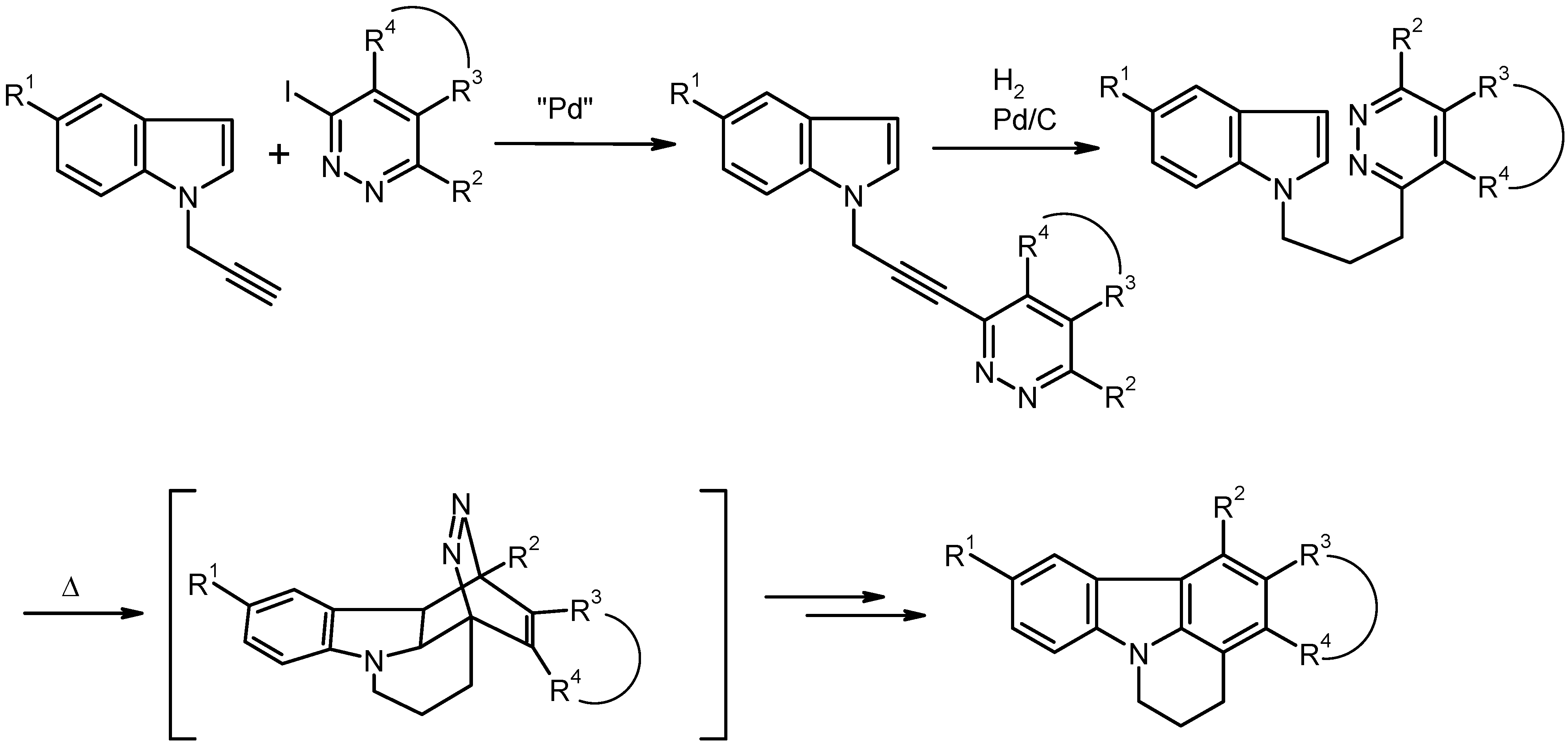
General
Melting points (uncorrected) were determined on a Kofler hot-stage microscope (Reichert). 1H-NMR spectra were recorded on a Bruker Avance DPX 200 (200 MHz) or on a Varian UnityPlus 300 (300 MHz) spectrometer. IR spectra were taken on a Perkin-Elmer 1605 FT-IR instrument. Mass spectra were obtained on a Shimadzu QP5050A DI 50 instrument, high-resolution mass spectra were recorded on a Finnigan MAT 8230 spectrometer at the Institute of Organic Chemistry, University of Vienna. Column chromatography was carried out on Merck Kieselgel 60, 0.063–0.200 mm, thin layer chromatography was done on Merck aluminium sheets pre-coated with Kieselgel F254. Microanalyses were performed at the Microanalytical Laboratory, Faculty of Chemistry, University of Vienna.
