Prodrugs in Cardiovascular Therapy

Abstract
:Introduction
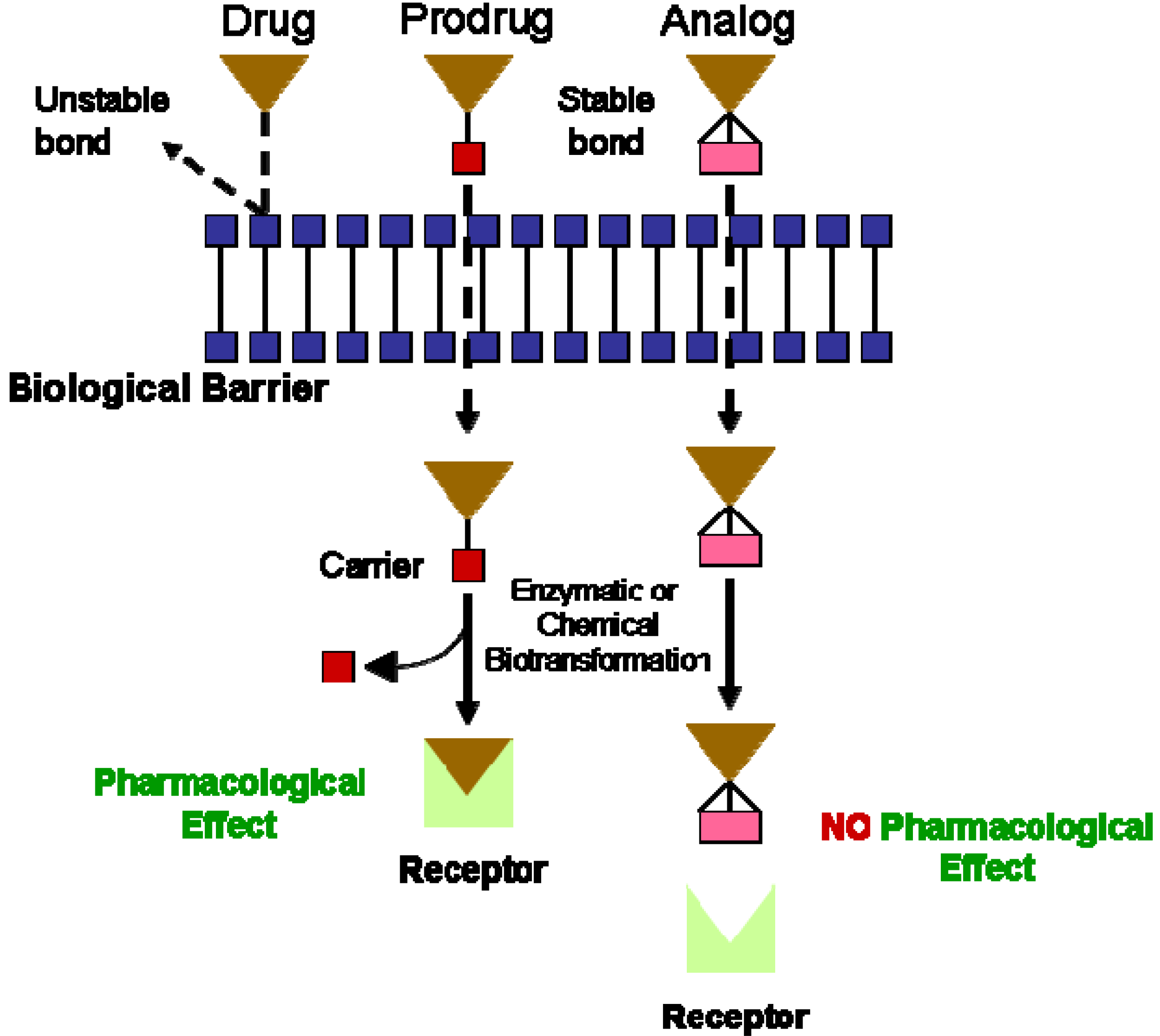
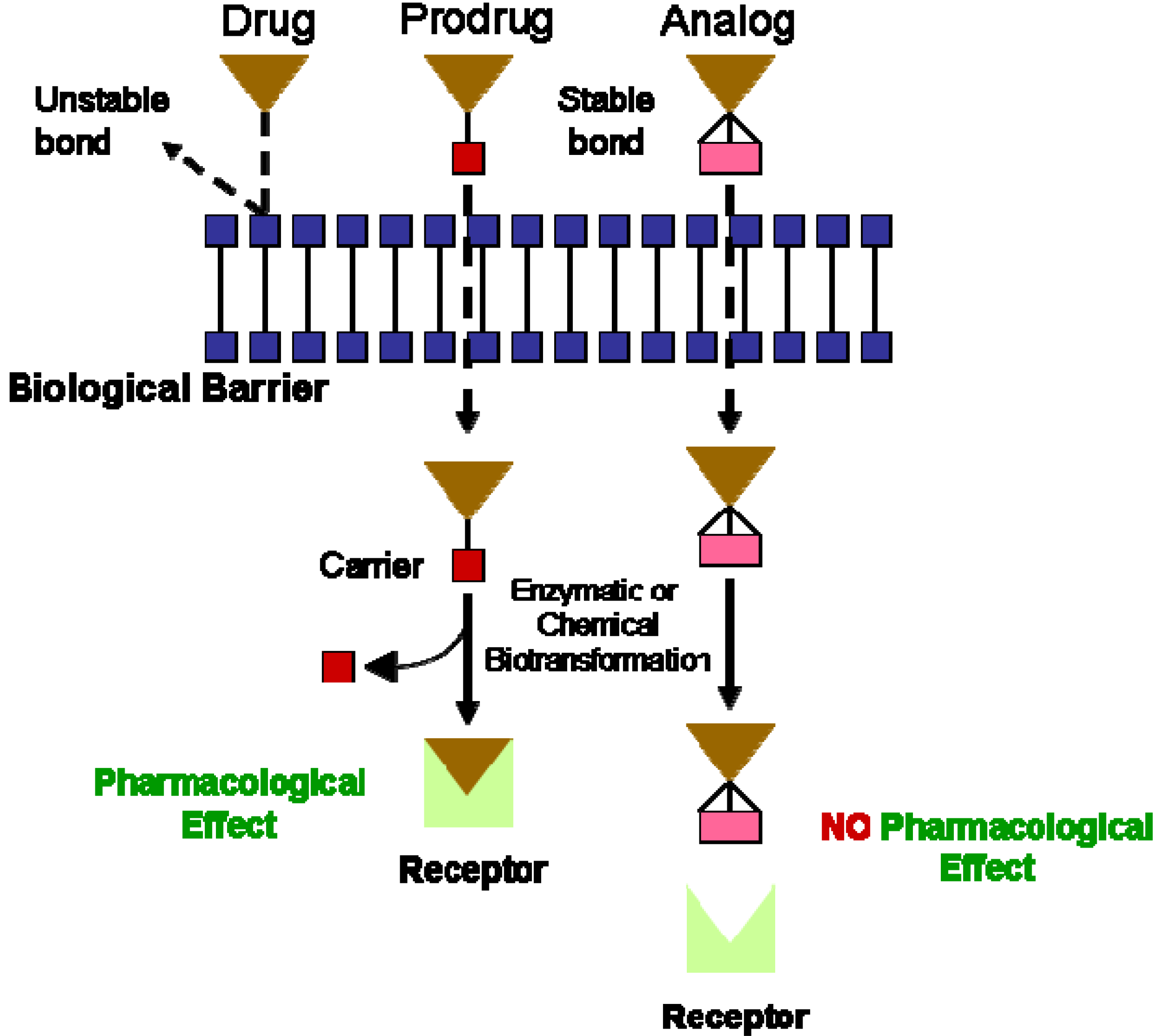
I. Prodrugs for the Treatment of Cardiovascular Diseases:
I.1. Prodrugs for Vascular Thrombosis
I.1.a. Antithrombin produgs
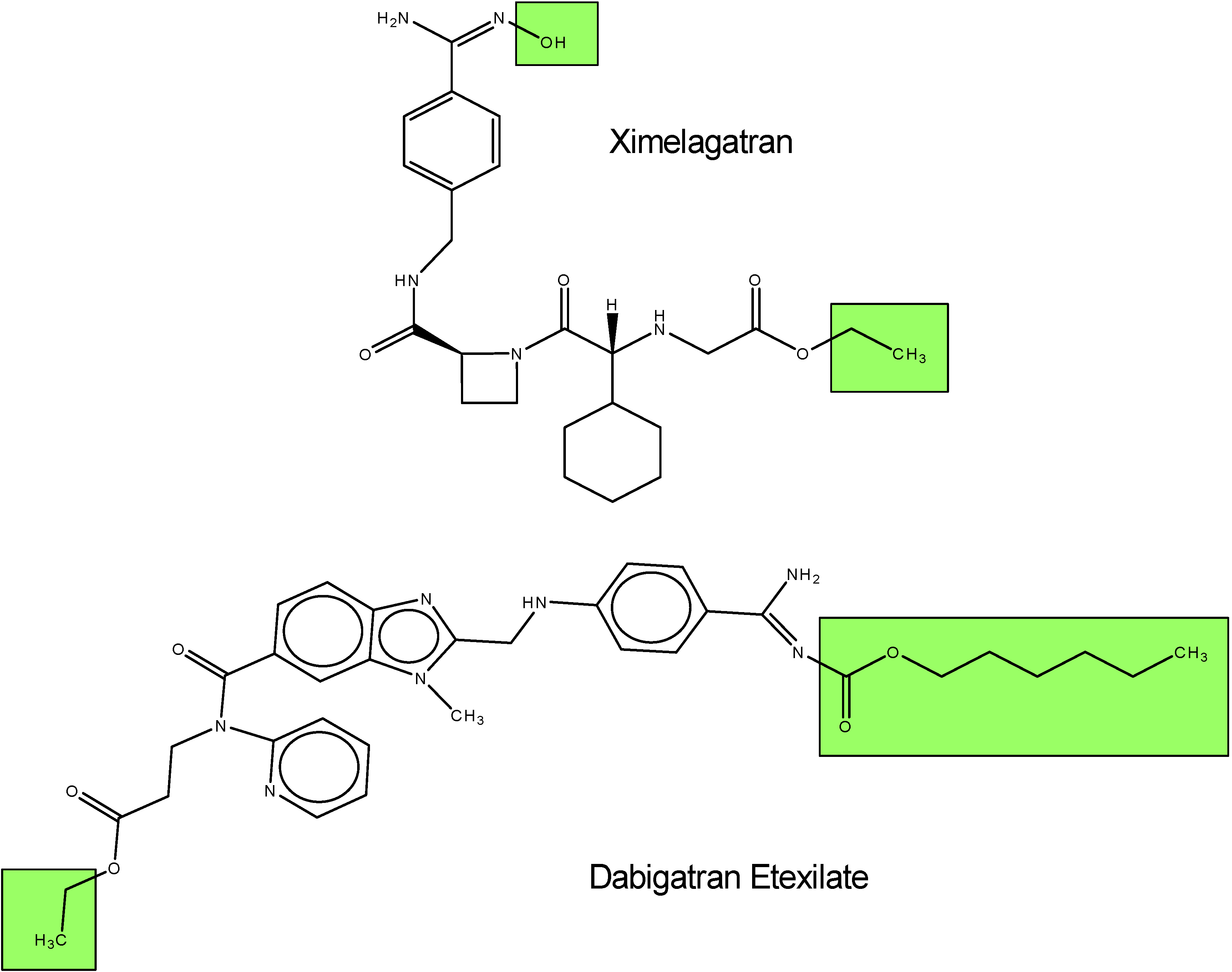
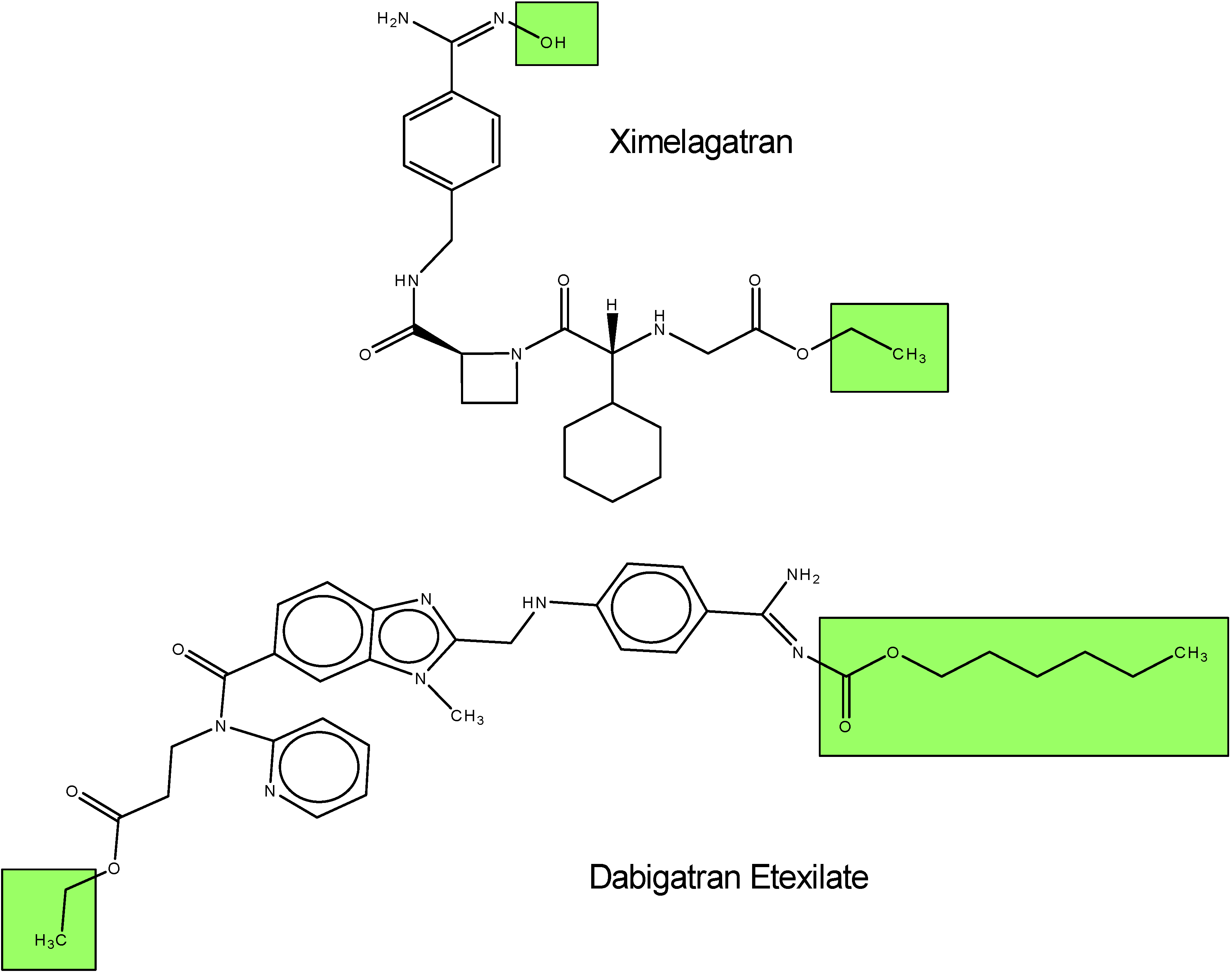
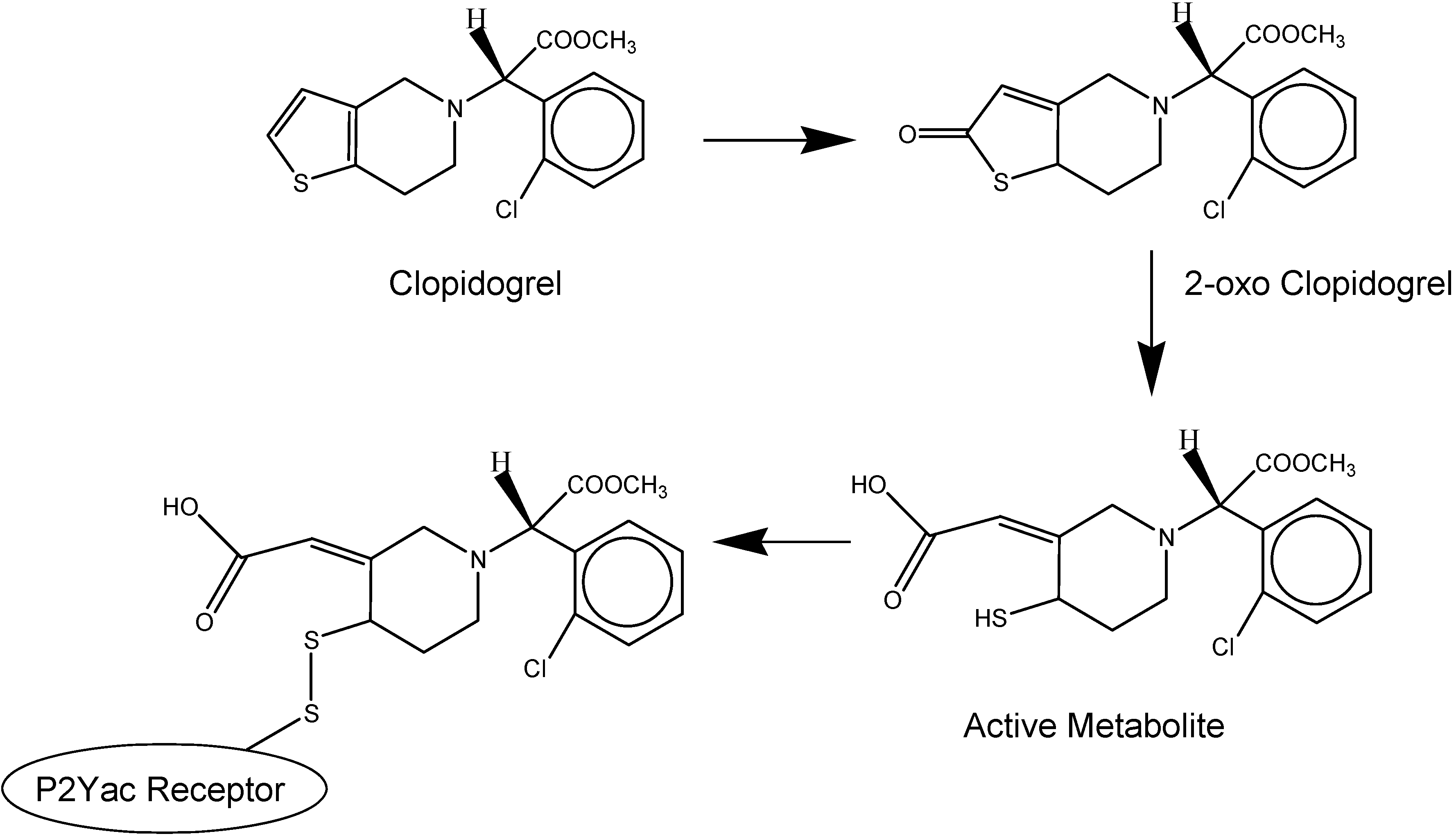
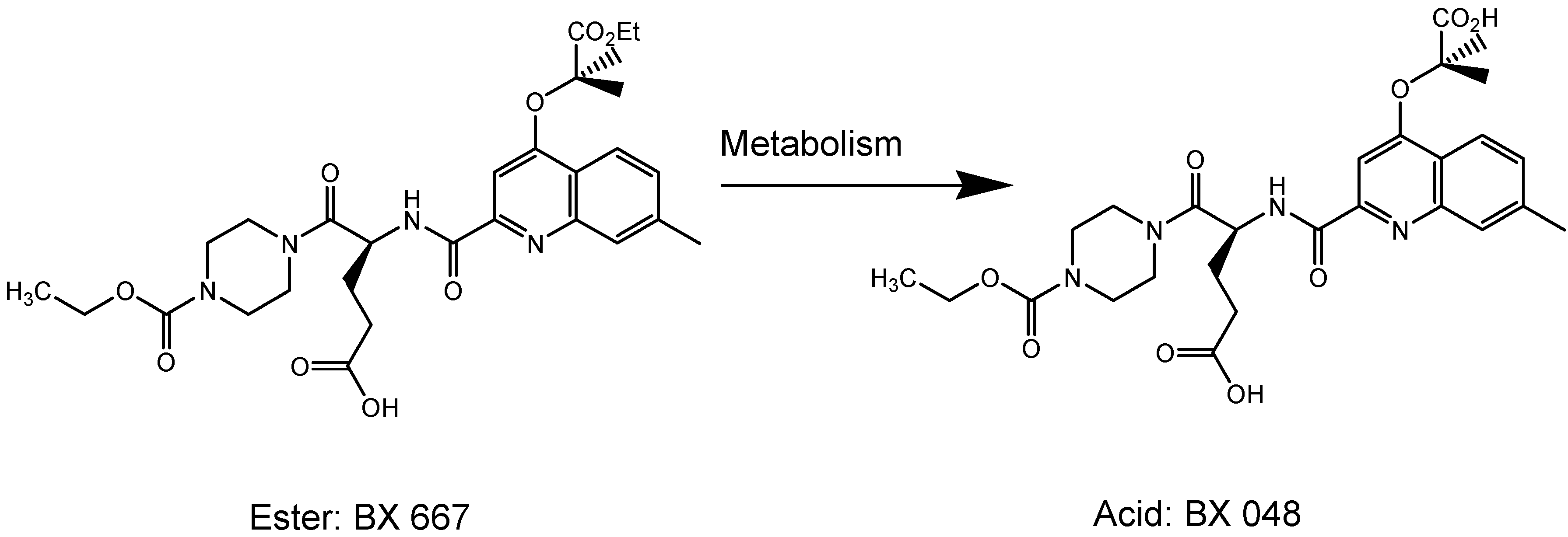
I.1.b. Antiplatelet prodrugs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prodrug | Type | Active Drug | % of Platelet Inhibition |
|---|---|---|---|
| Xemilofiban (4-pentynoic acid, 3-[[4-[[4-aminoimin-omethyl)phenyl] amino]-1,4 dioxobutyl]amino]-, ethyl ester, (3S)) | Nonpeptidic | SC-54701A | 50 % |
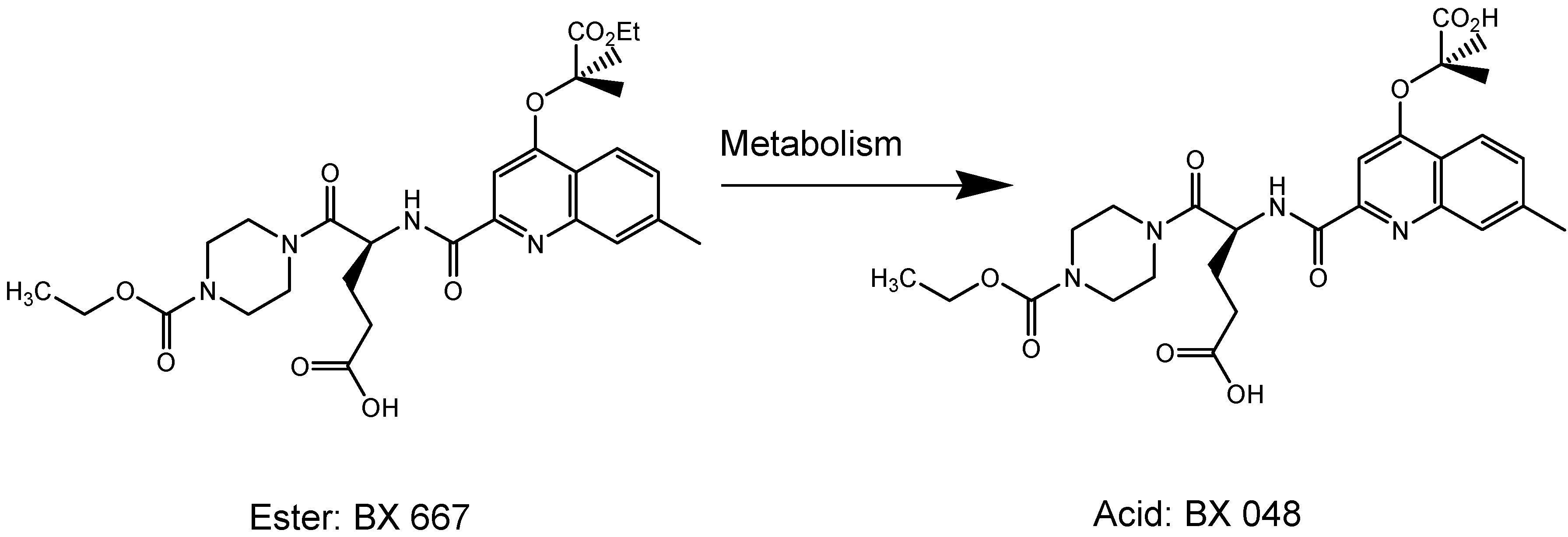
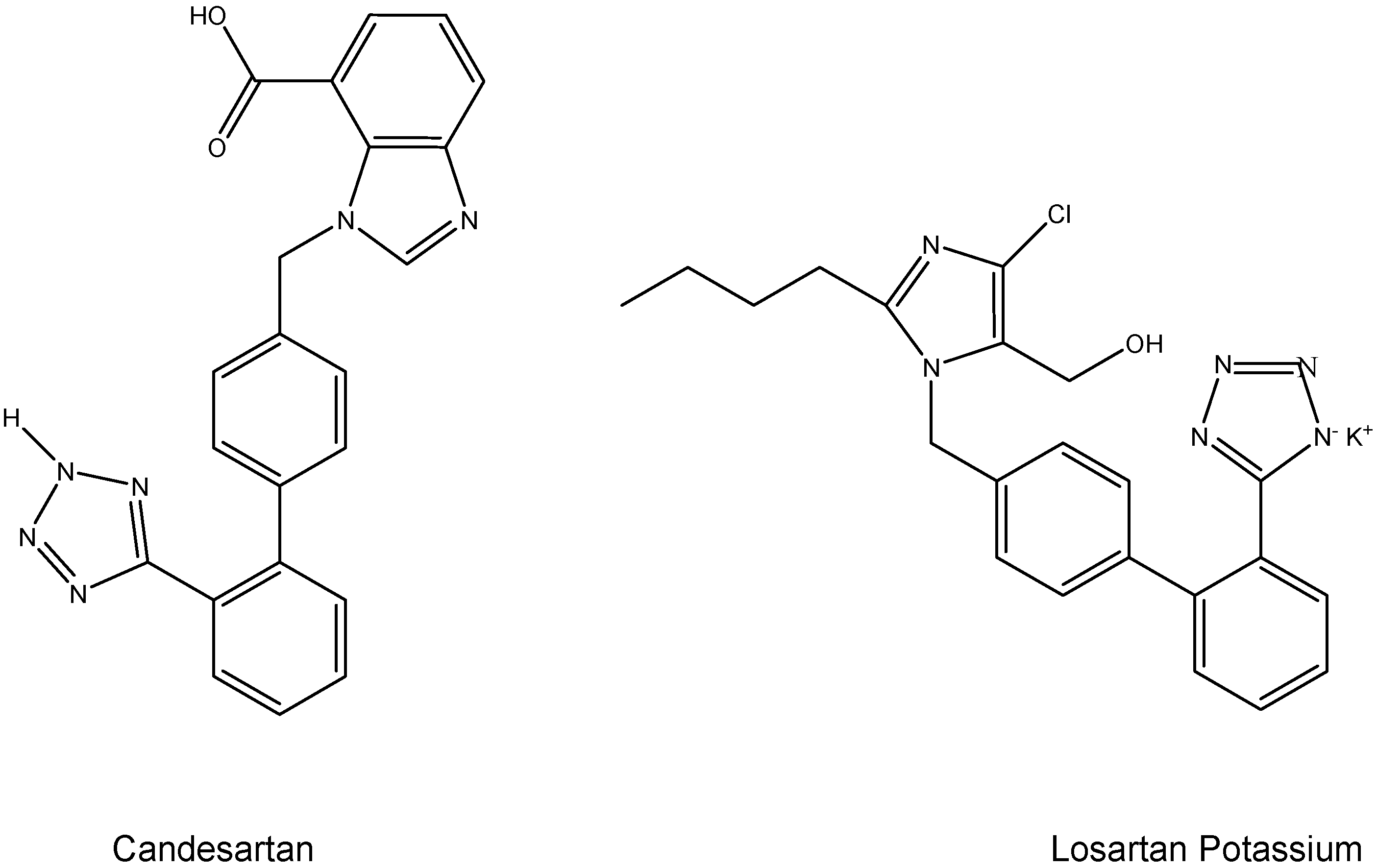
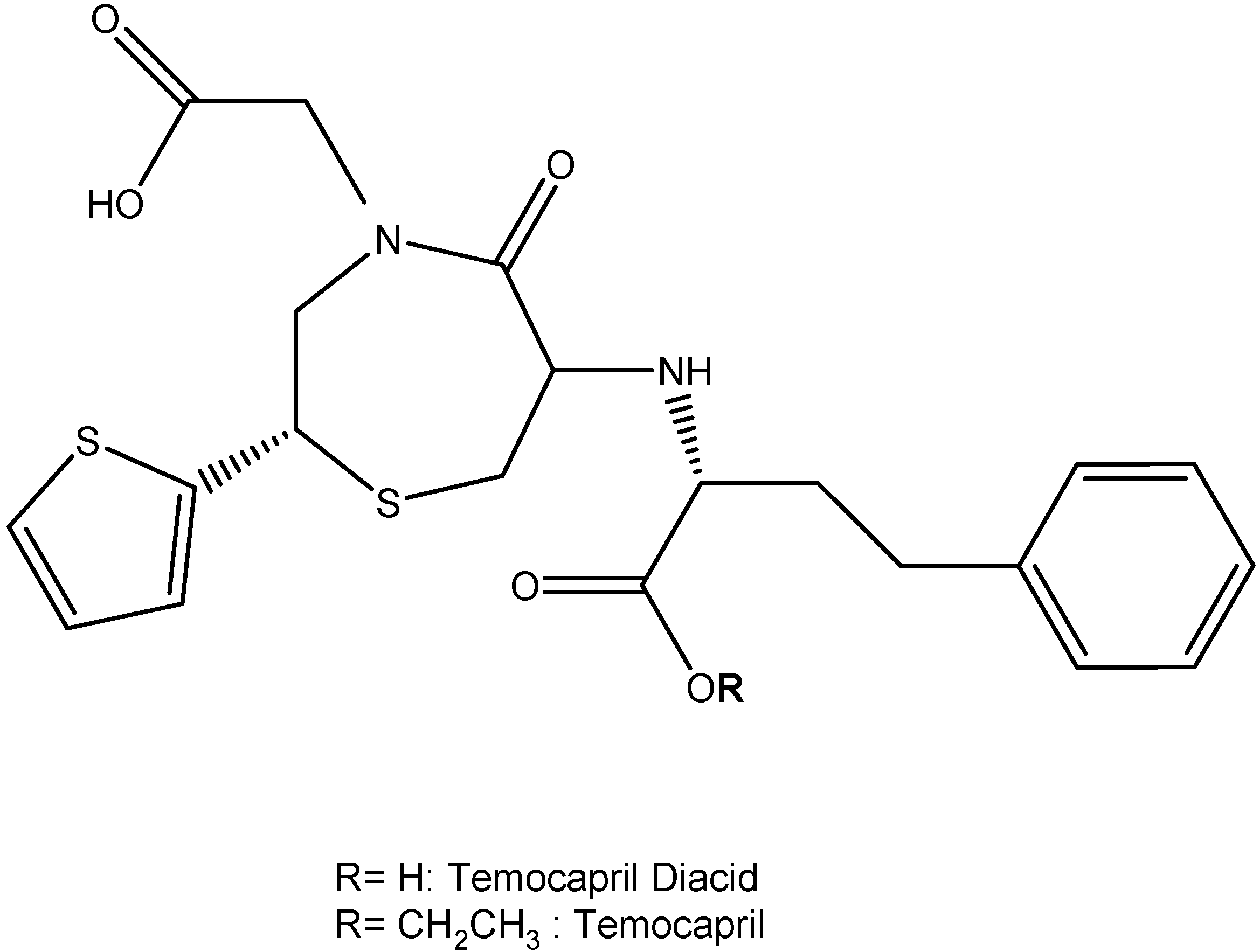
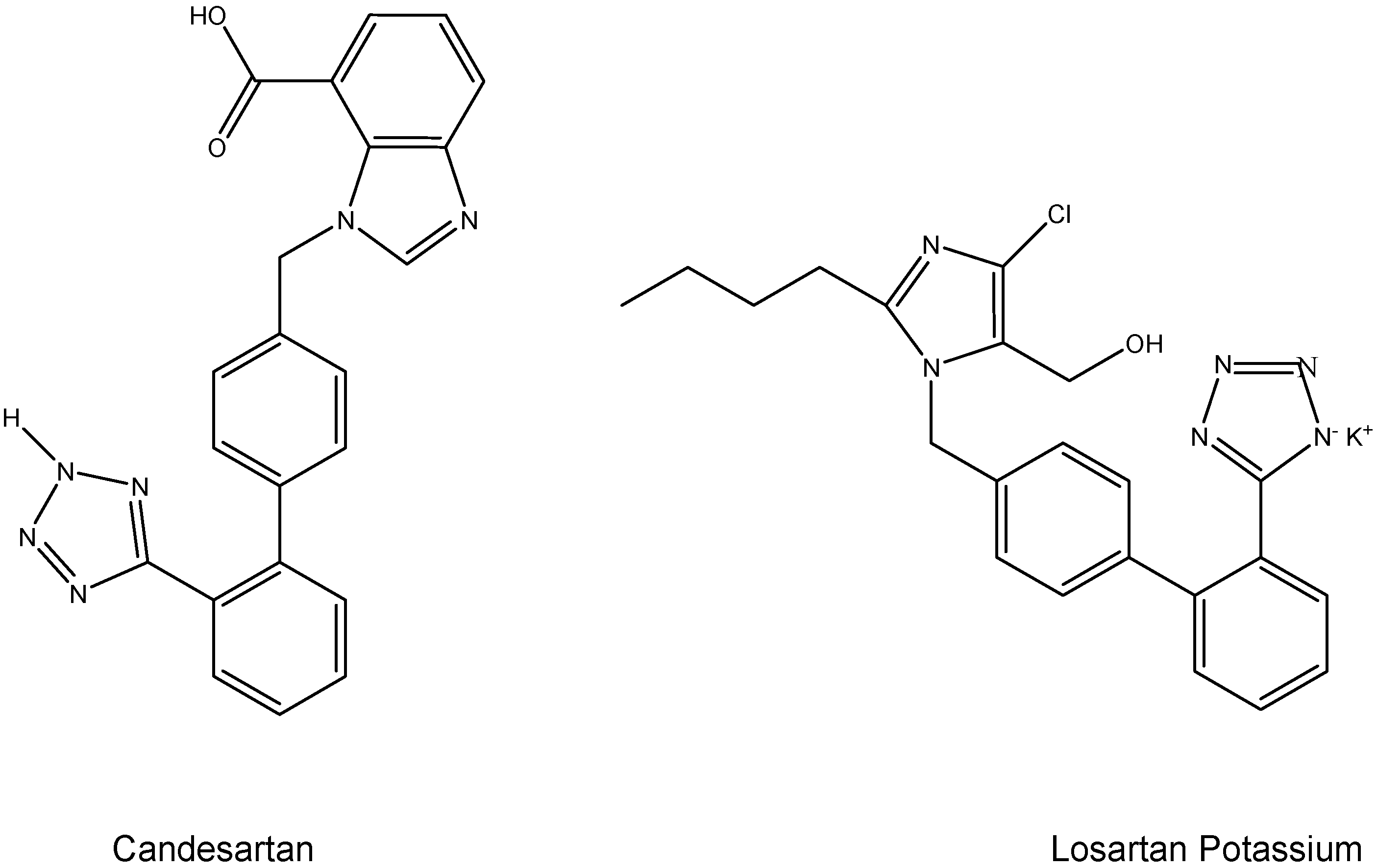
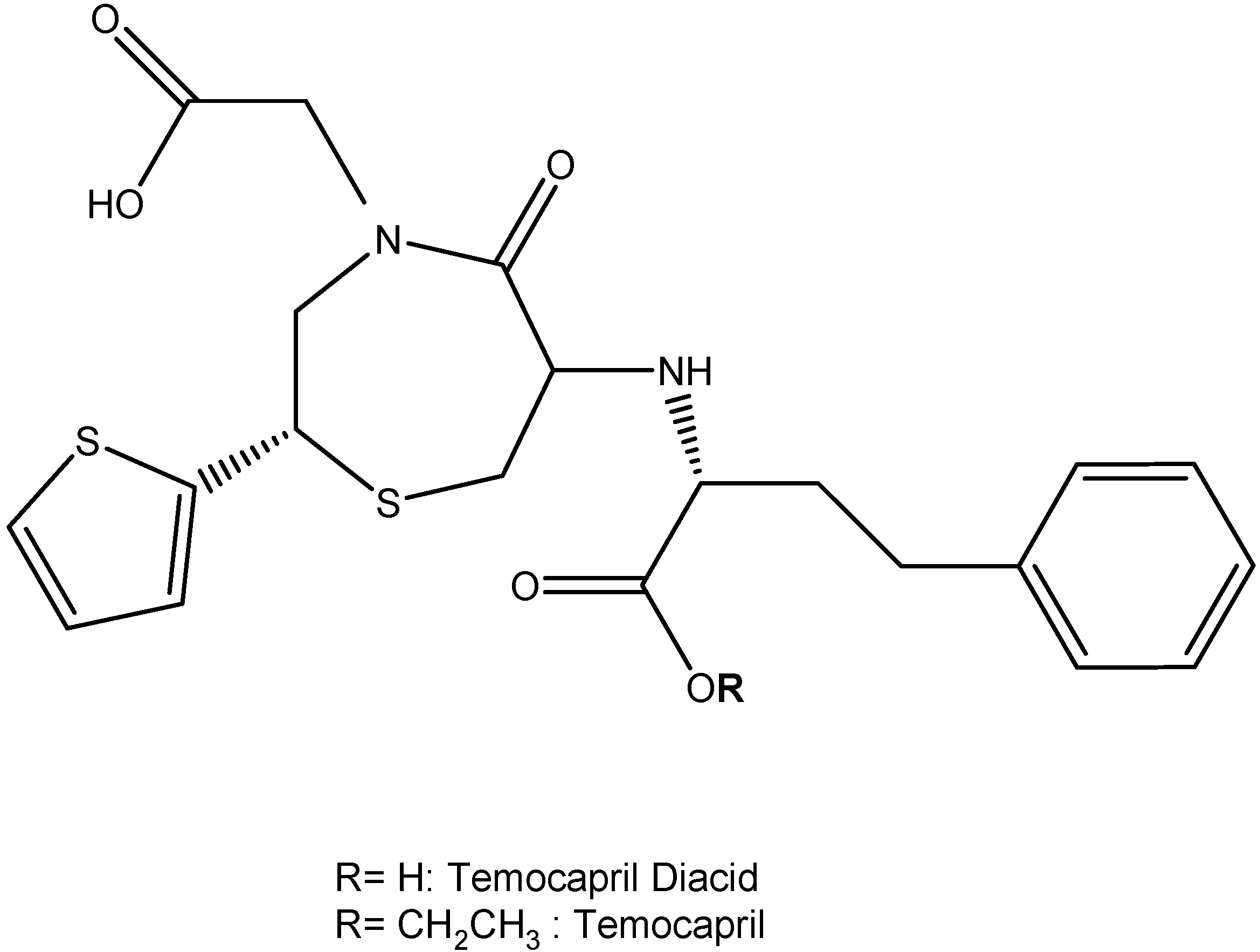
I.2. Hypertension Prodrugs
I.3. Pulmonary Hypertension Prodrugs
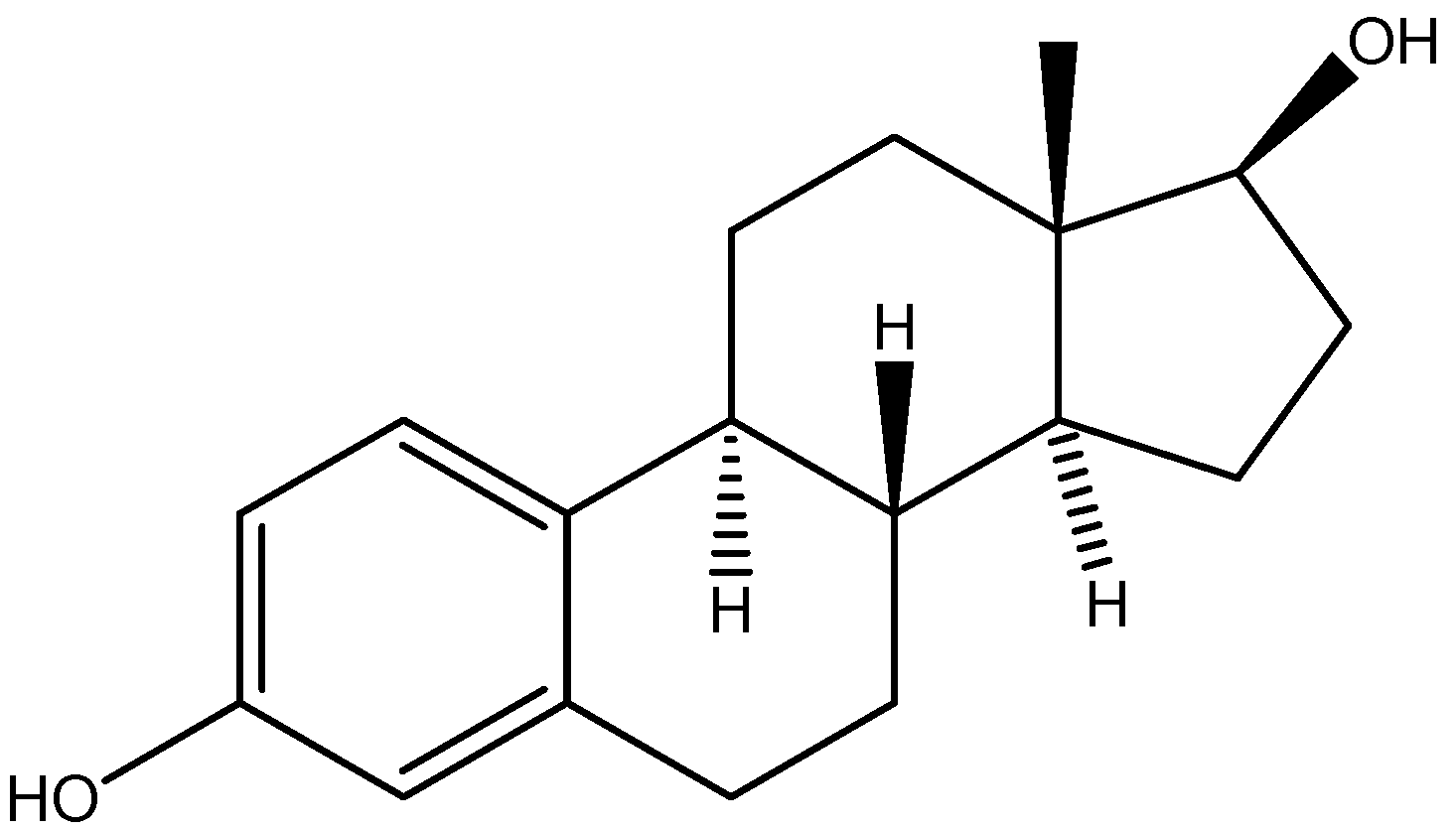
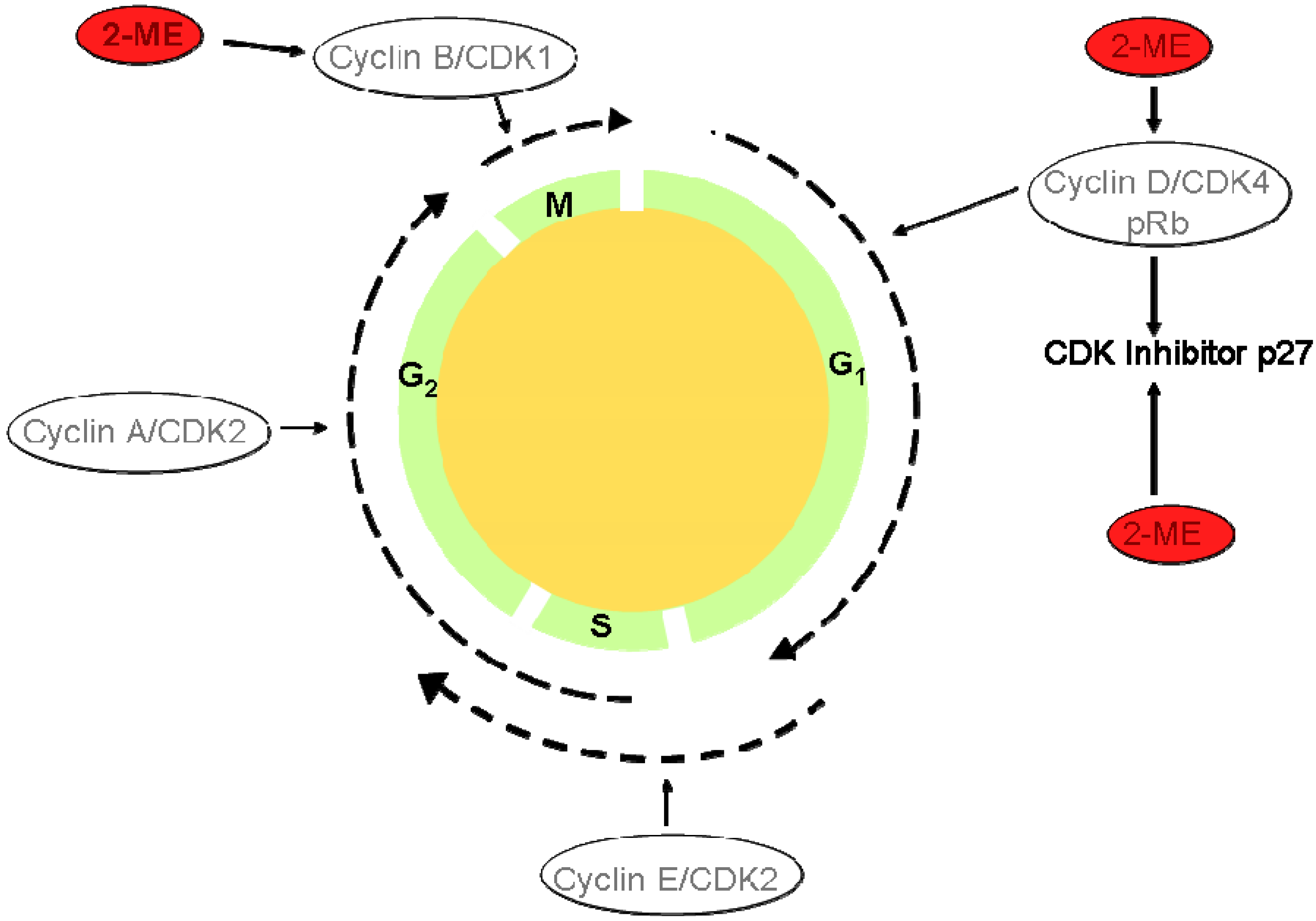
I.4. Atherosclerosis Prodrugs
II. Delivery Strategies
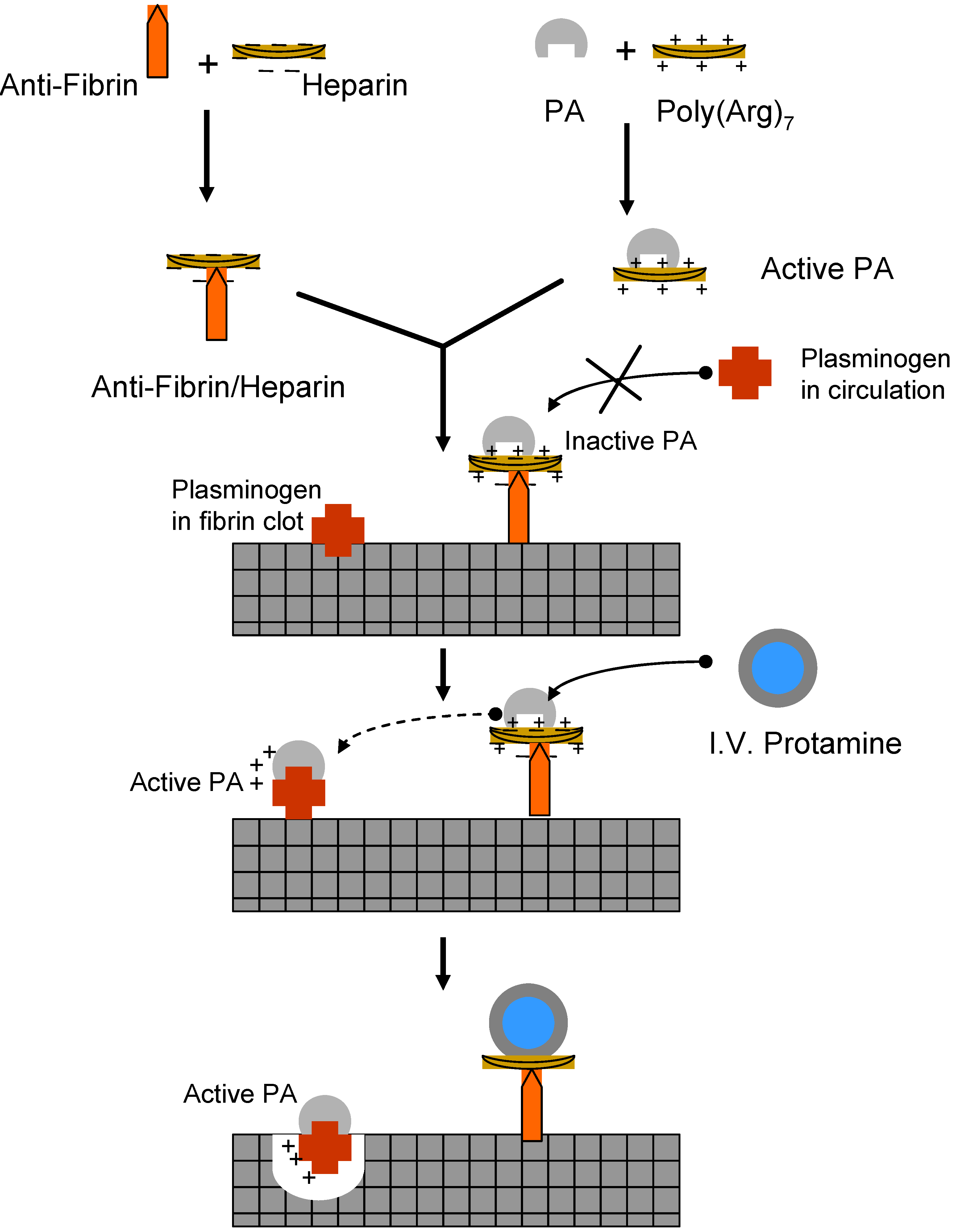
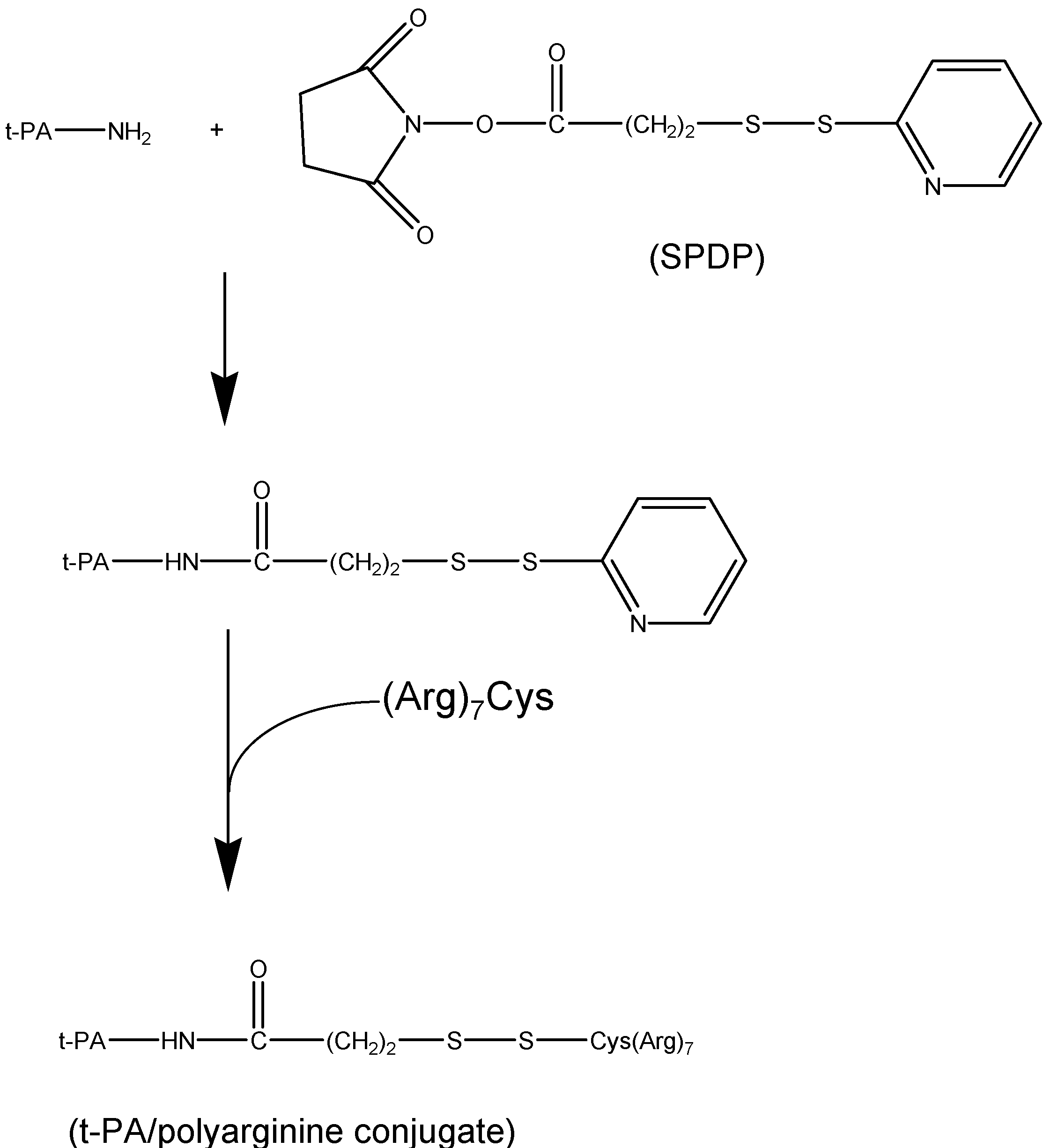
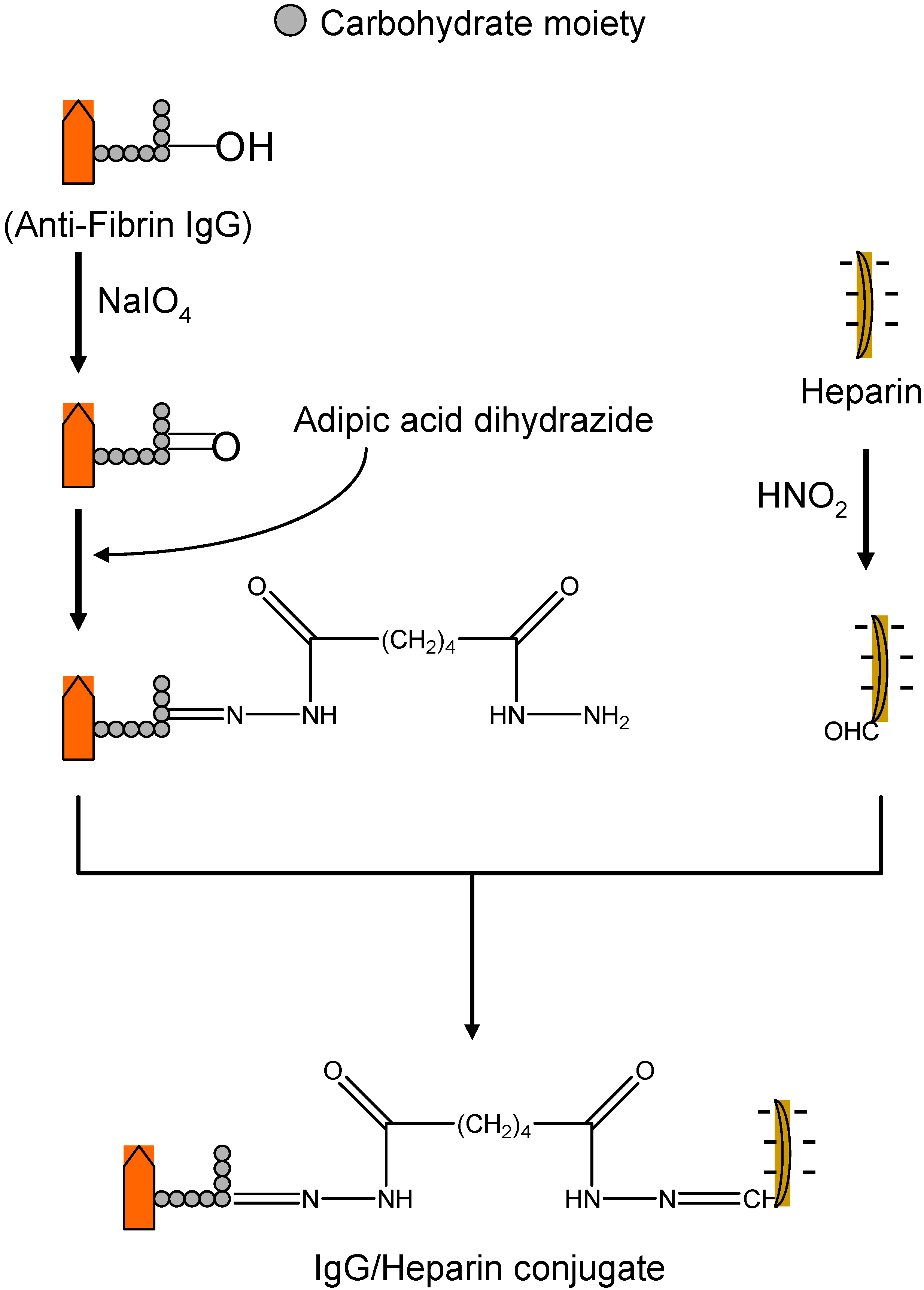
II.1. Antibody Targeted, Triggered, Electrically Modified Prodrug-Type Strategy (ATTEMPTS)
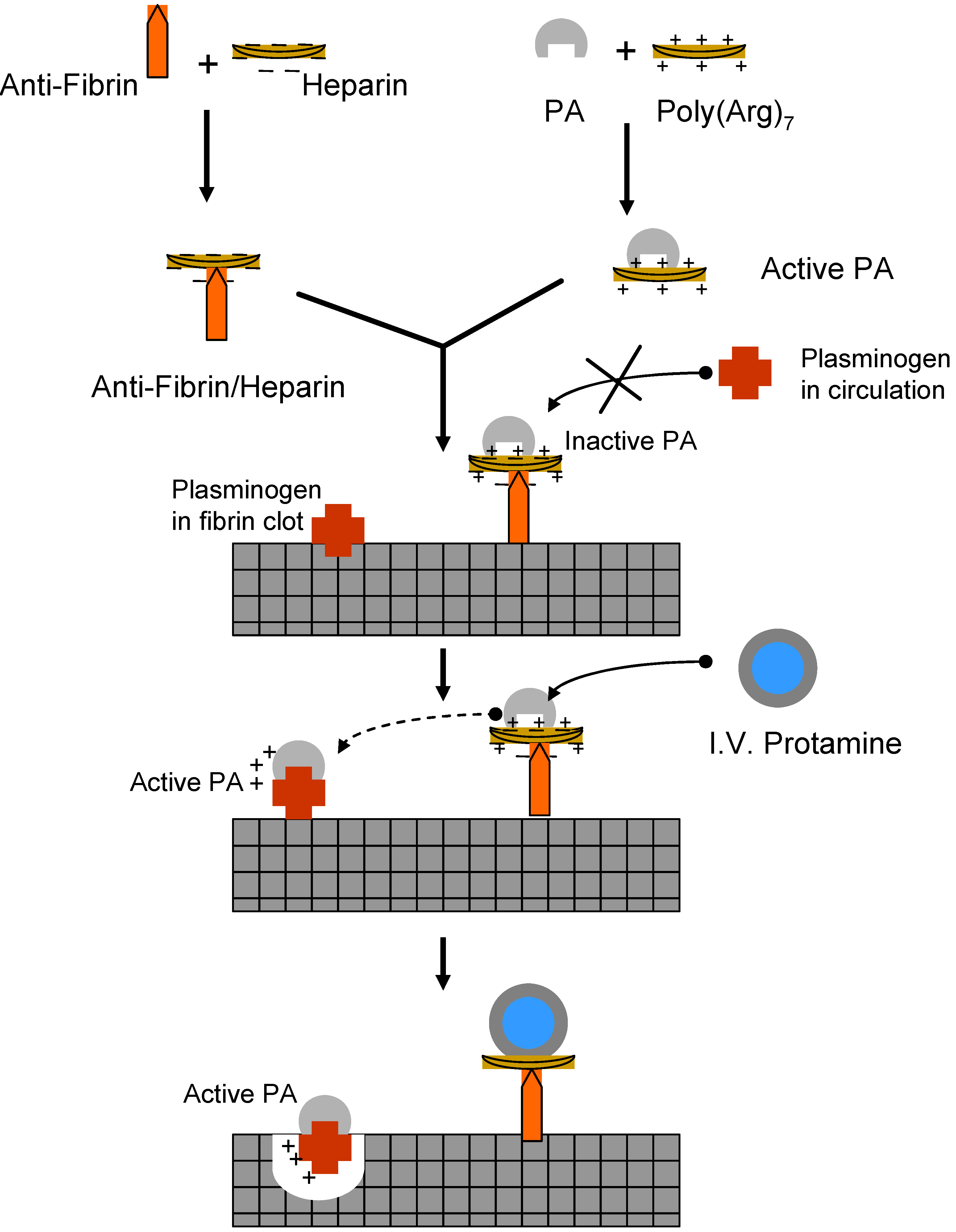
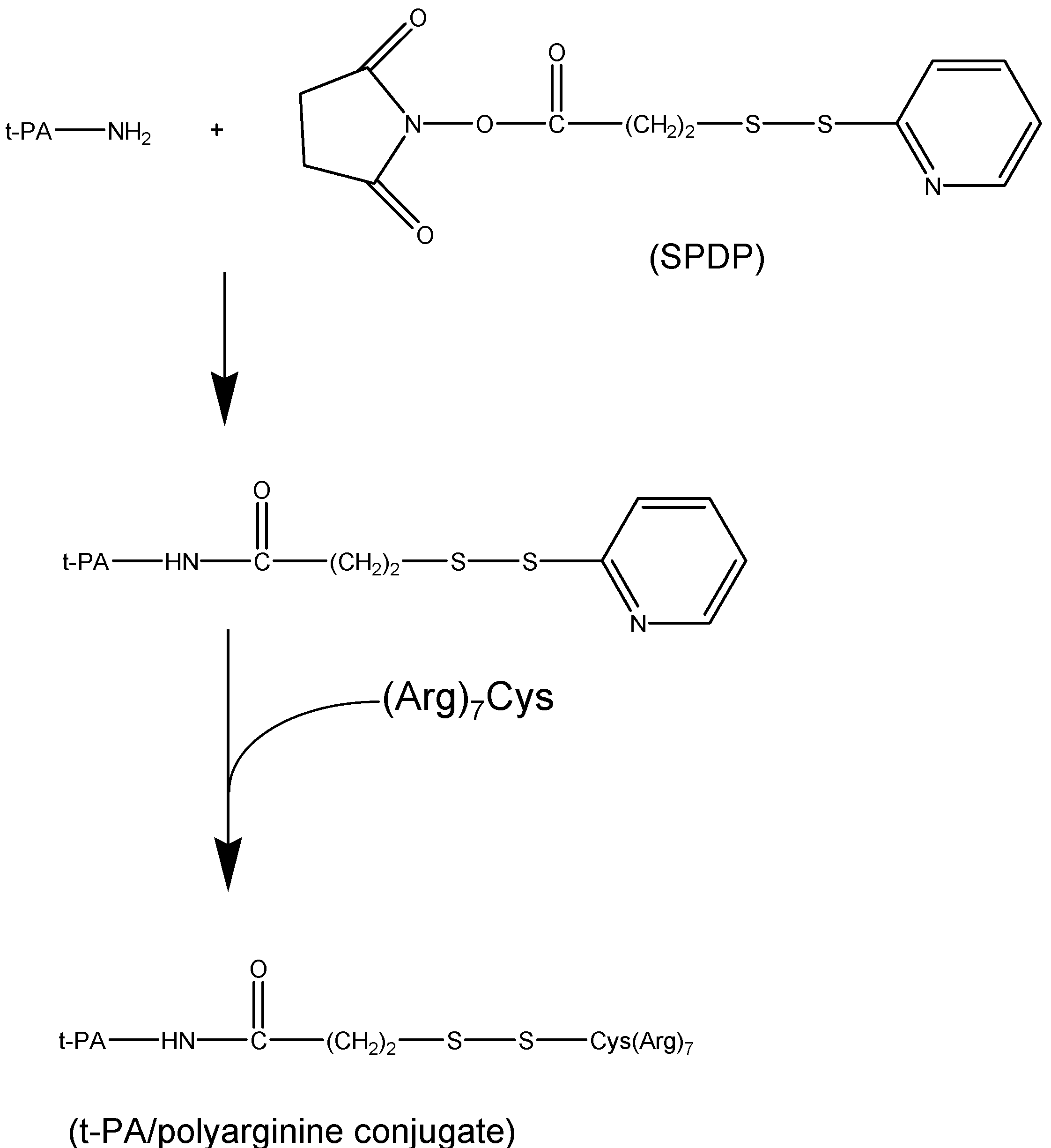
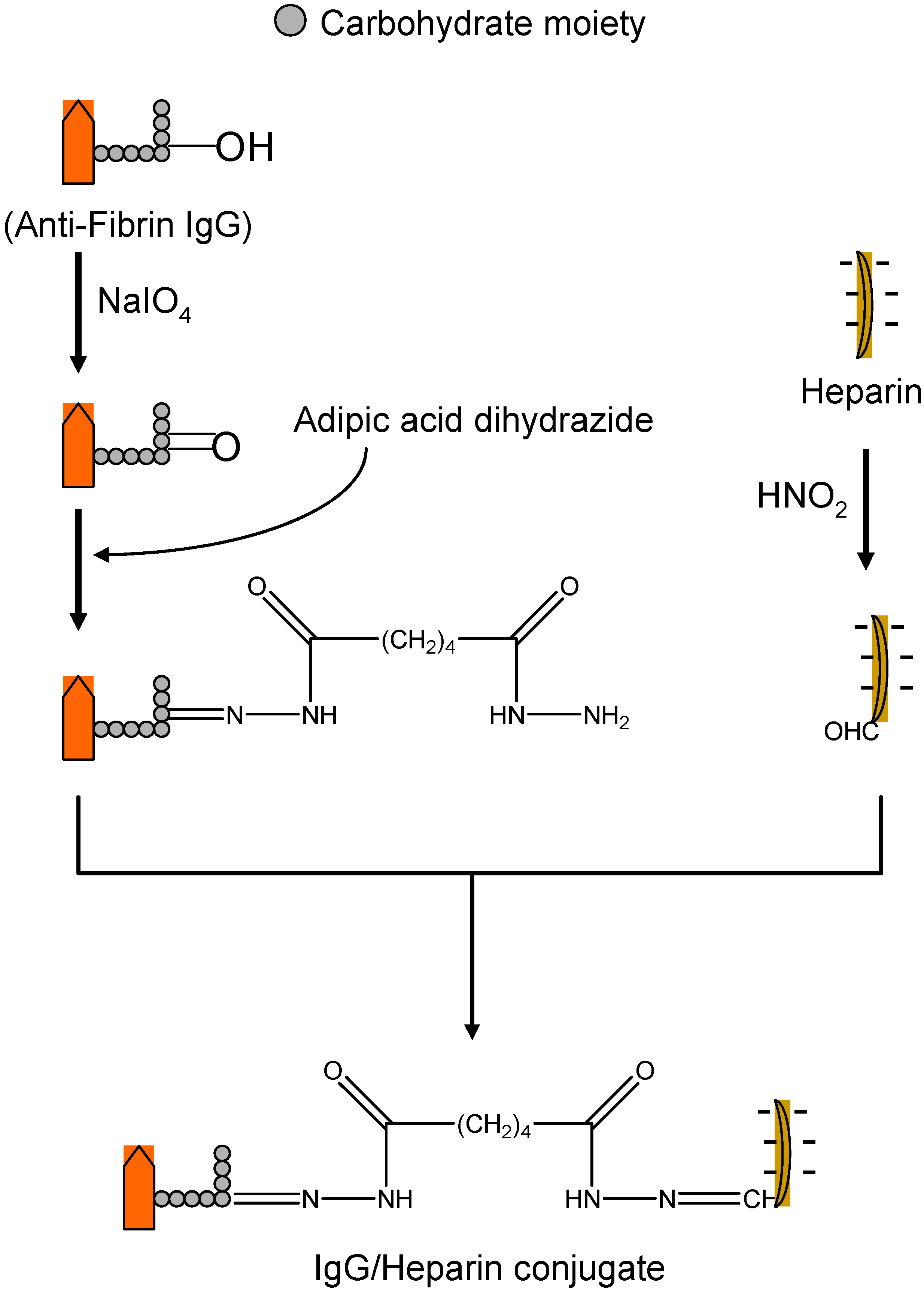
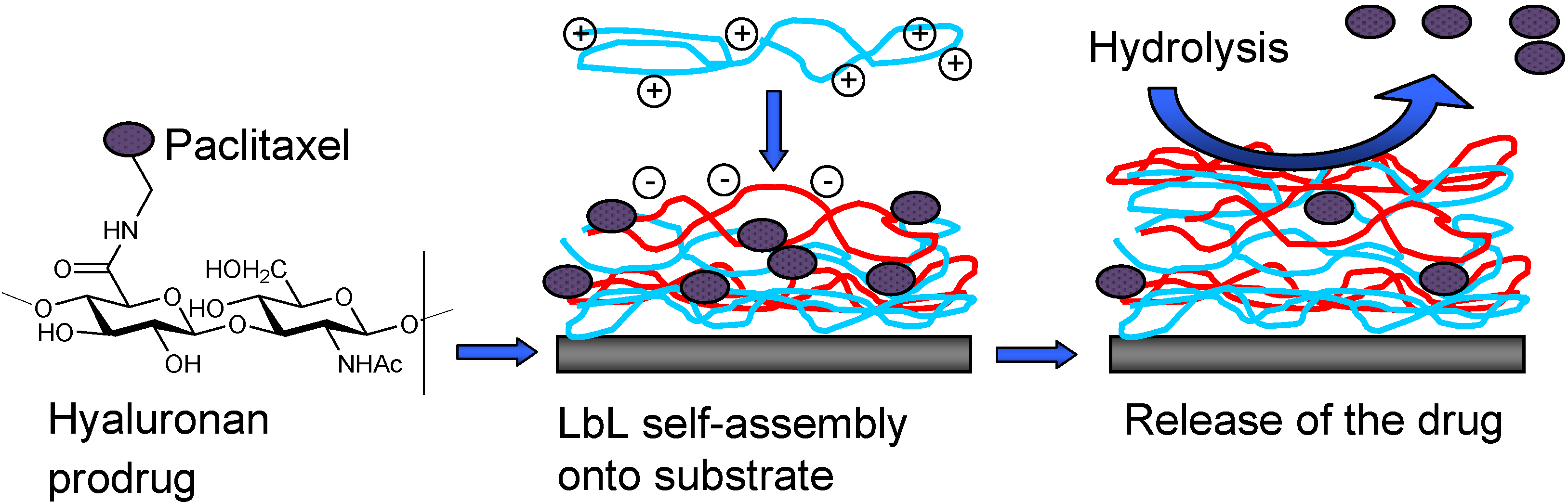
II.2. Prodrug Approach Combined with Layer by Layer Assembly
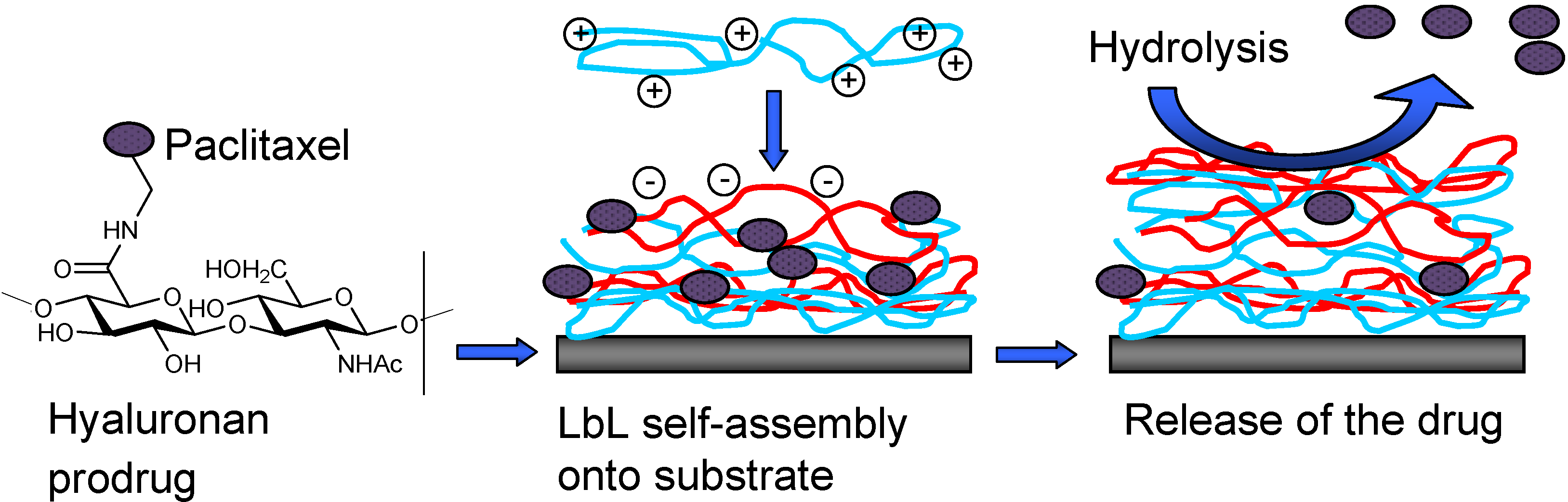
Conclusions
Acknowledgements
References
- Albert, A. Chemical Aspects of Selective Toxicity. Nature 1958, 182, 421–423. [Google Scholar] [CrossRef]
- de Albuquerque Silva, A.T.; Chung, M.C.; Castro, L.F.; Carvalho Guido, R.V.; Ferreira, E.I. Advances in Prodrug Design Mini Rev. Med. Chem. 2005, 5, 893–914. [Google Scholar]
- Morice, M.C.; Serruys, P.W.; Sousa, J.E.; Fajadet, J.; Ban Hayashi, E.; Perin, M.; Colombo, A.; Schuler, G.; Barragan, P.; Guagliumi, G.; Molnar, F.; Falotico, R. A Randomized Comparison of a Sirolimus-Eluting Stent with a Standard Stent for Coronary Revascularization. N. Engl. J. Med. 2002, 346, 1773–1780. [Google Scholar] [CrossRef]
- Thijssen, H.H.W.; Drieman, H.C. Prodrugs in Cardiovascular Medicine. Cardiovas. Drug Rev. 1990, 8, 386–400. [Google Scholar] [CrossRef]
- Weitz, J.I.; Buller, H.R. Direct Thrombin Inhibitors in Acute Coronary Syndromes: Present and Future. Circulation 2002, 105, 1004–1011. [Google Scholar] [CrossRef]
