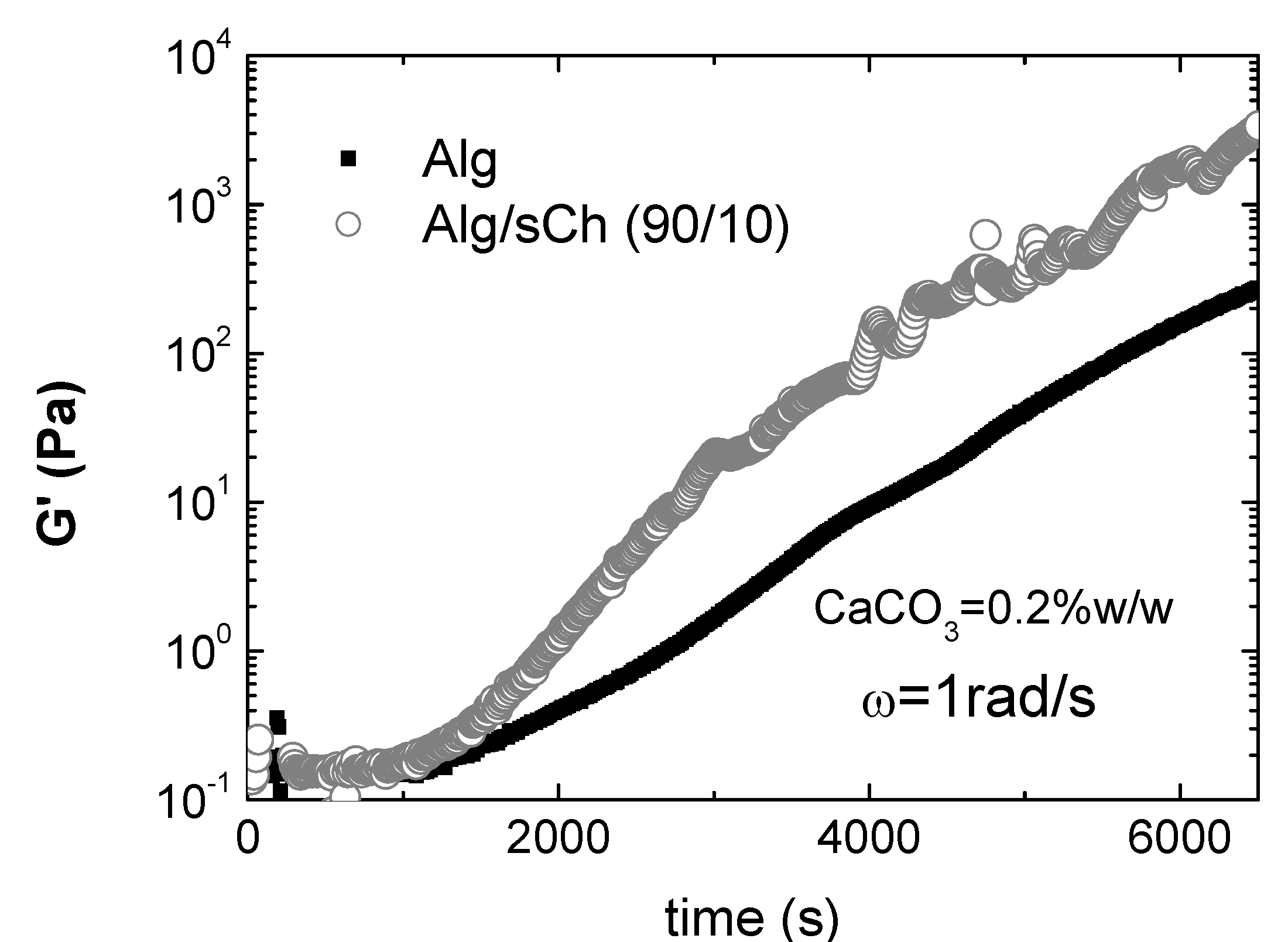
7. Composites and hydrogels based on N-succinylchitosan/alginate blends
| Sample | Yield strength | Young modulus |
|---|---|---|
| (MPa) | (MPa) | |
| CHS/Alg/sCh: 50/10/40 | 4.58 ± 1.63 | 427.4 ± 79.8 |
| CHS/Alg/sCh: 50/20/30 | 13.62 ± 0.41 | 873.9 ± 145.8 |
| CHS/Alg/sCh: 50/25/25 | 8.20 ± 0.15 | 370.8 ± 73.7 |
| CHS/Alg/sCh: 50/30/20 | 9.02 ± 2.62 | 437.9 ± 83.5 |
| CHS/Alg/sCh: 50/40/10 | 3.22 ± 0.59 | 444.2 ± 74.7 |
| CHS/Alg/Ch: 50/30/20 | 0.04 ± 0.01 | 9.9 ± 0.6 |
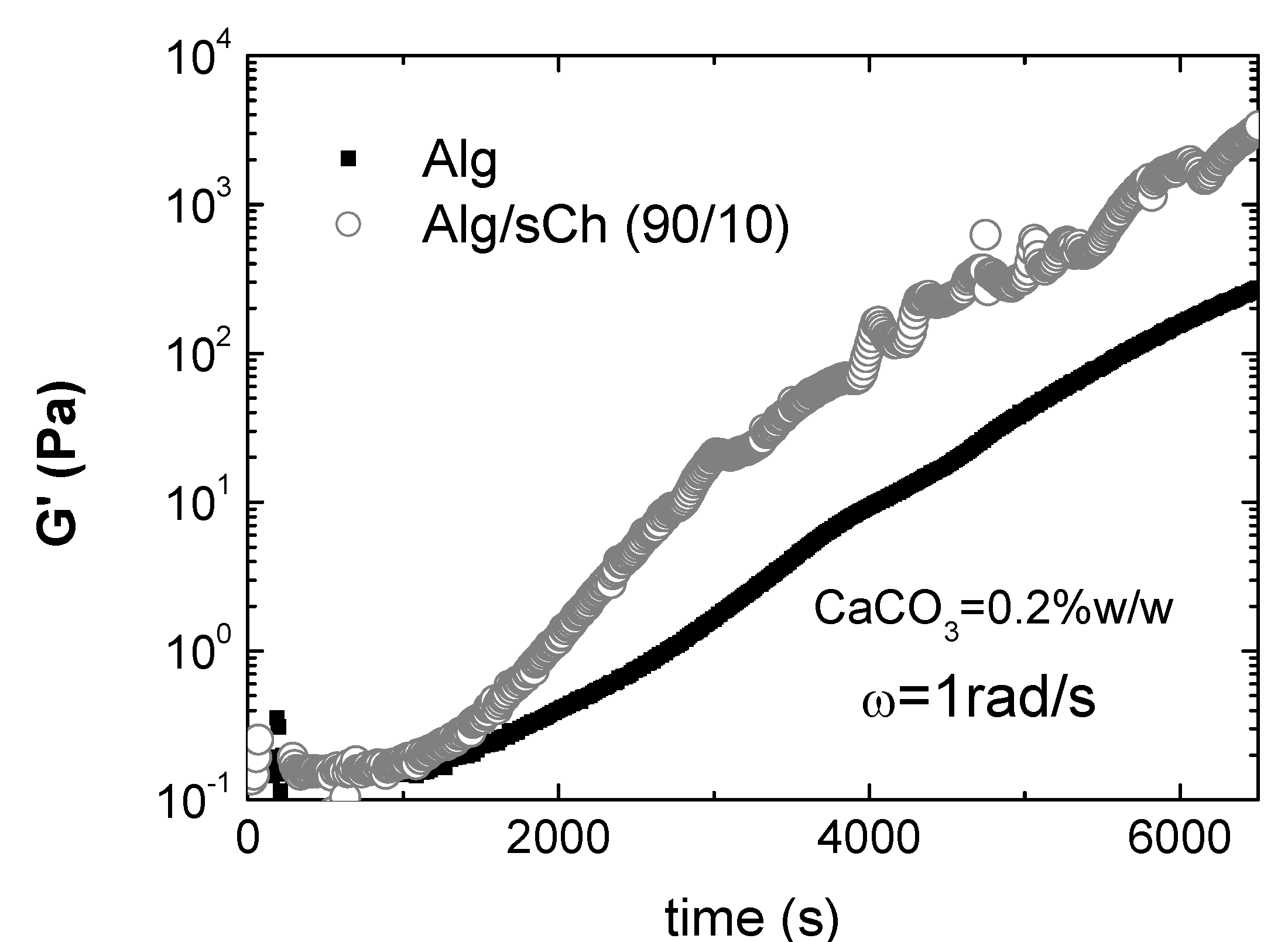
8. Conclusions
Acknowledgements
References
- Krishnan, G. Nature of tunicin and its interaction with other chemical components of the tunic of the ascidian, Polyclinum madrasensis Sebastian. Indian J. Exp. Biol. 1975, 13, 172–176. [Google Scholar]
- Belton, P.S.; Tanner, S.F.; Cartier, N.; C–hanzy, H. High resolution solid-state 13C Nuclear Magnetic Resonance Spectroscopy of tunicin, an animal cellulose. Macromolecules 1989, 22, 1615–1617. [Google Scholar] [CrossRef]
- Anglès, M.N.; Dufresne, A. Plasticized starch/tunicin whiskers nanocomposite materials. 2. Mechanical behaviour. Macromolecules 2001, 34, 2921–2931. [Google Scholar]
- Cima, F.; Ballarin, L.; Bressa, G.; Martinucci, G.; Burighel, P. Toxicity of organotin compounds on embryos of a marine invertebrate. Ecotoxicol. Environ. Safety 1996, 35, 174–182. [Google Scholar] [CrossRef]
- Mancuso Nichols, C.A.; Guezennec, J.; Bowman, J.P. Bacterial Exopolysaccharides from Extreme Marine Environments with special Consideration of the Southern Ocean, sea Ice, and deep-Sea Hydrothermal Vents: A Review. Marine Biotechnol. 2005, 7, 253–271. [Google Scholar] [CrossRef]
- Decho, A.W. Microbial exopolymer secretions in ocean environments: their role(s) in food webs and marine processes. In Oceanogr. Mar. Biol. Annu. Rev.; Barnes, M., Ed.; Aberdeen Univ. Press: Aberdeen, UK, 1990; pp. 73–153. [Google Scholar]
- Corpe, W.A. An acid polysaccharide produced by primary film forming bacteria. Dev. Ind. Microbiol. 1970, 16, 249–255. [Google Scholar]
- Ehrlich, H.; Maldonado, M.; Spindler, K-D; Eckert, C.; Hanke, T.; Born, R.; Goebel, C.; Simon, P.; Heinemann, S.; Worch, H. First evidence of chitin as a component of the skeletal fibers of marine sponges. Part I. Verongidae (Demospongia: Porifera). J. Exp. Zool. (Mol. Dev. Evol.) 2007, 308B, 347–356. [Google Scholar] [CrossRef]
- Ehrlich, H.; Krautter, M.; Hanke, T.; Simon, P.; Knieb, C.; Heinemann, S.; Worch, H. First evidence of the presence of chitin in skeletons of marine sponges. Part II. Glass sponges (Hexactinellida: Porifera). J. Exp. Zool. (Mol. Dev. Evol.) 2007, 308B, 473–483. [Google Scholar] [CrossRef]
- Paul, W.; Sharma, C.P. Chitosan and Alginate Wound Dressings: A Short Review. Trends Biomater. Artif. Organs 2004, 18, 18–23. [Google Scholar]
- Thiele, H.; Plohnke, K.; Brandt, E.; Moll, G. Ordnen von Polyelektrolyten durch Ionendiffusion. Colloid Polym. Sci. 1962, 182, 24–35. [Google Scholar]
- Thumbs, H.; Kohler, H.H. Capillaries in alginate gels as an example of dissipative structure formation. Chem. Phys. 1996, 208, 9–24. [Google Scholar] [CrossRef]
- Dittrich, R.; Tomandl, G.; Mangler, M. Preparation of Al2O3, TiO2 and Hydroxyapatite Ceramics with Pores Similar to a Honeycomb Structure. Adv. Eng. Mat. 2002, 4, 487–490. [Google Scholar] [CrossRef]
- Despang, F.; Börner, A.; Dittrich, R.; Tomandl, G.; Pompe, W.; Gelinsky, M. Alginate/calcium phosphate scaffolds with oriented, tube-like pores. Mat.-wiss. Werkstoff. 2005, 36, 761–767. [Google Scholar] [CrossRef]
- Dittrich, R.; Despang, F.; Tomandl, G.; Gelinsky, M.; Pompe, W. Mineralised scaffolds for hard tissue engineering by ionotrop gelation of alginate. J. Am. Ceram. Soc. 2007, 90, 1703–1708. [Google Scholar] [CrossRef]
- Dittrich, R.; Despang, F.; Bernhardt, A.; Mannschatz, A.; Hanke, T.; Tomandl, G.; Pompe, W.; Gelinsky, M. Mineralized Scaffolds for hard tissue engineering by ionotropic gelation of alginate; Vincenzini, P., Ed.; Trans Tech Publications Inc.: Zurich, Switzerland, 2006; pp. 159–164. [Google Scholar]
- Gelinsky, M.; Eckert, M.; Despang, F. Biphasic, but monolithic scaffolds for the therapy of osteochondral defects. Int. J. Mater. Res. 2007, 98, 749–755. [Google Scholar] [CrossRef]
- Ito, M.; Matahira, Y.; Sakai, K. The application of chitin-chitosan to bone filling materials. Kichin, Kitosan Kenkyu Publ.: Nippon Kichin, Kitosan Gakkai, 1998; Vol. 4, pp. 142–143. [Google Scholar]
- Felt, O.; Furrer, P.; Mayer, J.M.; Plazonnet, B.; Buri, P.; Gurn, R. Topical use of chitosan in ophtalmology: tolerance assessment and evaluation of precorneal retention. Int. J. Pharm. 1999, 180, 185–193. [Google Scholar] [CrossRef]
- Patashnik, S.; Rabinovich, L.; Golomb, G. Preparation and evaluation of chitosan microspheres containing biphosphonates. J. Drug Target 1997, 4, 371–380. [Google Scholar] [CrossRef]
- Song, J.S.; Such, C.H.; Park, Y.B.; Lee, S.H.; Yoo, N.C.; Lee, J.D.; Kim, K.H.; Lee, S.K. A phase I/IIa study on intra-articular injection of holmium-166-chitosan complex for the treatment of knee synovitis of rheumatoid arthritis. Eur. J. Nucl. Med. 2001, 28, 489–497. [Google Scholar]
- Muzzarelli, R.A.A. Human enzymatic activities related to the therapeutic administration of chitin derivatives. Cell Mol. Life Sci. 1997, 53, 131–140. [Google Scholar] [CrossRef]
