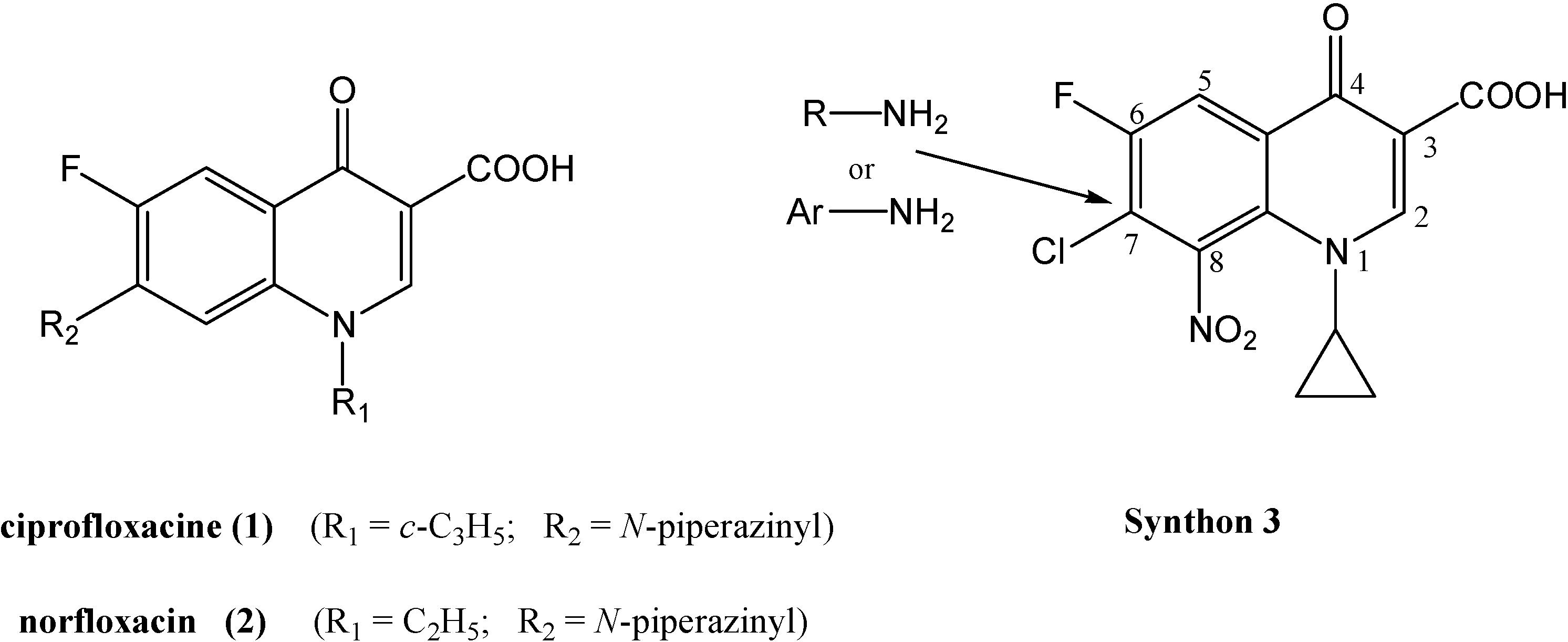
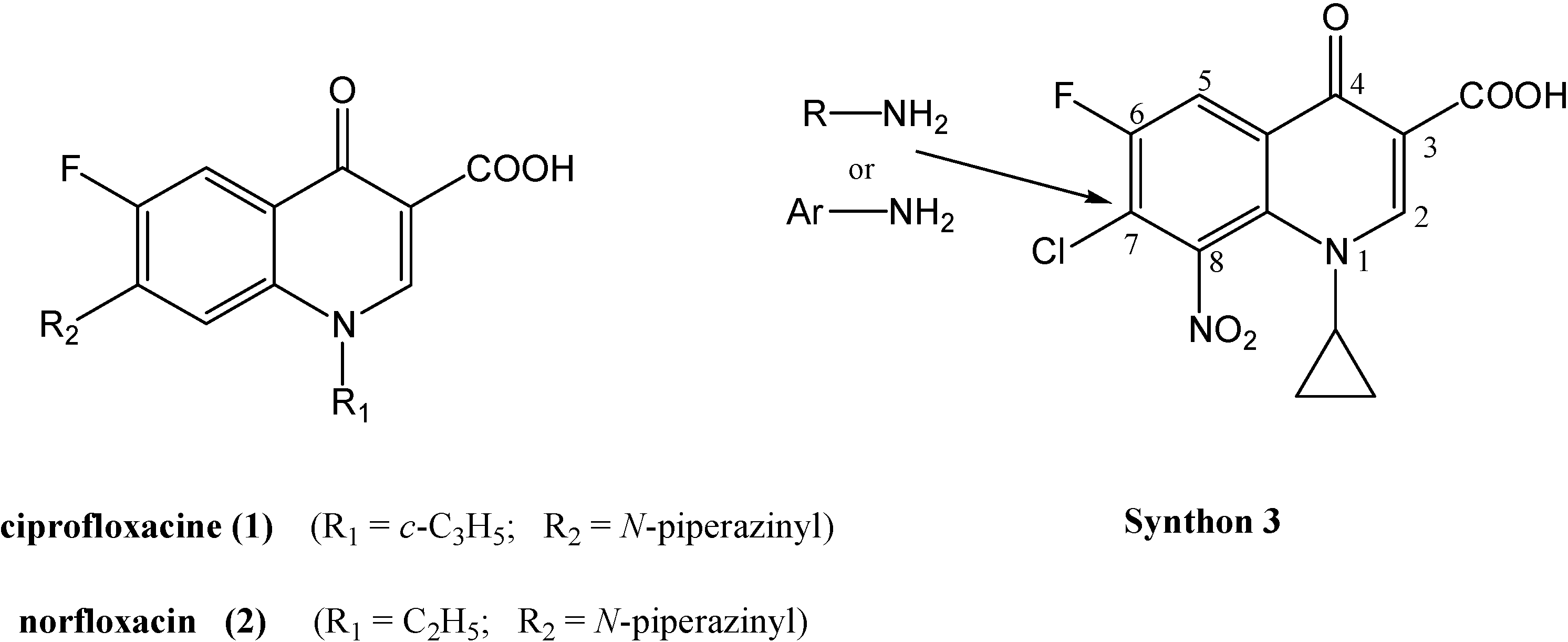
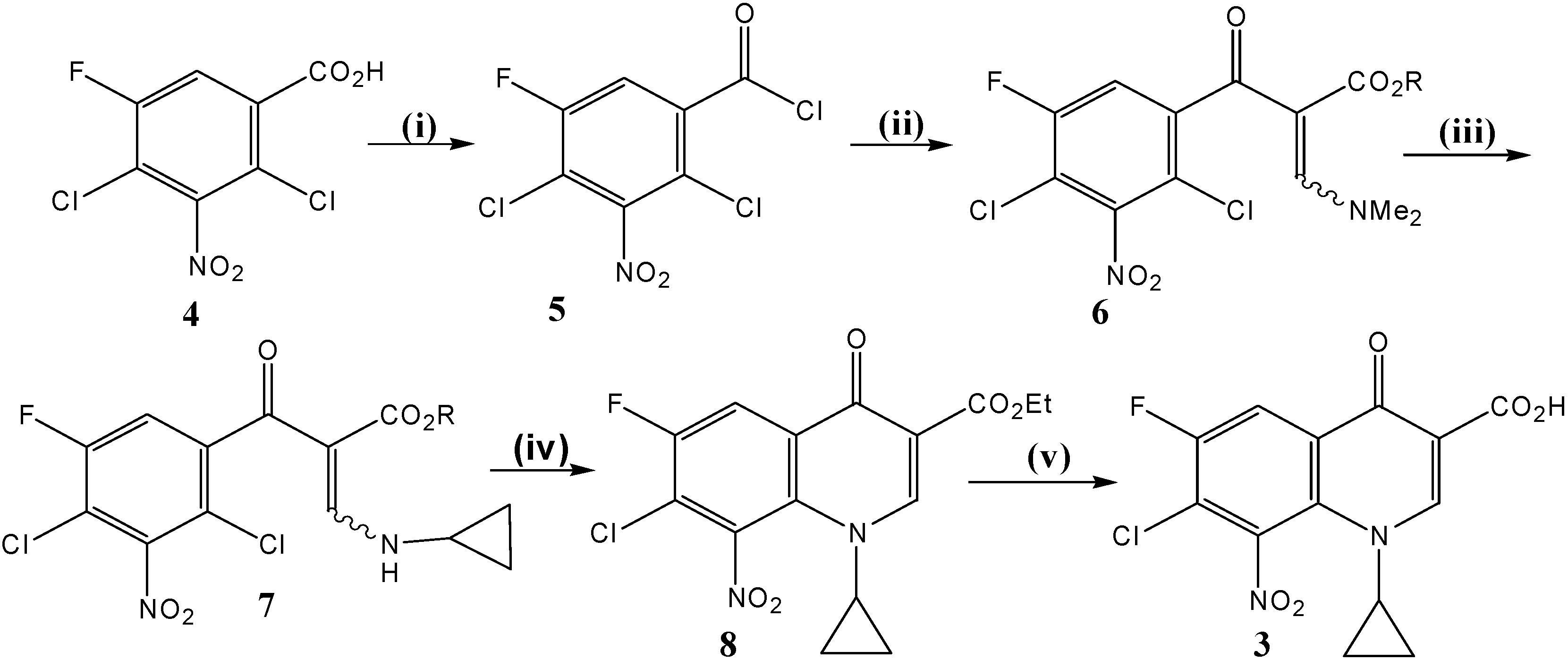
Synthesis of synthon 3 (Scheme 1)
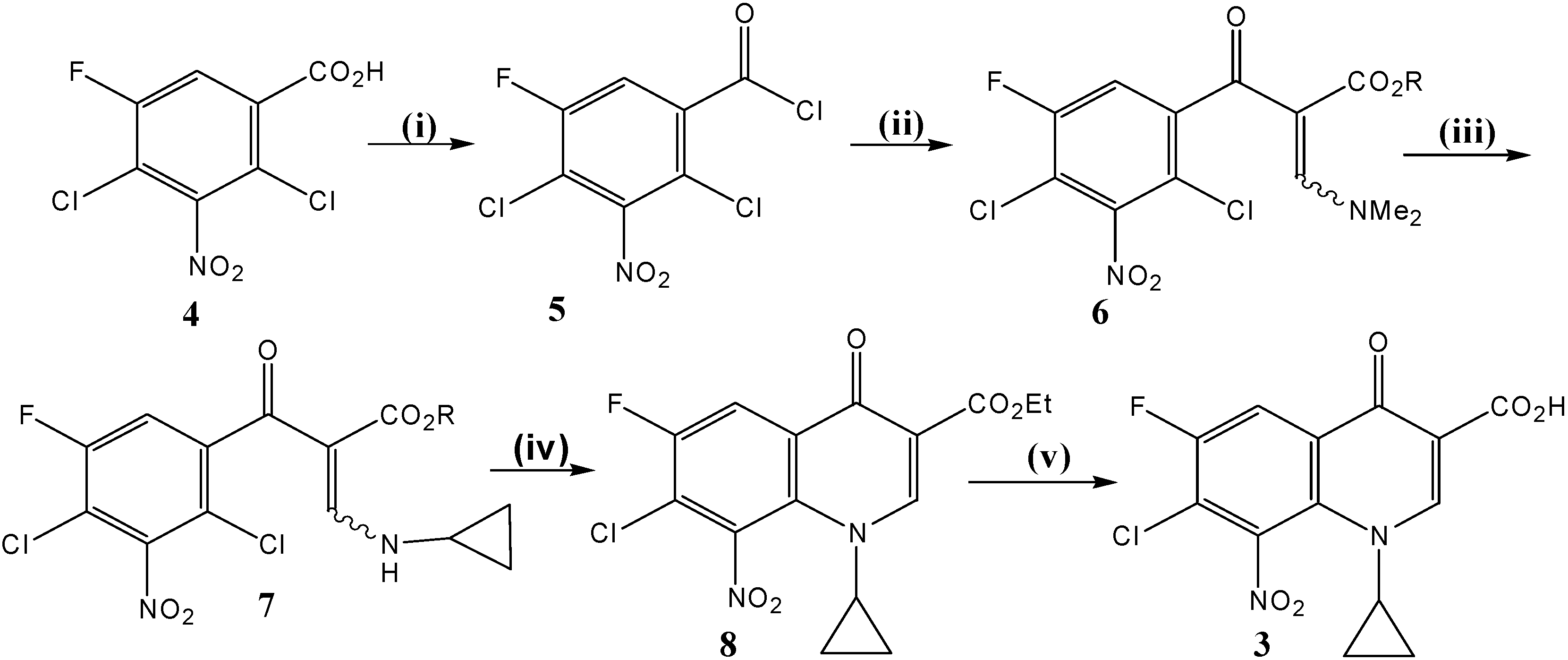
i) Ethyl- 3-(N,N-dimethylamino)-2-(2,4-dichloro-5-fluoro-3-nitrobenzoyl) acrylate (6). A mixture of 2,4-dichloro-5-fluoro-3-nitrobenzoic acid (4, 10.2 g, 40 mmol), and thionyl chloride (SOCl2) (19.0 g, 160 mmol), dissolved in dry benzene (120 mL) was refluxed at 75-80 °C for 3–4 h under anhydrous conditions. The mixture was then distilled off under reduced pressure to remove solvent and excess thionyl chloride. Dry benzene was then added twice (2 x 20 