- Boström, S.L.; Dagnelid, E.; Hansson, G.F.H.; Ulvinge, J.C. Inhibition of thrombin-induced feedback activation of factor V: a potential pathway for inhibition of thrombin generation by melagatran. Blood Coagul. Fibrinolysis 2004, 15, 25–30. [Google Scholar] [CrossRef]
- Gustafsson, D.; Antonsson, T.; Bylund, R.; Eriksson, U.; Gyzander, E.; Nilsson, I.; Elg, M.; Mattsson, C.; Deinum, J.; Pehrsson, S.; Karlsson, O.; Nilsson, A.; Sörensen, H. Effects of Melagatran, a New Low-molecular-weight Thrombin Inhibitor, on Thrombin and Fibrinolytic Enzymes. Thromb. Haemost. 1998, 79, 110–118. [Google Scholar]
- Eriksson, U.G.; Bredberg, U.; Hoffmann, K.J.; Thuresson, A.; Gabrielsson, M.; Ericsson, H.; Ahnoff, M.; Gislen, K.; Fager, G.; Gustafsson, D. Absorption, Distribution, Metabolism, and Excretion of Ximelagatran, an Oral Direct Thrombin Inhibitor, in Rats, Dogs, and Humans. Drug Metab. Dispos. 2003, 31, 294–305. [Google Scholar] [CrossRef]
- Wienen, W.; Nar, H.; Ries, U.; Priepke, H.; Hauel, N.; Stassen, J. Antithrombotic effects of the direct thrombin inhibitor BIBR953ZW and its orally active prodrug BIBR1048MS in a model of venous thrombosis in rabbits. Thromb. Haemost. 2001, 79, 110–118. [Google Scholar]
- Wienen, W.; Nar, H.; Ries, U.J.; Priepke, H.W.M.; Hauel, N.H. Effects of the direct thrombin inhibitor BIBR953ZW and its orally active prodrug BIBR1048MS on experimentally-induced clot formation and template bleeding time in rats. Thromb. Haemost. 2001, (Suppl.), P761. [Google Scholar]
- Gustafsson, D. Oral direct thrombin inhibitors in clinical development. J. Intern. Med. 2003, 254, 322–334. [Google Scholar] [CrossRef]
- Fuster, V.; Badimon, L.; Badimon, J.; Chesebro, J. Mechanisma of Disease - The Pathogenesis of Coronary-Artery Disease and the Acute Coronary Syndromes. N. Engl. J. Med. 1992, 326, 242–250. [Google Scholar] [CrossRef]
- Lefkovits, J.; Plow, E.F.; Topol, E.J. Platelet Glycoprotein IIb/IIIa Receptors in Cardiovascular Medicine. N. Engl. J. Med. 1995, 332, 1553–1559. [Google Scholar] [CrossRef]
- Verstraete, M. Synthetic Inhibitors of Platelet Glycoprotein IIb/IIIa in Clinical Development. Circulation 2000, 101, e76–e80. [Google Scholar] [CrossRef] [Green Version]
- Muller, T.H.; Weisenberger, H.; Brickl, R.; Narjes, H.; Himmelsbach, F.; Krause, J. Profound and Sustained Inhibition of Platelet Aggregation by Fradafiban, a Nonpeptide Platelet Glycoprotein IIb/IIIa Antagonist, and Its Orally Active Prodrug, Lefradafiban, in Men. Circulation 1997, 96, 1130–1138. [Google Scholar] [CrossRef]