5-Bromo-1-prop-2-yn-1-yl-1H-indole (1c). To a solution of 5-bromoindole (1.96 g, 10 mmol) and propargyl bromide (2.23 g of a 80% solution in toluene; 15 mmol) in toluene (30 mL) were added tetrabutylammonium bromide (0.161 g, 0.5 mmol) and 50% aqueous NaOH (6 mL). The two-phase system was stirred vigorously for 3 h at room temperature, then it was diluted with toluene (10 mL) and the phases were separated. The organic layer was washed several times with water and then with brine. It was dried over Na2SO4 and the solvent was removed under reduced pressure to give the product (2.179 g, 93%) as an almost colorless oil which darkened slowly on storage. IR (KBr): 3290, 2125, 1564, 1507, 1465, 1189, 1053, 898, 752 cm–1; MS (EI, 70 eV) m/z: 235 (M+, 46%), 233 (M+, 49), 196 (12), 194 (10), 154 (100), 127 (18), 115 (33), 88 (22), 62 (19); 1H-NMR (CDCl3) δ: 7.77 (d, J4–6 = 1.8 Hz, 1H, 4-H), 7.36–7.26 (m, 2H, 6-H, 7-H), 7.21 (d, J2–3 = 3.3 Hz, 2-H), 6.48 (d, J2–3 = 3.3 Hz, 1H, 3-H), 4.86 (d, J = 2.6 Hz, 2H, CH2), 2.42 (t, J = 2.6 Hz, 1H, C≡CH). HRMS (EI, 70 eV) m/z calcd. for C11H8BrN (M+): 232.9840. Found: 232.9844.
1-Iodophthalazine (
2a) [
11].
Modified Procedure. A mixture of 1-chlorophthalazine (2.75 g, 17 mmol), potassium iodide (5.0 g, 30 mmol), 57% hydroiodic acid (3.4 mL) and acetone (50 mL) was stirred in the dark at room temperature for 96 h. The yellow precipitate (
2a·HI) was collected by filtration, washed with diethyl ether, and dried