mL) into the reaction vessel and the mixture was re-distilled so as to remove traces of thionyl chloride. The resulting 2,4-dichloro-5-fluoro-3-nitrobenzoyl chloride (5), formed as thick oil, was used as such for the next step without further purification. To a stirred and cooled (5-10 °C) solution of ethyl 3-(N,N-dimethyl-amino)acrylate (6.3 g, 44 mmol) and triethylamine (4 mLml, 8.1 g, 80 mmol) in dry benzene (50 mLml), a solution of the crude acid chloride (prepared above) in dry benzene (25 mLml) was added drop by drop. The resulting mixture was stirred continuously for 2 h at room temperature under anhydrous conditions. Then, the solution was refluxed at 90 °C for 90 minutes. This crude product was evaporated to dryness, re-dissolved in chloroform; the chloroform was extracted with water (30 mL) and dried (with anhydrous MgSO4). The solvent, chloroform, was then evaporated to dryness under reduced pressure. The residual product (about 20 mL) was soaked in methanol (10 mL) whereby the title compound 6 was produced as yellowish powder that was collected by suction filtration and dried. Yield ≈ 13.8 g (91 %). 1H-NMR (CDCl3): δ 0.95 (t, J = 7.1 Hz, 3H, CH3), 2.97 (s, 3H) and 3.37 (s, 3H) [N (CH3)2], 3.94 (q, J = 7.1 Hz, 2H, CH2Me), 7.27 (d, 3JH-F = 8.2 Hz, 1H, H-6), 7.91 (br s, 1H, N-C(3′′)-H); 13C -NMR (CDCl3): δ 13.8 (CH3CH2), 43.3, 48.4 [N (CH3)2], 60.2 (CH2Me), 100.9 (C-2′′), 114.5 (d, 2JC-F = 23.3 Hz, C-4), 116.9 (d, 2JC-F = 23.1 Hz, C-6), 118.2 (d, 3JC-F = 4.5 Hz, C-1), 144.2 (d, 3JC-F = 6 Hz, C-3), 148.8 (br d, 4JC-F = 1.3 Hz, C-2), 156.6 (d, 1JC-F = 254 Hz, C-5), 160.5 (N-C-3′′), 166.5 (CO2Et), 185.1 (C=O); IR (KBr): ν 3431, 3073, 2987, 2928, 2864, 1689, 1619, 1553, 1455, 1421, 1374, 1344, 1321, 1278, 1205, 1177, 1129, 1030 cm-1.; Anal. Calcd. for C14H13Cl2FN2O5 (379.17): C, 44.35; H, 3.46; N, 7.39. Found: C, 44.30; H, 3.38; N, 7.62; mp = 139-141 °C (decomposition); Rf value in system 1 = 0.89 and in system 2 = 0.90.