- Akkerhuis, K.; van den Brand, M.J.B.M.; van der Zwaan, C.; Peels, H.; Suryapranata, H.; van der Wieken, L.R.; Stibbe, J.; Hoffmann, J.; Baardman, T.; Deckers, J.; Simoons, M. Pharmacodynamics and safety of lefradafiban, an oral platelet glycoprotein IIb/IIIa receptor antagonist, in patients with stable coronary artery disease undergoing elective angioplasty. Heart 2001, 85, 444–450. [Google Scholar] [CrossRef]
- Serebruany, V.L.; Malinin, A.I.; O'Connor, C.M.; Gurbel, P.A. Effects of roxifiban on platelet aggregation and major receptor expression in patients with coronary artery disease for the Roxifiban Oral Compound Kinetics Evaluation Trial-I (ROCKET-I Platelet Substudy). Am. Heart J. 2003, 146, 91–98. [Google Scholar] [CrossRef]
- Mousa, S.A.; Ahmad, S. Platelet GPIIb/IIIa antagonist, XV459, in heparin-induced thrombocytopenia. Am. J. Hematol. 2007, 82, 276–282. [Google Scholar] [CrossRef]
- Barrett, Y.C.; Ebling, W.; Pieniaszek, H.; Billheimer, J.; Seiffert, D. Validation and implementation of drug-dependent antibody assays in clinical trials for safety monitoring of patients dosed with roxifiban, an orally bioavailable glycoprotein IIb/IIIa antagonist. J. Pharm. Biomed. Anal. 2007, 44, 938–946. [Google Scholar] [CrossRef]
- Shiu, M.; Silverton, N.; Oakley, D.; Cumberland, D. Acute coronary occlusion during percutaneous transluminal coronary angioplasty. Br. Heart J. 1985, 54, 129–133. [Google Scholar] [CrossRef]
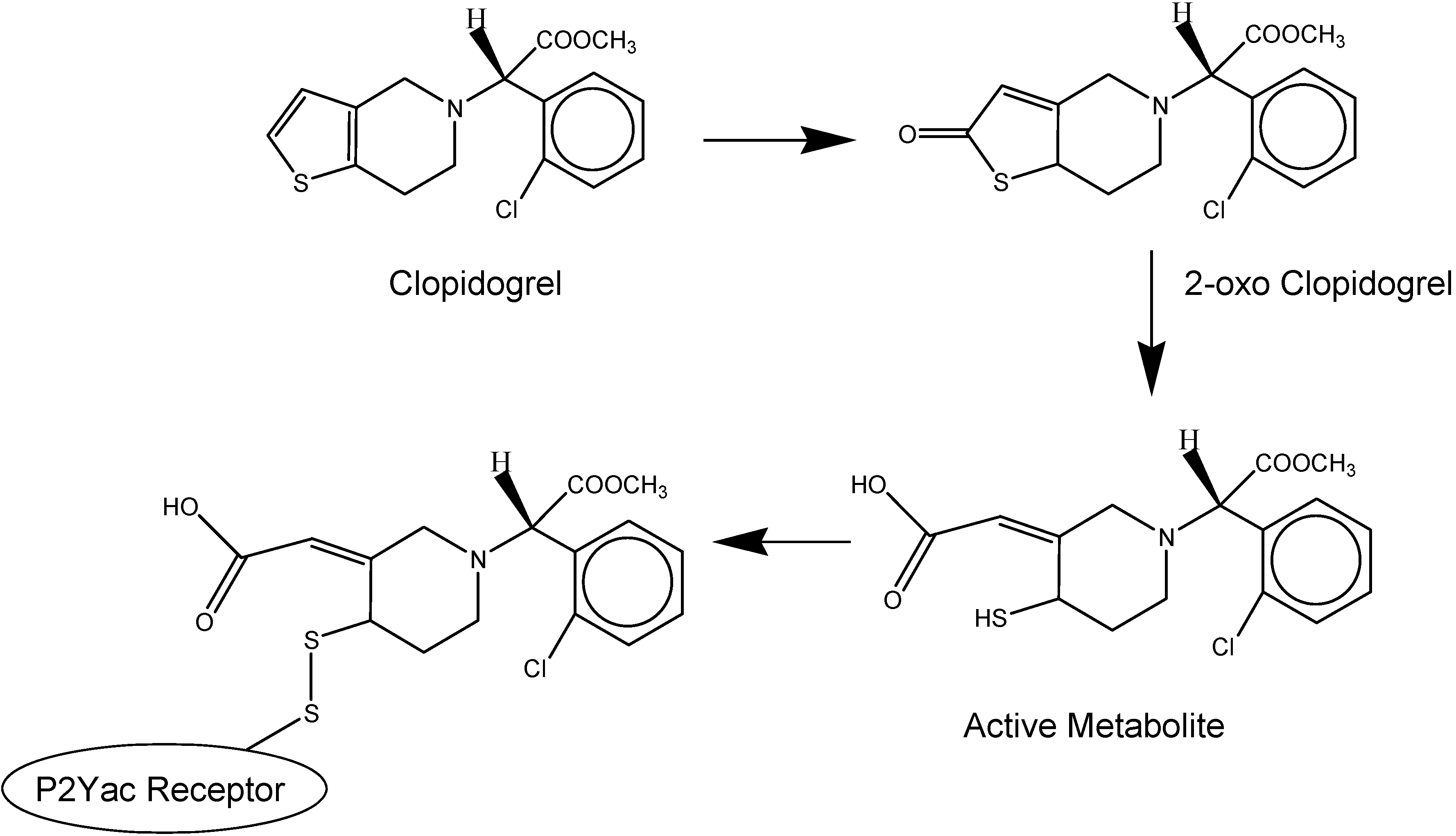
- Savi, P.; Herbert, J.M.; Pflieger, A.M.; Dol, F.; Delebassee, D.; Combalbert, J.; Defreyn, G.; Maffrand, J.P. Importance of hepatic metabolism in the antiaggregating activity of the thienopyridine clopidogrel. Biochem. Pharmacol. 1992, 44, 527–532. [Google Scholar] [CrossRef]
- Savi, P.; Combalbert, J.; Gaich, C.; Rouchon, M.C.; Maffrand, J.P.; Berger, Y.; Herbert, J.M. The antiaggregating activity of clopidogrel is due to a metabolic activation by the hepatic cytochrome P450-1A. Thromb. Haemost. 1994, 72, 313–317. [Google Scholar]
- Savi, P.; Pereillo, J.M.; Uzabiaga, M.F.; Combalbert, J.; Picard, C.; Maffrand, J.P.; Pascal, M.; Herbert, J.M. Identification and biological activity of the active metabolite of clopidogrel. Thromb. Haemost. 2000, 84, 891–896. [Google Scholar]
- Geiger, J.; Brich, J.; Honig-Liedl, P.; Eigenthaler, M.; Schanzenbacher, P.; Herbert, J.M.; Walter, U. Specific Impairment of Human Platelet P2YAC ADP Receptor–Mediated Signaling by the Antiplatelet Drug Clopidogrel. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2007–2011. [Google Scholar] [CrossRef]
- Heitzer, T.; Rudolph, V.; Schwedhelm, E.; Karstens, M.; Sydow, K.; Ortak, M.; Tschentscher, P.; Meinertz, T.; Boger, R.; Baldus, S. Clopidogrel Improves Systemic Endothelial Nitric Oxide Bioavailability in Patients With Coronary Artery Disease: Evidence for Antioxidant and Antiinflammatory Effects. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1648–1652. [Google Scholar] [CrossRef]