in vacuo. The material was then suspended in ice-water and the mixture was stirred for 15 min and made alkaline with dilute ammonium hydroxide. It was extracted several times with CH
2Cl
2 and the combined extracts were repeatedly washed with a solution of sodium thiosulfate (0.5 g) in 1% ammonium hydroxide, then with brine. Triethylamine (2 mL) was added to the CH
2Cl
2 extract, then it was dried over Na
2SO
4. Evaporation of the volatile components gave
2a (3.153 g, 74%) as yellow crystals, mp 87–96 °C (lit. [
11]: 78 °C), which were stored in a deep freezer and which were used for the subsequent steps without further purification.
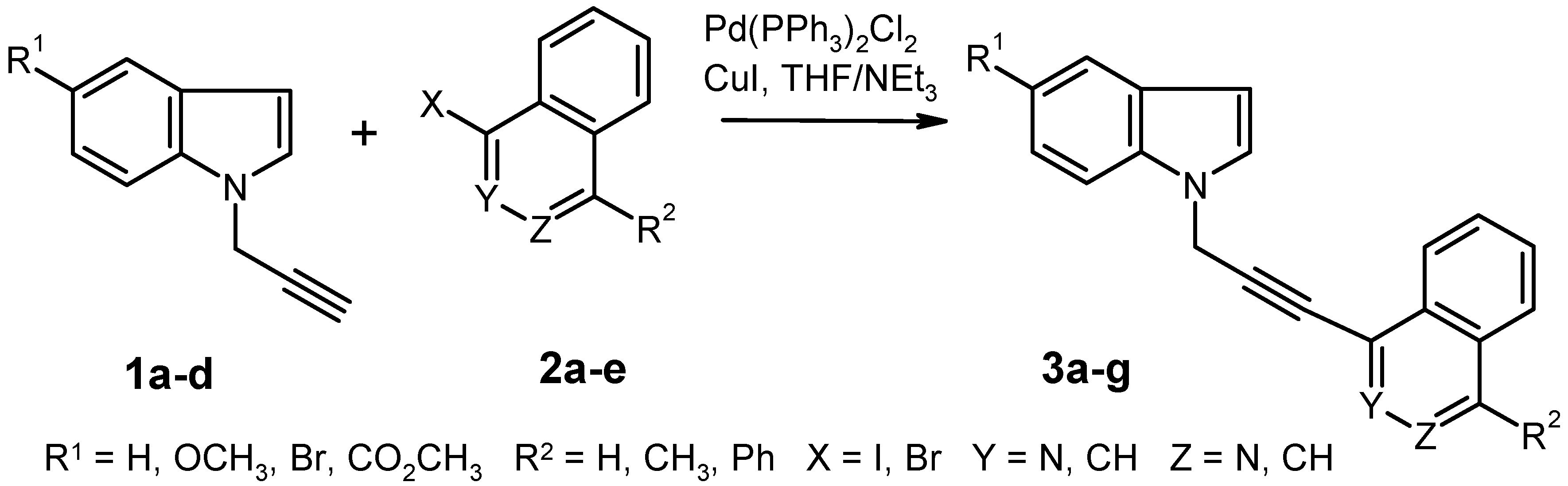
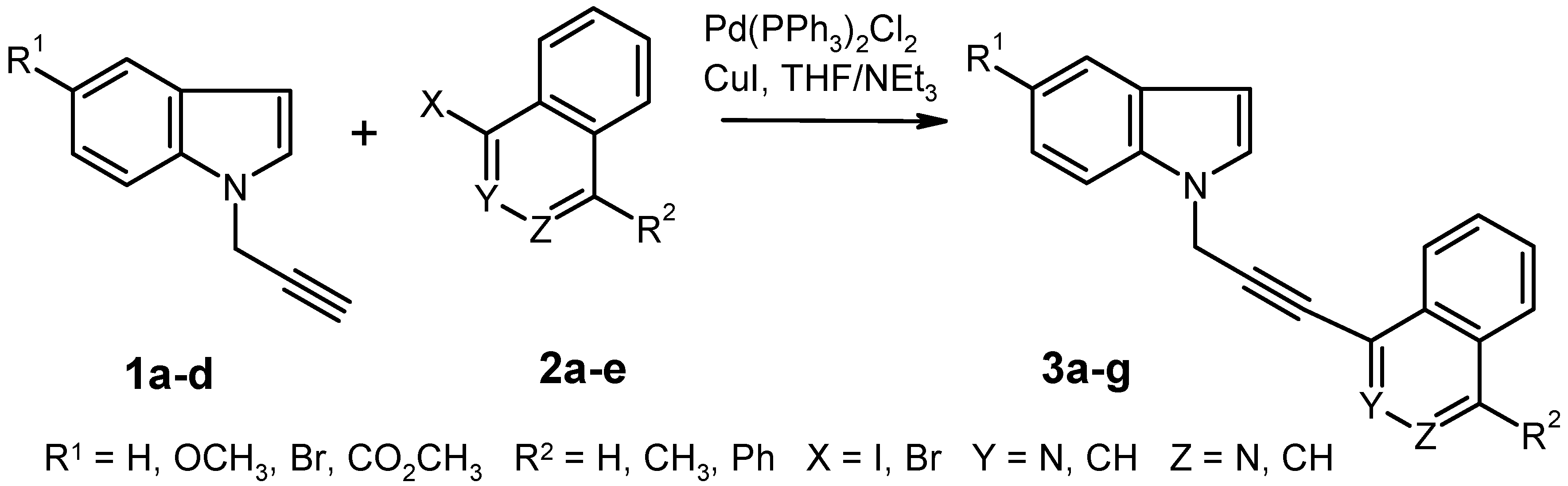
Synthesis of Compounds 3 by Pd-Catalyzed Cross-Coupling Reaction. General Procedure. To a solution of the aryl halide
2a,
2b,
2c,
2d, or
2e (2.6 mmol), respectively, and the appropriate alkyne
1a,
1b,
1c, or
1d (3.25 mmol), respectively, in dry THF (6 mL) were added triethylamine (1.0 mL, 7.2 mmol), CuI (0.015 g, 3 mol%) and Pd(PPh
3)
2Cl
2 (0.055 g, 3 mol%), and the mixture was flushed with argon. It was then stirred under an argon atmosphere under the conditions (room temperature or reflux) and for the time listed in
Table 1. The insoluble material was filtered off and washed carefully with THF. The combined filtrates were evaporated under reduced pressure and the residue was purified by column chromatography (eluent: ethyl acetate or ethyl acetate/light petroleum).