- Koga, D. Chitin enzymology chitinase. In Adv. Chitin Sci.; vol. 3 R. Chen, H.C., Ed.; 1998; pp. 16–23. [Google Scholar]
- Shahidi, F.; Abuzaytoun, R. Chitin, chitosan and co-products: chemistry, production, applications, and health effects. Adv. Food. Nutr. 2005, 49, 93–135. [Google Scholar] [CrossRef]
- He, P.; Davis, S.S.; Illum, L. In vitro evaluation of the mucoadhesive properties of chitosan microspheres. Int. J. Pharm. 1998, 166, 75–88. [Google Scholar] [CrossRef]
- Calvo, P.; Vila-Jato, J.L.; Alonso, M.J. Evaluation of cationic polymer-coated nanocapsules as ocular drug carriers. Int. J. Pharm. 1997, 153, 41–50. [Google Scholar] [CrossRef]
- Lee, J.I.; Ha, K.; Yoo, H.S. Quantum-dot-assisted fluorescence resonance energy transfer approach for intracellular trafficking of chitosan/DNA complex. Acta Biomat. 2008, 4, 791–798. [Google Scholar] [CrossRef]
- Sato, T.; Ishii, T.; Okahata, Y. In vitro gene delivery mediated by chitosan. Effect of pH, serum, and molecular mass of chitosan on the transfection efficiency. Biomaterials 2001, 22, 2075–2080. [Google Scholar] [CrossRef]
- Ge, Z.; Baguenard, S.; Lim, L.S.; Wee, A.; Khor, E. Hydroxyapatite-chitin materials as potential tissue-engineered bone substitutes. Biomaterials 2004, 25, 1049–1058. [Google Scholar]
- Shi, C.; Zhu, Y.; Ran, X.; Wang, M.; Su, Y.; Cheng, T. Therapeutic Potential of Chitosan and Its Derivatives in Regenerative Medicine. J. Surg. Res. 2006, 133, 185–192. [Google Scholar] [CrossRef]
- Nettles, D.L.; Elder, S.H.; Gilbert, J.A. Potential use of chitosan as a cell scaffold material for cartilage tissue engineering. Tissue Eng. 2002, 8, 1009–1020. [Google Scholar] [CrossRef]
- Green, D.W.; Leveque, I.; Walsh, D.; Howard, D.; Yang, X.; Partridge, K.; Mann, S.; Oreffo, R.O.C. Biomineralized Polysaccharide Capsules for Encapsulation, Organization, and Delivery of Human Cell Types and Growth Factors. Adv. Funct. Mat. 2005, 15, 917–923. [Google Scholar] [CrossRef]
- Ertesvag, H.; Valla, S. Biosynthesis and applications of alginates. Polym. Degrad. Stabil. 1998, 59, 85–91. [Google Scholar] [CrossRef]
- Johnson, F.A.; Craig, D.Q.M.; Mercer, A.D. Characterization of the block structure and molecular weight of sodium alginates. J. Pharm. Pharmacol. 1997, 49, 639–643. [Google Scholar] [CrossRef]
- Draget, K.I.; Smidsrod, O.; Skjàk-Bræk, G. Alginate from algae. In Polysaccharide and Polyamides in the food Industry; Steinbüchel, A., Rhee, S.K., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, 2005; pp. 1–30. [Google Scholar]
- Ertesvåg, H.; Valla, S.; Skjåk-Bræk, G. Genetics and biosynthesis of alginates. Carbohyd. Europe 1996, 14, 14–18. [Google Scholar]
- Grant, G.T.; Morris, E.S.; Rees, D.A.; Smith, P.J.C.; Thom, D. Biological interactions between polysaccharides and divalent cations: the egg-box model. Febs. Letts. 1973, 32, 195–198. [Google Scholar] [CrossRef]
- Serp, D.; Cantana, E.; Heinzen, C.; Stockar, U.V.; Marison, I.W. Characterization of an encapsulation device for the production of monodisperse alginate beads for cell immobilization. Biotech. Bioeng. 2000, 70, 41–53. [Google Scholar] [CrossRef]
- Smidsrod, O.; Haug, A. Dependence upon uronic acid composition of some ion-exchange properties of alginates. Acta Chem. Scand. 1968, 22, 1989–1997. [Google Scholar] [CrossRef]
- Dumitriu, S. Bioencapsulation of living cells by entrapment in polysaccharide gels. In Polysaccharides: Structural Diversity and Functional Versatility; Marcel Dekker, Inc.: New York, 1998; pp. 755–757. [Google Scholar]
- Amsden, B.; Turner, N. Diffusion characteristics of calcium alginate gels. Biotech. Bioeng. 1999, 65, 605–610. [Google Scholar] [CrossRef]
- Rassis, D.K.; Saguy, I.S.; Nussinovitch, A. Food hydrocolloids 2002, 16, 139–151. [CrossRef]
- Onsøyen, E. Alginates. In Thickening and gelling agents for food; Imeson, A., Ed.; Blackie Academic & Professional: London, 1992; pp. 1–24. [Google Scholar]
- Kuo, C.K.; Ma, P.X. Ionically crosslinked alginate hydrogels as scaffolds for tissue engineering: Part 1. Structure, gelation rate and mechanical properties. Biomaterials 2001, 22, 511–521. [Google Scholar] [CrossRef]
- Smidsrod, O.; Draget, K.I. Chemistry and physical properties of alginates. Carbohyd. Eur. 1996, 14, 6–13. [Google Scholar]
- Yeom, C.K.; Lee, K.-H. Characterization of sodium alginate membrane crosslinked with glutaraldehyde in pervaporation separation. J. Appl. Polym. Sci. 1998, 67, 209–219. [Google Scholar] [CrossRef]
- Smeds, K.A.; Grinstaff, M.W. Photocrosslinkable polysaccharides for in situ hydrogel formation. J. Biomed. Mat. Res. 2001, 54, 115–121. [Google Scholar] [CrossRef]
- Smidsrod, O.; Skjak-Braek, G. Alginate as immobilization matrix for cells. Trends Biotech. 1990, 8, 71–78. [Google Scholar] [CrossRef]
- Linker, A.; Jones, R.S. A polysaccharide resembling alginic acid from Pseudomonas micro-organism. Nature 1964, 204, 187–188. [Google Scholar] [CrossRef]
- Albarghouthi, M.; Fara, D.A.; Saleem, M.; El-Thaher, T.; Matalka, K.; Badwan, A. Immobilization of antibodies on alginate-chitosan beads. Int. J. Pharm. 2000, 206, 23–34. [Google Scholar] [CrossRef]
- Rowley, J.A.; Madlambayan, G.; Mooney, D.J. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 1999, 20, 45–53. [Google Scholar] [CrossRef]