- Lau, W.C.; Waskell, L.A.; Watkins, P.B.; Neer, C.J.; Horowitz, K.; Hopp, A.S.; Tait, A.R.; Carville, D.G.M.; Guyer, K.E.; Bates, E.R. Atorvastatin Reduces the Ability of Clopidogrel to Inhibit Platelet Aggregation: A New Drug-Drug Interaction. Circulation 2003, 107, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X.; Vincelette, J.; da Cunha, V.; Martin-McNulty, B.; Mallari, C.; Fitch, R.M.; Alexander, S.; Islam, I.; Buckman, B.O.; Yuan, S.; Post, J.M.; Subramanyam, B.; Vergona, R.; Sullivan, M.E.; Dole, W.P.; Morser, J.; Bryant, J. A novel P2Y(12) adenosine diphosphate receptor antagonist that inhibits platelet aggregation and thrombus formation in rat and dog models. Thromb. Haemost. 2007, 97, 847–855. [Google Scholar]
- Burnier, M.; Brunner, H.R. Angiotensin II receptor antagonists in hypertension. Kidney Int. 1998, 54, S107–S111. [Google Scholar] [CrossRef]
- Awan, N.A.; Mason, D.T. Direct selective blockade of the vascular angiotensin II receptors in therapy for hypertension and severe congestive heart failure. Am. Heart J. 1996, 131, 177–185. [Google Scholar] [CrossRef]
- Ellis, M.L.; Patterson, J.H. A new class of antihypertensive therapy : Angiotensin II receptor antagonists. Pharmacotherapy 1996, 16, 849–860. [Google Scholar]
- Lo, M.W.; Goldberg, M.R.; McCrea, J.B.; Lu, H.; Furtek, C.I.; Bjornsson, T.D. Pharmacokinetics of losartan, an angiotensin II receptor antagonist, and its active metabolite EXP3174 in humans. Clin. Pharmacol. Ther. 1995, 58, 641–649. [Google Scholar] [CrossRef]
- Oparil, S. Newly emerging pharmacologic differences in angiotensin II receptor blockers. Am. J. Hypertens. 2000, 13, S18–S24. [Google Scholar] [CrossRef]
- Suzuki, H.; Kawaratani, T.; Shioya, H.; Uji, Y.; Saruta, T. Study on pharmacokinetics of a new biliary excreted oral angiotensin converting enzyme inhibitor, temocapril (CS-622) in humans. Biopharm. Drug Dispos. 1993, 14, 41–50. [Google Scholar] [CrossRef]
- Yasunari, K.; Maeda, K.; Nakamura, M.; Watanabe, T.; Yoshikawa, J.; Asada, A. Pharmacological and Clinical Studies with Temocapril, an Angiotensin Converting Enzyme Inhibitor that is Excreted in the Bile. Cardiovasc. Drug Rev. 2004, 22, 189–198. [Google Scholar]
- Kim, S.; Izumi, Y.; Izumiya, Y.; Zhan, Y.; Taniguchi, M.; Iwao, H. Beneficial Effects of Combined Blockade of ACE and AT1 Receptor on Intimal Hyperplasia in Balloon-Injured Rat Artery. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1299–1304. [Google Scholar]