1-[3-(5-Methoxy-1H-indol-1-yl)prop-1-yn-1-yl]-4-methylphthalazine (3a). Prepared from 5-methoxy-1-prop-2-yn-1-yl-1H-indole (1a) and 1-iodo-4-methylphthalazine (2b); yield: 0.408 g (47%), recrystallization from ethyl acetate/light petroleum gave almost colorless crystals, mp 166–168 °C. IR (KBr): 2921, 2231, 1620, 1484, 1383, 1239, 1153, 1029, 848, 799, 771, 619 cm–1; MS (EI, 70 eV) m/z: 327 (M+, 19%), 312 (10), 296 (2), 284 (6), 164 (6), 152 (15), 142 (9), 127 (7), 103 (4), 77 (7), 70 (13), 61 (14), 45 (24), 43 (100); 1H-NMR (CDCl3) δ: 8.09 (d, J = 8.7 Hz, 1H, phthalazine 8-H), 8.06 (d, J = 9.3 Hz, 1H, phthalazine 5-H, shows positive NOE on irradiation at 3.02 ppm), 7.92–7.80 (m, 2H, phthalazine 6-H, 7-H), 7.47 (d, J6–7 = 9.0 Hz, 1H, indole 7-H), 7.31 (d, J2–3 = 3.0 Hz, 1H, indole 2-H), 7.15 (d, J4–6 = 2.1 Hz, 1H, indole 4-H), 6.96 (dd, J6–7 = 9.0 Hz, J4–6 = 2.4 Hz, 1H, indole 6-H), 6.52 (d, J2–3 = 3.0 Hz, 1H, indole 3-H), 5.28 (s, 2H, CH2), 3.88 (s, 3H, OCH3), 3.02 (s, 3H, 4-CH3). Anal. calcd. for C21H17N3 O · 0.25 H2O: C, 76.00; H, 5.31; N, 12.60. Found: C, 76.04; H, 5.22; N, 12.45.
1-[3-(5-Methoxy-1H-indol-1-yl)prop-1-yn-1-yl]-4-phenylphthalazine (3b). Prepared from 5-methoxy-1-prop-2-yn-1-yl-1H-indole (1a) and 1-iodo-4-phenylphthalazine (2c); yield: 0.552 g (55%), recrystallization from ethyl acetate gave almost colorless crystals, mp 150–152 °C. IR (KBr): 3057, 2939, 2236, 1619, 1576, 1485, 1386, 1239, 1151, 1028, 702, 653 cm–1; MS (EI, 70 eV) m/z: 389 (M+, 100%), 374 (24), 358 (10), 346 (22), 244 (20), 213 (50), 194 (56), 187 (42), 173 (33), 165 (12), 147 (27), 132 (34), 104 (19), 77 (18), 51 (17), 43 (20); 1H-NMR (CDCl3) δ: 8.19–8.16 (m, 1H, phthalazine 8-H), 8.09–8.05 (m, 1H, phthalazine 5-H, shows positive NOE on irradiation at 7.75 ppm), 7.88–7.83 (m, 2H, phthalazine 6-H, 7-H), 7.77–7.74 (m, 2H, phenyl 2-H, 6-H), 7.60–7.56 (m, 3H, phenyl 3-H, 4-H, 5-H, shows positive NOE on irradiation at 7.75 ppm), 7.49 (d, J6–7 = 8.9 Hz, 1H, indole 7-H), 7.33 (d, J2–3 = 3.3 Hz, 1H, indole 2-H), 7.16 (d, J4–6 = 2.4 Hz, 1H, indole 4-H), 6.97 (dd, J6–7 = 8.9 Hz, J4–6 = 2.4 Hz, 1H, indole 6-H), 6.54 (d, J2–3 = 3.3 Hz, 1H, indole 3-H), 5.31 (s, 2H, CH2), 3.88 (s, 3H, OCH3). Anal. calcd. for C26H19N3 O: C, 80.19; H, 4.92; N, 10.79. Found: C, 79.98; H, 4.99; N, 10.55.