- Kunioka, M.; Furusawa, K. Poly(γ-glutamic acid) hydrogel prepared from microbial poly(γ-glutamic acid) and alkanediamine with water-soluble carbodiimide. J. Appl. Polym. Sci. 1997, 65, 1889–1896. [Google Scholar] [CrossRef]
- Gomez, C.G.; Chambat, G.; Heyraud, H.; Villar, M.; Auzély-Velty, R. Synthesis and characterization of a β-CD-alginate conjugate. Polymer 2006, 47, 8509–8516. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate chemistry; Academic Press: San Diego, 1996; pp. 169–173. [Google Scholar]
- Carrè, M.C.; Delestre, C.; Hubert, P.; Dellacherie, E. Covalent coupling of a short polyether on sodium alginate: synthesis and characterization of the resulting amphiphilic derivative. Carbohyd. Polym. 1991, 16, 367–379. [Google Scholar] [CrossRef]
- Kang, H.A.; Jeon, G.J.; Lee, M.Y.; Yang, J.W. Effectiveness test of alginate-derived polymeric surfactants. J. Chem. Techn. Biotechn. 2002, 77, 205–208. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Lesieur, S.; Labarre, D.; Jayakrishnan, A. Periodate oxidation of sodium alginate in water and in ethanol-water mixture: a comparative study. Carbohyd. Res. 2005, 340, 1425–1429. [Google Scholar] [CrossRef]
- Balkrishnan, B.; Jayakrishnan, A. Self-cross-linking biopolymers as injectable in situ forming biodegradable scaffolds. Biomaterials 2005, 26, 3941–3951. [Google Scholar] [CrossRef]
- Painter, T.; Larsen, B. Formation of hemiacetals between neighbouring hexuronic acid residues during the periodate oxidation of alginate. Acta Chem. Scand. 1970, 24, 813–833. [Google Scholar] [CrossRef]
- Han, D.K.; Park, H.D.; Han, K.-D.; Jeong, S.V.; Kim, Y.H. Preparation and surface characterization of PEO-grafted and heparin-immobilized polyurethanes. J. Biom. Mater. Res. Appl. Biomat. 1989, 23(S13), 87–104. [Google Scholar]
- Tu, R.; Lu, C.L.; Thyagarajan, K.; Wang, E.; Nguyen, H.; Shen, S.; Hata, C.; Quijano, R.C. Kinetic study of collagen fixation with polyepoxy fixatives. J. Biom. Mater. Res. 1993, 27, 3–9. [Google Scholar] [CrossRef]
- Chen, JP.; Chu, I.M.; Shiao, M.Y.; Hsu, B.R.; Fu, S.H. Microencapsulation of islets in PEG-amine modified alginate-poly(l-lysine)-alginate microcapsules for constructing bioartificial pancreas. J. Ferment. Bioeng. 1998, 86, 185–190. [Google Scholar] [CrossRef]
- Chandy, T.; Mooradian, D.L.; Rao, G.H.R. Chitosan/polyethylene glycol-alginate microcapsules for oral delivery of hirudin. J. Appl. Polym. Sci. 1998, 70, 2143–2153. [Google Scholar] [CrossRef]
- Pluemsab, W.; Sakairi, N.; Furuike, T. Synthesis and inclusion properties of a α-cyclodextrin-linked alginate. Polymer 2005, 46, 9778–9783. [Google Scholar]
- Bayomi, M.A.; Al-Suwayeh, S.A.; El-Helw, A.-R.M. Excipient-excipient interaction in the design of sustained-release theophylline tablets: in vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2001, 27, 499–506. [Google Scholar] [CrossRef]
- Jerry, N.; Anitha, Y.; Sharma, C.P.; Sony, P. In vivo absorption studies of insulin from an oral delivery system. Drug Deliv. 2001, 8, 19–23. [Google Scholar] [CrossRef]
- Fragonas, E.; Valente, M.; Pozzi-Mucelli, M.; Toffanin, R.; Rizzo, R.; Silvestri, F.; Vittur, F. Articular cartilage repair in rabbits by using suspensions of allogenic chondrocytes in alginate. Biomaterials 2000, 21, 795–801. [Google Scholar]
- Cohen, S.; Lobel, E.; Trevgoda, A.; Peled, Y. A novel in situ-forming ophthalmic drug delivery system from alginates undergoing gelation in the eye. J. Control. Release 1997, 44, 201–208. [Google Scholar] [CrossRef]
- Miyazaki, S.; Kubo, W.; Attwood, D. Oral sustained delivery of theophylline using in-situ gelation of sodium alginate. J. Control. Release 2000, 67, 275–280. [Google Scholar] [CrossRef]
- Hodsdon, A.C.; Mitchell, J.R.; Davies, M.C.; Melia, C.D. Structure and behaviour in hydrophilic matrix sustained release dosage forms: 3. The influence of pH on the sustained-release performance and internal gel structure of sodium alginate matrices. J. Control. Release 1995, 33, 143–152. [Google Scholar] [CrossRef]
- Yong, C.S.; Jung, J.-H.; Rhee, J.-D.; Kim, C.-K.; Choi, H.-G. Physicochemical characterization and evaluation of buccal adhesive tablets containing omeprazole. Drug Dev. Ind. Pharm. 2001, 27, 447–455. [Google Scholar] [CrossRef]
- Srivastavas, R.; Brown, J.Q.; Zhu, H.; McShane, M.J. Stabilization of glucose oxidase in alginate microspheres with photoreactive diazoresin nanofilm coatings. Biotechn. Bioengin. 2005, 91, 124–131. [Google Scholar] [CrossRef]
- Miyazaki, S.; Nakayama, A.; Oda, M.; Takada, M.; Attwood, D. Drug release from oral mucosal adhesive tablets of chitosan and sodium alginate. Int. I. Pharm. 1995, 118, 257–263. [Google Scholar] [CrossRef]
- Strand, B.L.; Gaserod, O.; Kulseng, B.; Espevik, T.; Skjak-Braek, G. Alginate-polylisine-alginate microcapsules: effect of size reduction on capsule properties. J. Microencapsul. 2002, 19, 612–630. [Google Scholar]
- Murata, Y.; Sasaki, N.; Miyamoto, E.; Kawashima, S. Use of floating alginate gel beads for stomach-specific drug delivery. Eur. J. Pharm. Biopharm. 2000, 50, 221–226. [Google Scholar] [CrossRef]
- Choi, B.Y.; Park, H.J.; Hwang, S.J.; Park, J.B. Preparation of alginate beads for floating durg delivery system: effect of CO2 gas-forming agents. Int. J. Pharm. 2002, 239, 81–91. [Google Scholar] [CrossRef]