- Mizuno, M.; Sada, T.; Ikeda, M.; Fukuda, N.; Miyamoto, M.; Yanagisawa, H.; Koike, H. Pharmacology of CS-866, a novel nonpeptide angiotensin II receptor antagonist. Eur. J. Pharmacol. 1995, 285, 181–188. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Simonneau, G. Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar]
- Franklin, T.J. Current approaches to the therapy of fibrotic diseases. Biochem. Pharmacol. 1995, 49, 267–273. [Google Scholar] [CrossRef]
- Simon, P.M.; Pachence, J.; Belinka, B.; Poiani, G.J.; Lu, S.E.; Tozzi, C.A.; Riley, D.J. Prodrug of proline analogue reduces hypoxic pulmonary hypertension in rats. Pulm. Pharmacol. Ther. 2006, 19, 242–250. [Google Scholar] [CrossRef]
- Brosnihan, K.B.; Senanayake, P.; Li, P.; Ferrario, C.M. Bi-directional actions of estrogen on the renin-angiotensin system. Braz J. Med. Biol. Res. 1999, 32, 373–381. [Google Scholar]
- Seely, E.W.; Brosnihan, K.B.; Jeunemaitre, X.; Okamura, K.; Williams, G.H.; Hollenberg, N.K.; Herrington, D.M. Effects of conjugated oestrogen and droloxifene on the renin-angiotensin system, blood pressure and renal blood flow in postmenopausal women. Clin. Endocrinol. 2004, 60, 315–321. [Google Scholar] [CrossRef]
- Moncada, S.; Martin, J.; Higgs, A. Symposium on regression of atherosclerosis. Eur. J. Clin. Invest. 1993, 23, 385–398. [Google Scholar] [CrossRef]
- Brunner, H.; Cockcroft, J.; Deanfield, J.; Donald, A.; Ferrannini, E.; Halcox, J.; Kiowski, W.; Luscher, T.; Mancia, G.; Natali, A.; Oliver, J.; Pessina, A.; Rizzoni, D.; Rossi, G.; Salvetti, A.; Spieker, L.; Taddei, S.; Webb, D. Endothelial function and dysfunction. Part II: Association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J. Hypertens. 2005, 23, 233–246. [Google Scholar]
- Writing Group for the Women's Health Initiative Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women: Principal Results From the Women's Health Initiative Randomized Controlled Trial. J. Am. Med. Assoc. 2002, 288, 321–333. [CrossRef]
- Hodgin, J.B.; Maeda, N. Minireview: Estrogen and Mouse Models of Atherosclerosis. Endocrinology 2002, 143, 4495–4501. [Google Scholar] [CrossRef]
- Dubey, R.K.; Jackson, E.K.; Keller, P.J.; Imthurn, B.; Rosselli, M. Estradiol Metabolites Inhibit Endothelin Synthesis by an Estrogen Receptor-Independent Mechanism. Hypertension 2001, 37, 640–644. [Google Scholar]
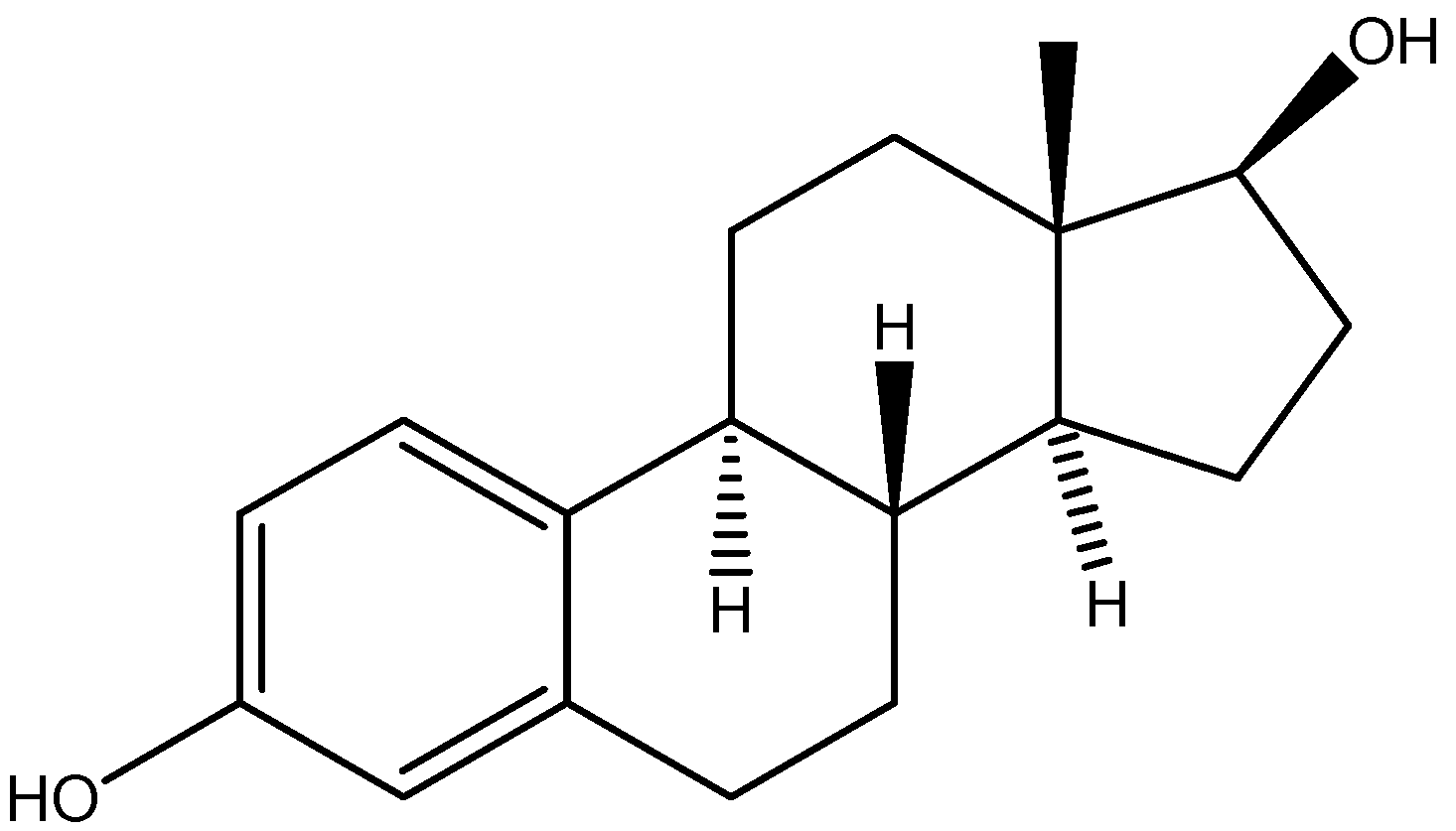
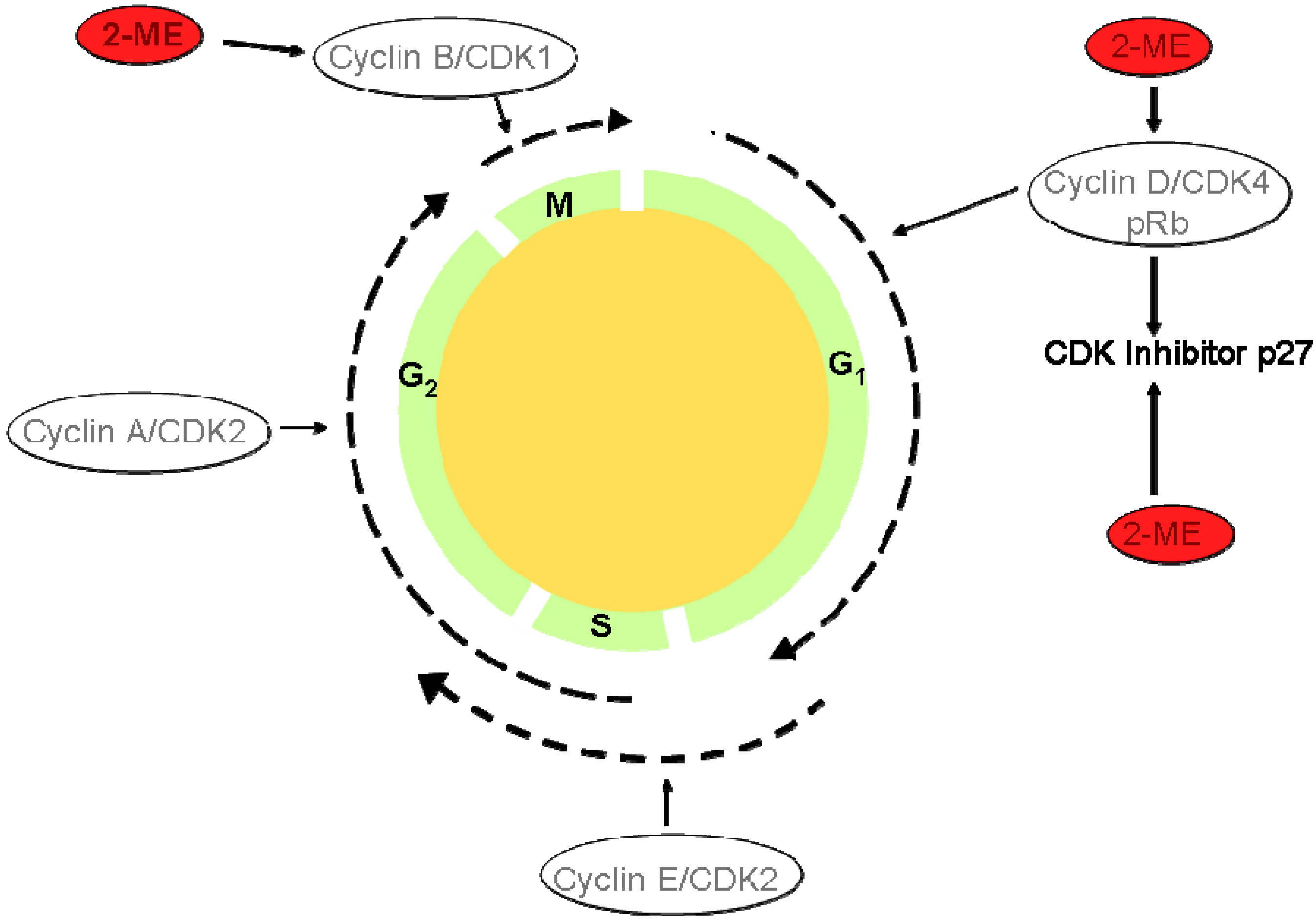
- Barchiesi, F.; Jackson, E. K.; Fingerle, J.; Gillespie, D. G.; Odermatt, B.; Dubey, R.K. 2-Methoxyestradiol, an Estradiol Metabolite, Inhibits Neointima Formation and Smooth Muscle Cell Growth via Double Blockade of the Cell Cycle. Circ. Res. 2006, 99, 266–274. [Google Scholar] [CrossRef]