4-[3-(5-Methoxy-1H-indol-1-yl)prop-1-yn-1-yl]isoquinoline (3c). Prepared from 5-methoxy-1-prop-2-yn-1-yl-1H-indole (1a) and 4-bromoisoquinoline (2e); yield: 0.284 g (34%), recrystallization from ethyl acetate/light petroleum gave brownish crystals, mp 131–133 °C. IR (KBr): 2903, 2825, 2244, 1620, 1488, 1241, 1150, 794, 753, 715, 580 cm–1; MS (EI, 70 eV) m/z: 312 (M+, 60%), 296 (2), 281 (3), 201 (4), 166 (100), 156 (9), 139 (36), 103 (5), 89 (6), 76 (6), 43 (14); 1H NMR (CDCl3) δ: 9.28 (br s, 1H, isoquinoline 1-H), 8.75 (br s, 1H, isoquinoline 3-H), 8.11 (d, J7–8 = 8.4 Hz, 1H, isoquinoline 8-H), 7.98 (d, J5–6 = 7.8 Hz, 1H, isoquinoline 5-H), 7.74 (t, J = 7.7 Hz, 1H, isoquinoline 7-H, shows positive NOE on irradiation at 8.11 ppm), 7.65 (t, J = 7.3 Hz, 1H, isoquinoline 6-H), 7.46 (d, J6–7 = 8.9 Hz, 1H, indole 7-H, shows positive NOE on irradiation at 5.25 ppm), 7.32 (d, J2–3 = 3.3 Hz, 1H, indole 2-H, shows positive NOE on irradiation at 5.25 ppm), 7.15 (d, J4–6 = 2.4 Hz, 1H, indole 4-H), 6.96 (dd, J6–7 = 8.9 Hz, J4–6 = 2.4 Hz, 1H, indole 6-H), 6.52 (d, J2–3 = 3.3 Hz, 1H, indole 3-H), 5.25 (s, 2H, CH2), 3.88 (s, 3H, OCH3). Anal. calcd for C21H16N2O · 0.25 H2O: C, 79.60; H, 5.25; N, 8.84. Found: C, 79.66; H, 5.50; N, 8.46.
1-[3-(5-Methoxy-1H-indol-1-yl)prop-1-yn-1-yl]isoquinoline (3d). Prepared from 5-methoxy-1-prop-2-yn-1-yl-1H-indole (1a) and 1-iodoisoquinoline (2d); yield: 0.620 g (75%), recrystallization from ethyl acetate/light petroleum gave almost colorless crystals, mp 155–157 °C. IR (KBr): 3048, 2953, 2235, 1620, 1578, 1551, 1486, 1350, 1242, 1153, 1028, 830, 747, 723, 666 cm–1; MS (EI, 70 eV) m/z: 312 (M+, 34%), 297 (6), 281 (2), 269 (9), 184 (11), 166 (44), 146 (11), 139 (38), 134 (15), 103 (7), 89 (6), 76 (9), 61 (13), 45 (26), 43 (100); 1H-NMR (CDCl3) δ: 8.51 (d, J3–4 = 5.4 Hz, 1H, isoquinoline 3-H), 8.22 (dd, J7–8 = 8.4 Hz, J6–8 = 1.2 Hz, 1H, isoquinoline 8-H), 7.82 (d, J5–6 = 8.1 Hz, 1H, isoquinoline 5-H), 7.72–7.69 (m, 1H, isoquinoline 6-H, shows positive NOE on irradiation at 7.82 ppm), 7.67–7.63 (m, 1H, isoquinoline 4-H, shows positive NOE on irradiation at 8.51 ppm), 7.61–7.55 (m, 1H, isoquinoline 7-H, shows positive NOE on irradiation at 8.22 ppm), 7.47 (d, J6–7 = 8.9 Hz, 1H, indole 7-H), 7.32 (d, J2–3 = 3.3 Hz, 1H, indole 2-H), 7.14 (d, J4–6 = 2.4 Hz, 1H, indole 4-H), 6.96 (dd, J6–7 = 8.9 Hz, J4–6 = 2.4 Hz, 1H, indole 6-H), 6.51 (d, J2–3 = 3.3 Hz, 1H, indole 3-H), 5.26 (s, 2H, CH2), 3.88 (s, 3H, OCH3). Anal. calcd. for C21H16N2O · 0.2 H2O: C, 79.83; H, 5.23; N, 8.87. Found: C, 79.82; H, 5.22; N, 8.59.