- Yazdani-Pedram, M.; Lagos, A.; Jaime Reweri, P. Study of the effect of reaction variables on grafting of polyacrylamide onto chitosan. Polym. Bull. 2002, 48, 93–98. [Google Scholar] [CrossRef]
- Hu, Y.; Jiang, X.; Ding, Y.; Ge, H.; Yuan, Y.; Yang, C. Synthesis and characterization of chitosan-polyacrylic acid nanoparticles. Biomaterials 2002, 23, 3193–3201. [Google Scholar] [CrossRef]
- Chun, M.K.; Cho, C.S.; Choi, H.K. Mucoadhesive drug carrier based on interpolymer complex of poly(vinyl pyrrolidone) and poly(acrylic acid) prepared by template polymerization. J. Control. Release 2002, 81, 327–334. [Google Scholar] [CrossRef]
- Ahn, J.S.; Choi, H.K.; Cho, C.S. A novel mucoadhesive polymer prepared by template polymerization of acrylic acid in the presence of chitosan. Biomaterials 2001, 22, 923–928. [Google Scholar] [CrossRef]
- Shojaei, A.H.; Paulson, J.; Honary, S. Evaluation of poly(acrylic acid-co-ethylhexyl acrylate) films for mucoadhesive transbuccal drug delivery: factors affecting the force of mucoadhesion. J. Control. Release 2000, 67, 223–232. [Google Scholar] [CrossRef]
- Peniche, C.; Argüelles-Monal, W.; Davidenko, N.; Sastre, R.; Gallardo, A.; San Román, J. Self-curing membranes of chitosan/PAA IPNs obtained by radical polymerization: preparation, characterization and interpolymer complexation. Biomaterials 1999, 20, 1869–1878. [Google Scholar] [CrossRef]
- Mumper, R.J.; Hoffman, A.S.; Puolakkainen, P.A.; Bouchard, L.S.; Gombotz, W.R. Calcium-alginate beads for the oral delivery of transforming growth factor-β1 (TGF-β1): stabilization of TGF-β1 by the addition of polyacrylic acid within acid-treated beads. J. Control. Release 1994, 30, 241–251. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical applications. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. Hydrogels as mucoadhesive and bioadhesive materials: a review. Biomaterials 1996, 17, 1553–1561. [Google Scholar] [CrossRef]
- Limer, A.J.; Rullay, A.K.; Miguel, V.S.; Peinado, C.; Keely, S.; Fitzpatrick, E.; Carrington, F.D.; Brayden, D.; Haddleton, D.M. Fluorescently tagged star polymers by living radical polymerisation for mucoadhesion and bioadhesion. React. Funct. Polym. 2006, 66, 51–64. [Google Scholar] [CrossRef]
- Laurienzo, P.; Malinconico, M.; Mattia, G.; Russo, R.; La Rotonda, M.I.; Quaglia, F.; Capitani, D.; Mannina, L. Novel alginate-acrylic polymers as a platform for drug delivery. J. Biomed. Mater. Res., Part A 2006, 78, 523–531. [Google Scholar]
- Matsumoto, M.; Takakura, K.; Okaya, T. Radical polmerizations in solution. In Polymerization processes; Schildknecht, C.E., Skeist, I., Eds.; John Wiley & Sons: New York, 1977; pp. 198–227. [Google Scholar]
- Raghuram Reddy, C.; George, A.; Rami Reddy, C. Immobilization of trypsin on poly(alginic acid-co-glycidyl methacrylate-co-hydroxy ethyl methacrylate). Angew. Makromol. Chem. 1986, 144, 183–192. [Google Scholar] [CrossRef]
- Raghuram Reddy, C.; George, A.; Rami Reddy, C. Immobilization of trypsin on poly(alginic acid-co-glycidyl methacrylate-co-methyl methacrylate). Angew. Makromol. Chem. 1987, 149, 101–110. [Google Scholar] [CrossRef]
- Morris, K.F.; Johnson, C.S. Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar] [CrossRef]
- Morris, K.F.; Stilbs, P.; Johnson, C.S. Analysis of mixtures based on molecular size and hydrophobicity by means of diffusion-ordered 2D NMR. Anal. Chem. 1994, 66, 211–215. [Google Scholar] [CrossRef]
- Morris, K.F.; Johnson, C.S. Resolution of discrete and continuous molecular size distributions by means of diffusion-ordered 2D NMR spectroscopy. J. Am. Chem. Soc. 1993, 115, 4291–4299. [Google Scholar] [CrossRef]
- Viel, S.; Capitani, D.; Mannina, L.; Segre, A.L. Diffusion-Ordered NMR Spectroscopy: A versatile tool for the molecular weight determination of uncharged polysaccharides. Biomacromolecules 2003, 4, 1843–1847. [Google Scholar] [CrossRef]
- Scott, J.E.; Tigwell, M.J.; Phelps, C.F.; Nieduszynski, I.A. On the mechanism of scission of alginate chains by periodate. Carbohyd. Res. 1976, 47, 105–117. [Google Scholar] [CrossRef]
- Tonnesen, H.H.; Karlsen, J. Alginate in drug delivery systems. Drug Dev Ind Pharm. 2002, 28, 621–30. [Google Scholar] [CrossRef]
- Seifert, D.B.; Phillips, J.A. Porous alginate-poly(ethylene glycol) entrapment system for the cultivation of mammalian cells. Biotechnol. Prog. 1997, 13, 569–576. [Google Scholar] [CrossRef]
- Laurienzo, P.; Malinconico, M.; Motta, A.; Vicinanza, A. Synthesis of a novel alginate-poly(ethylene glycol) graft copolymer for cell immobilization. Carbohyd. Polym. 2005, 62, 274–282. [Google Scholar] [CrossRef]
- Allan, C.R.; Hadwiger, L.A. The fungicidal effect of chitosan on fungi of varying cell wall composition. Exp. Mycol. 1979, 3, 285–287. [Google Scholar] [CrossRef]
- Knorr, D. Use of chitinous polymers in food. A challenge for food research and development. Food Technol. 1984, 38, 85–97. [Google Scholar]
- Muzzarelli, R.A.A. Chitin; Pergamon: Oxford, 1977. [Google Scholar]
- Ashford, N.A.; Hattis, D.; Murray, A.E. Industrial prospects for chitin and protein from shellfish wastes. MIT Sea Grant Report MISG 77–3; MIT: Cambridge, MA, 1977. [Google Scholar]
- Shigemasa, Y.; Minami, S. Applications of chitin and chitosan for biomaterials. Biotech. Gen. Eng. Rev. 1995, 13, 383–415. [Google Scholar] [CrossRef]