ii) Ethyl 3-(N-cyclopropylamino)-2-(2,4-dichloro-5-fluoro-3-nitrobenzoyl) acrylate (7). A stirred solution of ethyl- 3-(N,N-dimethylamino)-2-(2,4-dichloro-5-fluoro-3-nitrobenzoyl) acrylate (6, 14.4 g, 38 mmol) in methanol (10 mL) was treated drop-wise with cyclopropyl amine (3.2 g, 56 mmol). Methanol (50 mL) was then added and the reaction mixture was stirred at R.T. for 1–2 h. The precipitated white solid product was filtered, washed with cold ethanol (95 %, 10 mL) and dried. Yield ≈ 11.0 g; a second crop of 7 (1.7 g) was obtained upon concentration of the mother liquor. All yield ≈ 12.7 g (93 %). 1H-NMR (CDCl3): δ0.88 (m, 4H, H2-2′/H2-3′), 1.01, 0.92 (Z/E, 2t, J = 7.1 Hz, 3H, CH3), 3.00 (m, 1H, H-1′), 3.99, 3.73 (Z/E, 2q, J = 7.1 Hz, 2H, CH2Me), 7.10, 7.16 (Z/E, 2d, 3JH-F = 8.1 Hz, 1H, H-6), 8.26, 8.35 (Z/E, 2d, J = 14 Hz, 1H, N-C(3′′)-H), 11.01, 9.77 (Z/E, 2 br d, J = 14 Hz, 1H, exchangeable N-H); 13C-NMR (CDCl3): δ 6.7 (C-2′/C-3′), 13.9, 13.3 (Z/E, CH3), 31.0, 30.3 (Z/E, C-1′), 60.2, 60.1 (Z/E, CH2Me), 100.3, 99.8 (Z/E, C-2′′), 114.1, 114.0 (Z/E, 2d, 2JC-F = 23.3 Hz, C-4), 115.7, 116.0 (Z/E, 2d, 2JC-F = 23.3 Hz, C-6), 117.7, 117.9 (Z/E, 2d, 3JC-F = 4.7 Hz, C-1), 143.9, 143.7 (Z/E, 2d, 3JC-F = 6.2 Hz, C-3), 148.6 (br d, 4JC-F = 1.4 Hz, C-2), 156.7 (d, 1JC-F = 254 Hz, C-5), 161.9, 161.4 (Z/E, N-C-3′′), 165.7, 167.8 (Z/E, CO2Et), 188.4, 186.2 (Z/E, C = O); IR (KBr): ν 3431, 3196, 3068, 3032, 2985, 2906, 1679, 1617, 1545, 1426, 1367, 1316, 1253, 1192, 1151, 1112, 1063, 1021 cm--1. ; Anal. Calcd. for C15H13Cl2FN2O5 (390.18): C, 46.06; H, 3.35; Cl, 18.13; N, 7.16. Found: C, 46.34; H, 3.26; N, 7.19; mp = 142-145 °C (decomposition); Rf value in system 1 = 0.94 and in system 2 = 0.925.