1-[3-(2,3-Dihydro-1H-indol-1-yl)prop-1-yn-1-yl]phthalazine (3e). Preparation from 1-prop-2-yn-1-ylindoline (1d) and 1-iodophthalazine (2a); yield: 0.592 g (80%), recrystallization from ethyl acetate gave brownish crystals, mp 94–95 °C. IR (KBr): 3043, 2830, 2227, 1606, 1484, 1353, 1237, 1138, 757, 594 cm–1; MS (EI, 70 eV) m/z: 285 (M+, 2%), 168 (100), 141 (13), 114 (11), 91 (23), 77 (6), 65 (16), 51 (5); 1H-NMR (CDCl3) δ: 9.43 (s, 1H, phthalazine 4-H), 7.93–7.73 (m, 4H, phthalazine 5-H, 6-H, 7-H, 8-H), 7.21–7.16 (m, 2H, indoline 4-H, 6-H, shows positive NOE on irradiation at 3.05 ppm), 6.85–6.76 (m, 2H, indoline 5-H, indoline 7-H), 4.38 (s, 2H, C≡CCH2), 3.60 (t, J = 8.1 Hz, 2H, indoline 2-H, shows positive NOE on irradiation at 3.05 ppm), 3.05 (t, J = 8.1 Hz, 2H, indoline 3-H). Anal. calcd. for C19H15N3: C, 79.98; H, 5.30; N, 14.73. Found: C, 79.93; H, 5.47; N, 14.68.
1-[3-(5-Bromo-1H-indol-1-yl)prop-1-yn-1-yl]phthalazine (3f). Preparation from 5-bromo-1-prop-2-yn-1-yl-1H-indole (1b) and 1-iodophthalazine (2a); yield: 0.281 g (30%), recrystallization from ethyl acetate gave almost colorless crystals, mp 154–158 °C. IR (KBr): 3097, 2955, 2239, 1465, 1394, 1353, 1282, 1185, 1051, 899, 758, 594 cm–1; MS (EI, 70 eV) m/z: 363 (M+, 13%), 361 (M+, 12), 282 (8), 195 (14), 167 (7), 141 (34), 127 (36), 114 (15), 97 (11), 84 (20), 69 (25), 58 (93), 57 (39), 43 (100); 1H NMR (CDCl3) δ: 9.48 (s, 1H, phthalazine 4-H), 8.06 (d, J7–8 = 8.1 Hz, 1H, phthalazine 8-H), 7.99–7.86 (m, 3H, phthalazine 5-H, 6-H, 7-H), 7.81 (d, J4–6 = 1.6 Hz, 1H, indole 4-H), 7.45 (d, J6–7 = 8.7 Hz, 1H, indole 7-H), 7.38 (dd, J6–7 = 8.7 Hz, J4–6 = 1.6 Hz, 1H, indole 6-H), 7.34 (d, J2–3 = 3.3 Hz, 1H, indole 2-H), 6.55 (d, J2–3 = 3.3 Hz, 1H, indole 3-H), 5.31 (s, 2H, CH2). Anal. calcd. for C19H12N3Br: C, 63.00; H, 3.34; N, 11.60. Found: C, 62.75; H, 3.34; N, 11.34.
Methyl 1-(3-Phthalazin-1-ylprop-2-yn-1-yl)-1H-indole-5-carboxylate (3g). Preparation from methyl 1-prop-2-yn-1-yl-1H-indole-5-carboxylate (1c) and 1-iodophthalazine (2a); modified work-up procedure: after completion of the reaction, the solid material (3g + triethylammonium iodide) was filtered off and washed with diethyl ether. It was then dissolved in CH2Cl2 and this solution was washed with 0.5 N HCl, then with water. The extract was dried over Na2SO4 and evaporated under reduced pressure to give the product (0.133 g, 15%) as brownish crystals, mp 97 °C (decomposition). IR (KBr): 2948, 2241, 1710, 1610, 1434, 1310, 1196, 1097, 754, 595 cm–1; MS (EI, 70 eV) m/z: 341 (M+, 23%), 282 (10), 175 (50), 165 (10), 144 (100), 141 (36), 116 (73), 89 (39), 72 (29), 58 (66), 44 (81); 1H-NMR (CDCl3) δ: 9.49 (s, 1H, phthalazine 4-H), 8.45 (d, J4–6 = 0.9 Hz, 1H, indole 4-H), 8.10–7.85 (m, 5H, indole 6-H, phthalazine 5-H, 6-H, 7-H, 8-H), 7.58 (d, J6–7 = 8.7 Hz, 1H, indole 7-H, shows positive NOE on irradiation at 5.35 ppm), 7.41 (d, J2–3 = 3.4 Hz, 1H, indole 2-H, shows positive NOE on irradiation at 5.35 ppm), 6.71 (d, J2–3 = 3.4 Hz, 1H, indole 3-H), 5.35 (s, 2H, CH2), 3.95 (s, 3H, OCH3). HRMS (EI, 70 eV) m/z calcd. for C21H15N3O2 (M+): 341.1164. Found: 341.1167.



{kind=link}
{kind=link}
{kind=link}