- Kifune, K. Clinical application of chitin artificial skin (Beschitin W). In Advances in chitin and chitosan; Brine, C.J., Sandford, P.A., Zikakis, J.P., Eds.; Elsevier: London, 1992; pp. 9–15. [Google Scholar]
- Hirota, Y.; Tanioka, S.; Tanigawa, T.; Tanaka, Y.; Ojima, R. Clinical applications of chitin and chitosan to human decubitus. In Advances in chitin science; Domard, A., Jeuniaux, C., Muzzarelli, R., Roberts, G., Eds.; Jacques Andre Ed: Lyon, 1996; Vol.1, pp. 407–413. [Google Scholar]
- Freier, T.; Montenegro, R.; Koh, H.S.; Shoichet, M.S. Chitin-based tubes for tissue engineering in the nervous system. Biomaterials 2005, 26, 4624–4632. [Google Scholar] [CrossRef]
- No, H.K.; Meyers, S.P. Utilization of Crawfish Processing Wastes as Carotenoids, Chitin, and Chitosan Souces. J. Kor. Soc. Food Nutr. 1992, 21, 319–326. [Google Scholar]
- Tolaimate, A.; Desbrieres, J.; Rhazi, M.; Alagui, A.; Vincendon, M.; Vottero, P. On the influence of deacetylation process on the physicochemical characteristics of chitosan from squid chitin. Polymer 2000, 41, 2463–2469. [Google Scholar] [CrossRef]
- Rudall, K.M. Advances in insect physiology; Beament, J.W.L., Treherne, J.E., Wigglesworth, V.B., Eds.; Academic Press: London, 1963; p. 257. [Google Scholar]
- Knaul, J.Z.; Hudson, S.M.; Creber, K.A.M. Crosslinking of chitosan fibers with dialdehydes: Proposal of a new reaction mechanism. J. Polym. Sci. B: Polym. Phys. 1999, 37, 1079–1094. [Google Scholar] [CrossRef]
- No, H.K.; Meyers, S.P. Preparation and Characterization of Chitin and Chitosan-A Review. J. Aquat. Food Prod. Technol. 1995, 4, 27–52. [Google Scholar] [CrossRef]
- No, H.K.; Cho, Y.I.; Kim, H.R.; Meyers, S.P. Effective Deacetylation of Chitin under Conditions of 15 psi/121ºC. J. Agric. Food Chem. 2000, 48, 2625–2627. [Google Scholar] [CrossRef]
- Khan, T.; Peh, K.; Ch’ng, H.S. Reporting degree of deacetylation values of chitosan: the influence of analytical methods. J. Pharm. Pharmaceut. Sci. 2002, 5, 205–212. [Google Scholar]
- Moore, G.K.; Roberts, G.A.F. Studies on the acetylation of Chitosan. In Proceedings of the First International Conference on Chitin/Chitosan; Muzzarelli, R.A.A., Pariser, E.R., Eds.; MIT Sea Grant Program: Cambridge, MA, 1978; pp. 421–425. [Google Scholar]
- Li, Q.; Dunn, E.T.; Grandmaison, E.W.; Goosen, M.F.A. Applications and properties of chitosan. J. Bioact. Compat. Polym. 1992, 7, 370–397. [Google Scholar] [CrossRef]
- Sini, T.K.; Santhosh, S.; Mathew, P.T. Study on the influence of processing parameters for the preparation of carboxymethylchitin. Polymer 2005, 46, 3128–3131. [Google Scholar] [CrossRef]
- Hudson, S.M.; Jenkins, D.W. Chitin and chitosan. In EPSTMark, H.F., Ed.; Wiley: New York, 2003; Vol.1, (3rd ed.); pp. 569–580. [Google Scholar]
- Gupta, K.C.; Jabrail, F.H. Glutaraldheyde cross-linked chitosan microspheres for controlled release of centchroman. Carbohyd. Res. 2007, 342, 2244–2252. [Google Scholar] [CrossRef]
- Nemtsev, S.V.; Gamzazade, A.I.; Rogozhin, S.V.; Bykova, V.M.; Bykov, V.P. Deacetylation of chitin under homogeneous conditions. Appl. Biochem. Microb. 2002, 3, 8521–8526. [Google Scholar]
- Muzzarelli, R.A.A.; Muzzarelli, C. Chitosan Chemistry: Relevance to the Biomedical Sciences. Adv Polym Sci 2005, 186, 151–209. [Google Scholar] [CrossRef]
- Sashiwa, H.; Makimura, Y.; Shigemasa, Y.; Roy, R. Chemical modification of chitosan: preparation of chitosan-sialic acid branched polysaccharide hybrids. Chem Comm. 2000, 909–910. [Google Scholar]
- Muzzarelli, R.A.A. Carboxymethylated chitins and chitosans. Carbohydr. Polym. 1988, 8, 1–21. [Google Scholar] [CrossRef]
- Chen, S.C.; Wu, Y.C.; Mi, F.L.; Lin, Y.-H.; Yu, L.-C.; Sung, H.-W. A novel pH-sensitive hydrogel composed of N,O-carboxymethyl chitosan and alginate cross-linked by genipin for protein drug delivery. J. Control. Release 2004, 96, 285–300. [Google Scholar] [CrossRef]
- Thanou, M.; Nihot, M.T.; Jansen, M.; Verhoef, J.C.; Junginger, H.E. Mono-N-carboxymethyl chitosan (MCC), a polyampholytic chitosan derivative, enhances the intestinal absorption of low molecular weight heparin across intestinal epithelia in vitro and in vivo. J. Pharm. Sci. 2001, 90, 38–46. [Google Scholar] [CrossRef]
- Chen, Y.; Tan, H.-M. Crosslinked carboxymethylchitosan-g-poly(acrylic acid) copolymer as a novel superabsorbent polymer. Carbohydr. Res. 2006, 341, 887–896. [Google Scholar] [CrossRef]
- Sui, W.; Wang, S.; Chen, G.; Xu, G. Surface and aggregate properties of an amphiphilic derivative of carboxymethylchitosan. Carbohydr. Res. 2004, 339, 1113–1118. [Google Scholar] [CrossRef]
- Zhu, A.P.; Liu, J.H.; Ye, W.H. Effective loading and controlled release of camptothecin by O-carboxymethylchitosan aggregates. Carbohydr. Polym. 2006, 63, 89–96. [Google Scholar] [CrossRef]
- Kurita, K. Chitin and chitosan: functional biopolymers from marine crustaceans. Marine Biotechnol. 2006, 8, 203–226. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; Tanfani, F.; Emanuelli, M.; Mariotti, S. N-(Carboxymethylidene) chitosans and N-(carboxymethyl)chitosans: novel chelating polyampholytes obtained from chitosan glyoxylate. Carbohydr. Res. 1982, 107, 199–214. [Google Scholar] [CrossRef]
- Shigemasa, Y.; Ishida, A.; Sashiwa, H.; Saimoto, H.; Okamoto, Y.; Minami, S.; Matsuhashi, A. Synthesis of a new chitin derivative, (1-carboxyethyl)chitosan. Chem. Lett. 1995, 24, 623–624. [Google Scholar]