- Zacharia, L.C.; Piche, C.A.; Fielding, R.M.; Holland, K.M.; Allison, S.D.; Dubey, R.K.; Jackson, E.K. 2-Hydroxyestradiol Is a Prodrug of 2-Methoxyestradiol. J. Pharmacol. Exp. Ther. 2004, 309, 1093–1097. [Google Scholar] [CrossRef]
- Dubey, R.K.; Gillespie, D.G.; Zacharia, L.C.; Rosselli, M.; Korzekwa, K.R.; Fingerle, J.; Jackson, E.K. Methoxyestradiols Mediate the Antimitogenic Effects of Estradiol on Vascular Smooth Muscle Cells via Estrogen Receptor-Independent Mechanisms. Biochem.Bioph. Res. Co. 2000, 278, 27–33. [Google Scholar] [CrossRef]
- Zacharia, L.C.; Gogos, J.A.; Karayiorgou, M.; Jackson, E.K.; Gillespie, D.G.; Barchiesi, F.; Dubey, R.K. Methoxyestradiols Mediate the Antimitogenic Effects of 17{beta}-Estradiol: Direct Evidence From Catechol-O-Methyltransferase-Knockout Mice. Circulation 2003, 108, 2974–2978. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Dubey, R.; Salah, E.M.; Jackson, E.K. 2-Hydroxyestradiol Attenuates Renal Disease in Chronic Puromycin Aminonucleoside Nephropathy. J. Am. Soc. Nephrol. 2002, 13, 2737–2747. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Maddy, H.; Salah, E.M.; Jackson, E.K.; Melhem, M. Estradiol metabolites retard the progression of pulmonary hypertension-preclinical evidence for clinical development. Hypertension 2003b, 42, 416. [Google Scholar]
- Tofovic, S.P.; Maddy, H.; Jackson, E.K. Estradiol metabolites attenuate renal and cardiovascular injury induced by chronic angiotensin II administration. Hypertension 2003a, 42, 414. [Google Scholar]
- Dantas, A.P.V.; Sandberg, K. Does 2-Methoxyestradiol Represent the New and Improved Hormone Replacement Therapy for Atherosclerosis? Circ. Res. 2006, 99, 234–237. [Google Scholar] [CrossRef]
- Zovko, M.; Zorc, B.; Novak, P.; Tepes, P.; Cetina-Cizmek, B.; Horvat, M. Macromolecular prodrugs: XI. Synthesis and characterization of polymer-estradiol conjugate. Int. J. Pharm. 2004, 285, 35–41. [Google Scholar]
- Chandrasekar, B.; Sirois, M.G.; Geoffroy, P.; Lauzier, D.; Nattel, S.; Tanguay, J. F. Local delivery of 17 beta -estradiol improves reendothelialization and decreases inflammation after coronary stenting in a porcine model. Thromb. Haemost. 2005, 94, 1042–1047. [Google Scholar]
- Andresen, T.L.; Davidsen, J.; Begtrup, M.; Mouritsen, O.G.; Jorgensen, K. Enzymatic Release of Antitumor Ether Lipids by Specific Phospholipase A2 Activation of Liposome-Forming Prodrugs. J. Med. Chem. 2004, 47, 1694–1703. [Google Scholar] [CrossRef]