- Rinaudo, R.; Desbrieres, J.; Le Dung, P.; Binh, T.; Dong, N.T. 1NMR investigation of chitosan derivatives formed by the reaction of chitosan with levulinic acid. Carbohydr. Polym. 2001, 46, 339–348. [Google Scholar] [CrossRef]
- Biagini, G.; Bertani, A.; Muzzarelli, R.A.A; Damadei, A.; Di Benedetto, G.; Belligolli, A.; Riccoti, G.; Zucchini, C.; Rizzoli, C. Wound management with N-carboxybutylchitosan. Biomaterials 1991, 12, 281–286. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; Muzzarelli, C.; Tarsi, R.; Miliani, M.; Gobbanelli, F.; Cartolari, M. Fungistatic activity of modified chitosans against Saprolegnia parasitica. Biomacromolecules 2001, 2, 165–169. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Huang, K.-L.; Peng, D.-M.; Ding, P.; Li, G.-Y. Preparation and characterization of glutaraldehyde cross-linked O-carboxymethylchitosan microspheres for controlled delivery of pazufloxacin mesilate. Int. J. Biol. Macromol. 2007, 41, 87–93. [Google Scholar] [CrossRef]
- Alban, S.; Schauerte, A.; Franz, G. Anticoagulant sulfated polysaccharides: Part I. Synthesis and structure–activity relationships of new pullulan sulfates. Carbohydr. Polym. 2002, 47, 267–276. [Google Scholar] [CrossRef]
- Huang, R.H.; Du, Y.; Zhang, L.S.; Liu, H.; Fan, L.H. A new approach to chemically modified chitosan sulfates and study of their influences on the inhibition of Escherichia coli and Staphylococcus aureus growth. React. Funct. Polym. 2004, 59, 41–51. [Google Scholar] [CrossRef]
- Desai, U.R. New antithrombin-based anticoagulants. Med. Res. Rev. 2004, 24, 151–181. [Google Scholar] [CrossRef]
- Drozd, N.N.; Sher, A.I.; Makarov, V.A.; Vichoreva, G.A.; Gorbachiova, I.N.; Galbraich, L.S. Comparison of antitrombin activity of the polysulphate chitosan derivatives in vitro and in vivo system. Thromb. Res. 2001, 102, 445–455. [Google Scholar] [CrossRef]
- Horton, D.; Just, E.K. Preparation from Chitin of 2-Amino-2-deoxy-(1-64)-ß-glucopyranuronan and its 2-Sulfoamino Analog Having Blood-anticoagulant Properties. Carbohydr Res. 1973, 29, 173–180. [Google Scholar] [CrossRef]
- Vikhoreva, G.; Bannikova, G.; Stolbushkina, P.; Panov, A.; Drozd, N.; Makarov, V.; Varlamov, V.; Gal’braikh, L. Preparation and anticoagulant activity of a low-molecular-weight sulfated chitosan. Carbohydr. Polym. 2005, 62, 327–332. [Google Scholar] [CrossRef]
- Holme, K.R.; Perlin, A.S. Chitosan N-sulfate. A water-soluble polyelectrolyte. Carbohydr. Res. 1997, 302, 7–12. [Google Scholar] [CrossRef]
- Warner, D.T.; Coleman, L.L. Selective sulfonation of amino groups in amino alcohols. J. Org. Chem. 1958, 23, 1133–1135. [Google Scholar] [CrossRef]
- Zhang, C.; Ping, Q.; Zhang, H.; Shen, J. Preparation of N-alkyl-O-sulfate chitosan derivatives and micellar solubilization of taxol. Carbohydr. Polym. 2003, 54, 137–141. [Google Scholar] [CrossRef]
- Sashiwa, H.; Kawasaki, N.; Nakayama, A.; Muraki, E.; Yamamoto, N.; Zhu, H.; Nagano, H.; Omura, Y.; Saimoto, H.; Shigemasa, Y.; Aiba, S. Synthesis of organo-soluble, palladium adsorbable, and biodegradable chitosan derivatives toward the chemical plating on plastics. Biomacromolecules 2002b, 3, 1120–1125. [Google Scholar]
- Sashiwa, H.; Kawasaki, N.; Nakayama, A.; Muraki, E.; Yamamoto, N.; Aiba, S. Synthesis of water-soluble chitosan derivatives by simple acetylation. Biomacromolecules 2002c, 3, 1126–1128. [Google Scholar]
- Badawy, M.E.I.; Rabea, E.I.; Rogge, T.M; Stevens, C.V.; Smagghe, G.; Steurbaut, W.; Hofte, M. Synthesis and fungicidal activity of new N,O-acyl chitosan derivatives. Biomacromolecules 2004, 5, 589–595. [Google Scholar] [CrossRef]
- Lee, D.-W.; Baney, R.H. Detoxification of amitriptyline by oligochitosan derivatives. Biotechnol. Lett. 2004, 26, 713–716. [Google Scholar] [CrossRef]
- Lee, D.-W.; Powers, K.; Baney, R. Physicochemical properties and blood compatibility of acylated chitosan nanoparticles. Carbohyd. Polym. 2004, 58, 371–377. [Google Scholar] [CrossRef]
- Underhill, R.S.; Jovanovi, A.V.; Carino, S.R.; Varshney, M.; Shah, D.; Dennis, D.M.; Morey, T.E.; Duran, R.S. Oil-filled silica nanocapsules for lipophilic drug uptake: implication for drug detoxification therapy. Chem. Mater. 2002, 14, 4919–4925. [Google Scholar] [CrossRef]
- Aiedeh, K.; Taha, M.O. Synthesis of chitosan succinate and chitosan phthalate and their evaluation as suggested matrices in orally administered, colon-specific drug delivery systems. Arch. Pharm. 1999, 332, 103–107. [Google Scholar] [CrossRef]
- Hall, L.D.; Yalpani, M. Formation of branched-chain, soluble polysaccharides from chitosan. J. Chem. Soc. Chem. Comm. 1980, 1153–1154. [Google Scholar] [CrossRef]
- Yalpani, M.; Hall, L.D. Some chemical and analytical aspects of polysaccharide modifications.3.Formation of branched-chain, soluble chitosan derivatives. Macromolecules 1984, 17, 272–281. [Google Scholar] [CrossRef]
- Kato, Y.; Onishi, H.; Machida, Y. Biological characterization of lactosaminated N-succinyl-chitosan as a liver-specific drug carrier in mice. J. Control. Release 2001, 70, 295–307. [Google Scholar] [CrossRef]
- Park, I.K.; Yang, J.; Jeong, H.J.; Bom, H.S.; Harada, I.; Akaike, T.; Kim, S.; Cho, C.S. Galactosylated chitosan as a synthetic extracellular matrix for hepatocytes attachment. Biomaterials 2003, 24, 2331–2337. [Google Scholar] [CrossRef]
- Yang, T.C.; Chou, C.C.; Li, C.F. Preparation, water solubility and rheological property of the N-alkylated mono or disaccharide chitosan derivatives. Food Res. Inter. 2002, 35, 707–713. [Google Scholar] [CrossRef]
- Yang, T-C.; Chou, C.-C.; Li, C.F. Antibacterial activity of N-alkylated disaccharide chitosan derivatives. Int. J. Food Microb. 2005, 97, 237–245. [Google Scholar] [CrossRef]
- Sashiwa, H.; Shigemasa, Y. Chemical modification of chitin and chitosan: 2. Preparation and water soluble property of N-acylated or N-alkylated partially deacetylated chitins. Carbohyd. Polym. 1999, 39, 127–138. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y. Study on β-cyclodextrin grafting with chitosan and slow release of its inclusion complex with radioactive iodine. J. Appl. Polym. Sci. 2001, 82, 2414–2421. [Google Scholar] [CrossRef]
- Jung, B.O.; Kim, C.H.; Choi, K.S.; Lee, Y.M.; Kim, J.J. Preparation of amphiphilic chitosan and their antimicrobial activities. J. Appl. Polym. Sci. 1999, 72, 1713–1719. [Google Scholar] [CrossRef]
- Lueßen, H.L.; de Leeuw, B.J.; Perard, D.; Lehr, C.-M.; de Boer, A.G.; Verhoef, J.C.; Junginger, H.E. Mucoadhesive polymers in peroral peptide drug delivery. I. Influence of mucoadhesive excipients on the proteolytic activity of intestinal enzymes. Eur. J. Pharm Sci. 1996, 4, 117–128. [Google Scholar] [CrossRef]
- Thanou, M.; Verhoef, J.C.; Junginger, H.E. Oral drug absorption enhancement by chitosan and its derivatives. Adv. Drug Deliv. Rev. 2001, 52, 117–126. [Google Scholar] [CrossRef]
- Hoffman, A.S.; Chen, G.; Wu, X.; Ding, Z.; Kabra, B.; Randeri, K.; Schiller, L.; Ron, E.; Peppas, N.A.; Brazel, C. Graft copolymers of PEO-PPO-PEO triblock polyethers on bioadhesive polymer backbones: synthesis and properties. Polymer Preprints (American Chemical Society, Division of Polymer Chemistry) 1997, 38, 524–525. [Google Scholar]
- Tasker, R.A.; Connell, B.J.; Ross, S.J.; Elson, C.M. Development of an injectable sustained-release formulation of morphine: antinociceptive properties in rats. Lab. Anim. 1998, 32, 270–275. [Google Scholar] [CrossRef]
- Ono, K.; Saito, Y.; Yura, H.; Ishikawa, K.; Kurita, A.; Akaike, T.; Ishihara, M. Photo-crosslinkable chitosan as a biological adhesive. J. Biomed. Mat. Res. 2000, 49, 289–295. [Google Scholar] [CrossRef]
- Singh, D.K.; Ray, A.R. Characterization of grafted chitosan films. Carbohyd. Polym. 1998, 36, 251–255. [Google Scholar] [CrossRef]
- Luckachan, G.E.; Pillai, C.K.S. Chitosan/oligo L-lactide graft copolymers: Effect of hydrophobic side chains on the physico-chemical properties and biodegradability. Carbohyd. Polym. 2006, 64, 254–266. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, Y.; Yang, W.; Wang, C.; Hu, J.; Fu, S. Synthesis and characterization of a novel amphiphilic chitosan–polylactide graft copolymer. Carbohyd. Polym. 2005, 59, 165–171. [Google Scholar] [CrossRef]
- Jenkins, D.W.; Hudson, S.M. Review of vinyl graft copolymerization featuring recent advances toward controlled radical-based reactions and illustrated with chitin/chitosan trunk. Chem. Rev. 2001, 101, 3245–3274. [Google Scholar] [CrossRef]
- Bayramoglu, G.; Yilmaz, M.; Yakup Anca, M. Affinity dye-ligand poly(hydroxyethyl methacrylate)/chitosan composite membrane for adsorption lysozyme and kinetic properties. Biochem. Eng. J. 2003, 13, 35–42. [Google Scholar] [CrossRef]
- Lagos, A.; Reyes, J. Grafting onto chitosan. I.Graft copolymerization of methyl methacrylate onto chitosan with Fenton’s reagent (Fe2+–H2O2) as a redox initiator. J. Polym. Sci. A 1988, 26, 985–991. [Google Scholar] [CrossRef]
- Liang, J.; Ni, P.; Zhang, M.; Yu, Z. Graft copolymerization of (dimethylamino)ethyl methacrylate onto chitosan initiated by ceric ammonium nitrate. J. Macrom. Sci. Pure Appl. Chem. 2004, A41, 685–696. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, Y.; Shen, J.; Lin, S. Synthesis and characterization of chitosan grafted poly(N,N-dimethyl-N-meth-acryloxyethyl-N-(3-sulfopropyl)ammonium)initiated by ceric (IV) ion. Eur. Polym. J. 2003, 39, 847–850. [Google Scholar] [CrossRef]
- Cai, H.; Zhang, Z.; Sun, P.; Zhang, Y.; He, B. Preparation of hydrogels based on poly-N-isopropyl-acrylamide grafted chitosan via γ−radiation and their properties. Acta Polym. Sin. 2005, 5, 709–713. [Google Scholar]
- Shantha, K.L.; Bala, U.; Rao, K.P. Tailor-made chitosans for drug delivery. Eur. Polym. J. 1995, 31, 377–382. [Google Scholar] [CrossRef]
- Yao, F.; Chen, W.; Wang, H.; Liu, H.; Yao, K.; Sun, P.; Lin, H. A study on cytocompatible poly(chitosan-g-l-lactic acid). Polymer 2003, 44, 6435–6441. [Google Scholar] [CrossRef]
- Chunmeng, S.; Ying, Z.; Xinze, R.; Meng, W.; Yongping, S.; Tianmin, C. Therapeutic Potential of Chitosan and Its Derivatives in Regenerative Medicine. J. Surg. Res. 2006, 133, 185–192. [Google Scholar] [CrossRef]
- Mao, C.; Zhao, W.B.; Zhu, A.P.; Shen, J.; Lin, S.C. Introduction of photocrosslinkable chitosan to polyethylene film by radiation grafting and its blood compatibility. Mat. Sci. Eng. C 2004, 24, 479–485. [Google Scholar]
- Mao, C.; Zhao, W.B.; Zhu, A.P.; Shen, J.; Lin, S.C. A photochemical method for the surface modification of poly(vinyl chloride) with O-butyrylchitosan to improve blood compatibility. Process Biochem. 2004, 39, 1151–1157. [Google Scholar] [CrossRef]