- Brioschi, A.; Zenga, F.; Zara, G.; Gasco, M.; Ducati, A.; Mauro, A. Solid lipid nanoparticles: could they help to improve the efficacy of pharmacologic treatments for brain tumors? Neurol. Res. 2007, 29, 324–330. [Google Scholar] [CrossRef]
- Kucerova, L.; Altanerova, V.; Matuskova, M.; Tyciakova, S.; Altaner, C. Adipose Tissue-Derived Human Mesenchymal Stem Cells Mediated Prodrug Cancer Gene Therapy. Cancer Res. 2007, 67, 6304–6313. [Google Scholar]
- Niculescu-Duvaz, I.; Springer, C. J. Antibody-directed enzyme prodrug therapy (ADEPT): a review. Adv. Drug Deliv. Rev. 1997, 26, 151–172. [Google Scholar] [CrossRef]
- Bagshawe, K.D.; Sharma, S.K.; Springer, C.J.; Rogers, G.T. Antibody directed enzyme prodrug therapy (ADEPT): A review of some theoretical, experimental and clinical aspects. Ann. Oncol. 1994, 5, 879–891. [Google Scholar]
- Springer, C.; Bavetsias, V.; Jackman, A.; Boyle, F.; Marshall, D.; Pedely, R.; Bisset, G. Prodrugs of thymidylate synthase inhibitors: potential for antibody directed enzyme prodrug therapy (ADEPT). Anti-Cancer Drug Des. 1996, 11, 625–636. [Google Scholar]
- Sharma, S.; Boden, J.; Springer, C.; Burke, P.J.; Bagshawe, K. Antibody-directed enzyme prodrug therapy (ADEPT). A three-phase study in ovarian tumor xenografts. Cell Biophys. 1994, 24-25, 219–228. [Google Scholar] [CrossRef]
- Liang, J.F.; Park, Y.J.; Song, H.; Li, Y.T.; Yang, V. ATTEMPTS: A heparin/protamine-based prodrug approach for delivery of thrombolytic drugs. J. Control. Release 2001, 72, 145–156. [Google Scholar] [CrossRef]
- Naik, S.S.; Liang, J.F.; Park, Y.J.; Lee, W.K.; Yang, V.C. Application of "ATTEMPTS" for drug delivery. J. Control. Release 2005, 101, 35–45. [Google Scholar] [CrossRef]
- Park, Y.J.; Liang, J.F.; Song, H.; Li, Y.T.; Naik, S.; Yang, V.C. ATTEMPTS: a heparin/protamine-based triggered release system for the delivery of enzyme drugs without associated side-effects. Adv. Drug Deliv. Rev. 2003, 55, 251–265. [Google Scholar] [CrossRef]
- Brynda, E.; Houska, M. Multiple Alternating Molecular Layers of Albumin and Heparin on Solid Surfaces. J. Colloid Interface Sci. 1996, 183, 18–25. [Google Scholar] [CrossRef]
- Houska, M.; Brynda, E. Interactions of Proteins with Polyelectrolytes at Solid/Liquid Interfaces: Sequential Adsorption of Albumin and Heparin. J. Colloid Interface Sci. 1997, 188, 243–250. [Google Scholar] [CrossRef]
- Jian, J.; Qinggang, T.; Jiacong, S. Construction of albumin multilayer coating onto plasma treated poly(vinyl chloride) via electrostatic self-assembly. Polym. Advanc. Technol. 2004, 15, 490–494. [Google Scholar] [CrossRef]
- Ji, J.; Tan, Q.; Fan, D.Z.; Sun, F.Y.; Barbosa, M.A.; Shen, J. Fabrication of alternating polycation and albumin multilayer coating onto stainless steel by electrostatic layer-by-layer adsorption. Colloid. Surface. B. 2004, 34, 185–190. [Google Scholar] [CrossRef]
- Fu, J.; Ji, J.; Yuan, W.; Shen, J. Construction of anti-adhesive and antibacterial multilayer films via layer-by-layer assembly of heparin and chitosan. Biomaterials 2005, 26, 6684–6692. [Google Scholar] [CrossRef]
- Tan, Q.; Ji, J.; Barbosa, M.A.; Fonseca, C.; Shen, J. Constructing thromboresistant surface on biomedical stainless steel via layer-by-layer deposition anticoagulant. Biomaterials 2003, 24, 4699. [Google Scholar] [CrossRef]
- Serizawa, T.; Yamaguchi, M.; Akashi, M. Alternating Bioactivity of Polymeric Layer-by-Layer Assemblies: Anticoagulation vs Procoagulation of Human Blood. Biomacromolecules 2002, 3, 724–731. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Serizawa, T.; Akashi, M. Layer-by-Layer Assembly on Hydrogel Surfaces and Control of Human Whole Blood Coagulation. Chem. Lett. 2003, 32, 174–175. [Google Scholar] [CrossRef]
- Cai, K.; Rechtenbach, A.; Hao, J.; Bossert, J.; Jandt, K.D. Polysaccharide-protein surface modification of titanium via a layer-by-layer technique: Characterization and cell behaviour aspects. Biomaterials 2005, 26, 5960–5971. [Google Scholar] [CrossRef]
- Thierry, B.; Winnik, F.M.; Merhi, Y.; Silver, J.; Tabrizian, M. Bioactive Coatings of Endovascular Stents Based on Polyelectrolyte Multilayers. Biomacromolecules 2003, 4, 1564–1571. [Google Scholar] [CrossRef]
- Thierry, B.; Winnik, F.M.; Merhi, Y.; Tabrizian, M. Nanocoatings onto Arteries via Layer-by-Layer Deposition: Toward the in Vivo Repair of Damaged Blood Vessels. J. Am. Chem. Soc. 2003, 125, 7494–7495. [Google Scholar] [CrossRef]
- Thierry, B.; Kujawa, P.; Tkaczyk, C.; Winnik, F.M.; Bilodeau, L.; Tabrizian, M. Delivery Platform for Hydrophobic Drugs: Prodrug Approach Combined with Self-Assembled Multilayers. J. Am. Chem. Soc. 2005, 127, 1626–1627. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis is an inflammatory disease. Am. Heart J. 1999, 138, S419–S420. [Google Scholar] [CrossRef]
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sandros, M.G.; Sarraf, C.B.; Tabrizian, M. Prodrugs in Cardiovascular Therapy. Molecules 2008, 13, 1156-1178. https://doi.org/10.3390/molecules13051156
Sandros MG, Sarraf CB, Tabrizian M. Prodrugs in Cardiovascular Therapy. Molecules. 2008; 13(5):1156-1178. https://doi.org/10.3390/molecules13051156
Chicago/Turabian StyleSandros, Marinella G., Chady B. Sarraf, and Maryam Tabrizian. 2008. "Prodrugs in Cardiovascular Therapy" Molecules 13, no. 5: 1156-1178. https://doi.org/10.3390/molecules13051156