- Akkara, J.A.; Ayyagari, M.S.R.; Bruno, F.F. Enzymatic synthesis and modification of polymers in nonaqueous solvents. Trends Biotechnol. 1999, 17, 67–73. [Google Scholar] [CrossRef]
- Kaplan, D.L.; Dordick, J.S.; Gross, R.A.; Swift, G. Enzymes in polymer science: an introduction. Enzymes in polymer synthesis; ACS Symposium Series; Gross, R.A., Kaplan, D.L., Swift, G., Eds.; American Chemical Society: Washington, DC, 1998; Vol. 684, pp. 2–16. [Google Scholar]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Kumbar, S.G.; Kulkarni, A.R.; Aminabhavi, T.M. Crosslinked chitosan microspheres for encapsulation of diclofenac sodium: effect of crosslinking agent. J. Microencapsul. 2002, 19, 173–180. [Google Scholar] [CrossRef]
- Hejazi, R.; Amiji, M. Chitosan-based gastrointestinal delivery systems. J. Control. Release 2003, 89, 151–165. [Google Scholar] [CrossRef]
- Devi, D.A.; Smitha, B.; Sridhar, S.; Aminabhavi, T.M. Pervaporation separation of isopropanol/water mixtures through crosslinked chitosan membranes. J. Membr. Sci. 2005, 262, 91–99. [Google Scholar] [CrossRef]
- Mi, F.-L.; Sung, H.-W.; Shyu, S.-S.; Su, C.-C.; Peng, C.-K. Synthesis and characterization of biodegradable TPP/genipin cross-linked chitosan gel beads. Polymer 2003, 44, 6521–6530. [Google Scholar] [CrossRef]
- Neto, C.G.T.; Dantas, T.N.C.; Fonseca, J.L.C.; Pereira, M.R. Permeability studies in chitosan membranes. Effects of crosslinking and poly(ethylene oxide) addition. Carbohydr. Res. 2005, 340, 2630–2636. [Google Scholar] [CrossRef]
- Ngah, W.S.W.; Ghani, S.A.; Kamari, A. Adsorption behaviour of Fe(II) and Fe(III) ions in aqueous solution on chitosan and cross-linked chitosan beads. Biores. Technol. 2005, 96, 443–450. [Google Scholar] [CrossRef]
- Monteiro, O.A.C.; Airoldi, C. Some studies of crosslinking chitosan-glutaraldehyde interaction in a homogeneous system. Int. J. Biol. Macromol. 1999, 26, 119–128. [Google Scholar] [CrossRef]
- Gupta, K.C.; Jabrail, F.H. Glutaraldehyde cross-linked chitosan microspheres for controlled release of centchroman. Carbohyd. Res. 2007, 342, 2244–2252. [Google Scholar] [CrossRef]
- Kawashima, Y.; Handa, T.; Takenaka, H.; Lin, S.Y. Novel method for the preparation of controlled-release theophylline granules coated with a polyelectrolyte complex of sodium polyphosphate-chitosan. J. Pharm. Sci. 1985, 74, 264–268. [Google Scholar] [CrossRef]
- Kawashima, Y.; Handa, T.; Kasai, A.; Takenaka, H.; Lin, S.Y. The effect of thickness and hardness of the coating film on the drug release of theophylline granules. Chem. Pharm. Bull. 1985, 33, 2469–2474. [Google Scholar] [CrossRef]
- Bodmeier, R.; Oh, K.H.; Prama, Y. Preparation and evaluation of drug-containing chitosan beads. Drug Dev. Ind. Pharm. 1989, 15, 1475–1494. [Google Scholar] [CrossRef]
- Shu, X.Z.; Zhu, K.J. A novel approach to prepare Tripolyphosphate/chitosan complex beads for controlled release drug delivery. Int. J. Pharm. 2000, 201, 51–58. [Google Scholar]
- Gomez d’Ayala, G.; De Rosa, A.; Laurienzo, P.; Malinconico, M. Development of a new calcium sulphate-based composite using alginate and chemically modified chitosan for bone regeneration. J. Biomed. Mater. Res. A 2007, 81, 811–820. [Google Scholar]
- Sidqui, M.; Collin, P.; Vitte, C.; Forest, N. Osteoblast adherence and resorption activity of isolated osteoclasts on calcium sulfate hemihydrate. Biomaterials 1995, 16, 1327–1332. [Google Scholar]
- Murashima, Y.; Yoshikawa, G.; Wadachi, R.; Sawada, N.; Suda, H. Calcium sulphate as a bone substitute for various osseous defects in conjunction with apicectomy. Int. Endod. J. 2002, 35, 768–774. [Google Scholar]
- Pecora, G. L’uso del solfato di calcio nelle tecniche rigenerative multidisciplinari.; Ed. Elite Service s.r.l.: Rome, 2001; pp. 13–77. [Google Scholar]
- Pecora, G.; De Leonardis, D.; Ibrahim, N.; Bovi, M.; Cornelini, R. The use of calcium sulphate in the surgical treatment of a ‘‘through and through’’ periradicular lesion. Int. Endod. J. 2001, 34, 189–196. [Google Scholar] [CrossRef]
- De Leonardis, D.; Pecora, G. Augmentation of the maxillary sinus with calcium sulphate: One year clinical report from a prosective longitudinal study. Int. J. Oral Maxillofac. Implants 1999, 14, 869–879. [Google Scholar]
- Doadrio, J.C.; Arcos, D.; Cabanas, M.V.; Vallet-Regi, M. Calcium sulphate-based cements containing cephalexin. Biomaterials 2004, 25, 2629–2635. [Google Scholar]
- Nobile, M.R; Pirozzi, V.; Somma, E.; Gomez d’Ayala, G.; Laurienzo, P. Development and rheological investigation of novel alginate/N-succinylchitosan hydrogels. J. Polym. Sci. B 2008, 46, 1167–1182. [Google Scholar] [CrossRef]
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D’Ayala, G.G.; Malinconico, M.; Laurienzo, P. Marine Derived Polysaccharides for Biomedical Applications: Chemical Modification Approaches. Molecules 2008, 13, 2069-2106. https://doi.org/10.3390/molecules13092069
D’Ayala GG, Malinconico M, Laurienzo P. Marine Derived Polysaccharides for Biomedical Applications: Chemical Modification Approaches. Molecules. 2008; 13(9):2069-2106. https://doi.org/10.3390/molecules13092069
Chicago/Turabian StyleD’Ayala, Giovanna Gomez, Mario Malinconico, and Paola Laurienzo. 2008. "Marine Derived Polysaccharides for Biomedical Applications: Chemical Modification Approaches" Molecules 13, no. 9: 2069-2106. https://doi.org/10.3390/molecules13092069