iii) Ethyl 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylate (8). To the pure ethyl 3-(N-cyclopropylamino)-2-(2,4-dichloro-5-fluoro-3-nitrobenzoyl) acrylate (7, 11.7 g, 30 mmol) was added potassium carbonate (11.7 g, 85 mmol) in dimethyl-formamide (DMF, 50 mL) and the mixture was heated at 85 °C under reflux with continuous stirring. The progress of the cyclization reaction was monitored by TLC (eluent was AcOEt: n-hexane, 1:1 v/v) and was completed within 90-120 min. The reaction mixture was then poured slowly onto crushed ice (500 g) under vigorous stirring for 10-15 min. The gum layer formed was treated with little amount of MeOH. The precipitated pale yellow solid product 8 was collected, washed with water and left to dry in dark. Yield ≈ 9.5 g (89 %). 1H-NMR (CDCl3): δ 1.11 (m, 4H, H2-2′/H2-3′), 1.33 (t, J = 7.1 Hz, 3H, CH3CH2), 3.57 (m, 1H, H-1′), 4.30 (q, J = 7.1 Hz, 2H, OCH2Me), 8.27 (d, 3JH-F = 8.0 Hz, 1H, H-5), 8.56 (s, 1H, H-2); 13C- NMR (CDCl3): δ 11.1 (C-2′/C-3′), 14.4 (CH3CH2), 37.9 (C-1′), 61.4 (OCH2Me), 111.8 (C-3), 115.6 (d, 2JC-F = 23.0 Hz, C-5), 122.0 (d, 2JC-F = 23.7 Hz, C-7), 130.1 (d, 3JC-F = 5.0 Hz, C-4a), 130.8 (d, 4JC-F = 3.0 Hz, C-8a), 140.8 (d, 3JC-F = 1.6 Hz, C-8), 151.8 (C-2), 154.4 (d, 1JC-F = 258 Hz, C-6), 164.0 (CO2Et), 170.8 (d, 4JC-F = 2.0 Hz, C-4); EI MS m/z (%): 354(28, M+), 337(10), 309(35), 282(61), 265(42), 252(100), 236(57), 226(72), 196(72), 172(32), 160(17), 132(25), 117(7); HRMS: calcd. For C15H12ClFN2O5: 354.04184, found 354.04281; IR (KBr): ν 3225, 3091, 2983, 2909, 2361, 1729, 1634, 1603, 1545, 1463, 1426, 1387, 1344, 1271, 1238, 1173, 1129, 1046 cm-1.; Anal. Calcd. for C15H12ClFN2O5 (354.72): C, 50.79; H, 3.41; N, 7.90. Found: C, 50.96; H, 3.35; N, 7.71; mp = 165-167 °C (decomposition); Rf value in system 1 = 0.90 and in system 2 = 0.788.
iv) 7-Chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (
3). A vigorously stirred suspension of ethyl 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydro-quinoline-3-carboxylate (
8, 5.3 g, 15 mmol) in 12N HCl (100 mL) and ethanol (30 mL) was heated at 80-85 °C under reflux conditions. Progress of the ester hydrolysis was monitored by TLC and was completed within 36-48 h. Thereafter, the reaction mixture was cooled, poured onto crushed ice (250 g) and the resulting heavy faint yellow precipitate was collected, washed with cold water (2 x 20 mL), dried and recrystallized from a mixture of chloroform and methanol. Yield ≈ 4.5 g (92 %).
1H- NMR (DMSO-d
6):
δ1.02, 1.16 (2m, 4H, H
2-2′/H
2-3′), 3.71 (m, 1H, H-1′), 8.45 (d,
3JH-F = 8 Hz, 1H, H-5), 8.78 (s, 1H, H-2), 13.70 (s, 1H, CO
2H);
13C-NMR (DMSO-d
6):
δ 11.2 (C-2′/C-3′), 39.5 (C-1′), 109.7 (C-3), 115.3 (d,
2JC-F = 23 Hz, C-5), 122.7 (d,
2JC-F = 23.6 Hz, C-7), 128.3 (d,
3JC-F = 6.8 Hz, C-4a), 131.9 (d,
4JC-F = 2.3 Hz, C-8a), 141.1 (d,
3JC-F = 1.6 Hz, C-8), 153.4 (C-2), 154.5 (d,
1JC-F = 250 Hz, C-6), 164.8 (CO
2H), 175.4 (d,
4JC-F = 2.2 Hz, C-4);IR (KBr):
ν 3076, 2642, 1718, 1603, 1575, 1541, 1441, 1357, 1329, 1262, 1230, 1195, 1125, 1051 cm
-1.; Anal. Calcd. for C
13H
8ClFN
2O
5 (326.66): C, 47.80; H, 2.47; N, 8.58. Found: C, 47.39; H, 2.43; N, 8.98; mp = 243–250 °C(decomposition) (Lit. [
9] M. p. =261 °C, decomposition);
Rf value in system 1 = 0.72 and in system 2 = 0.363.
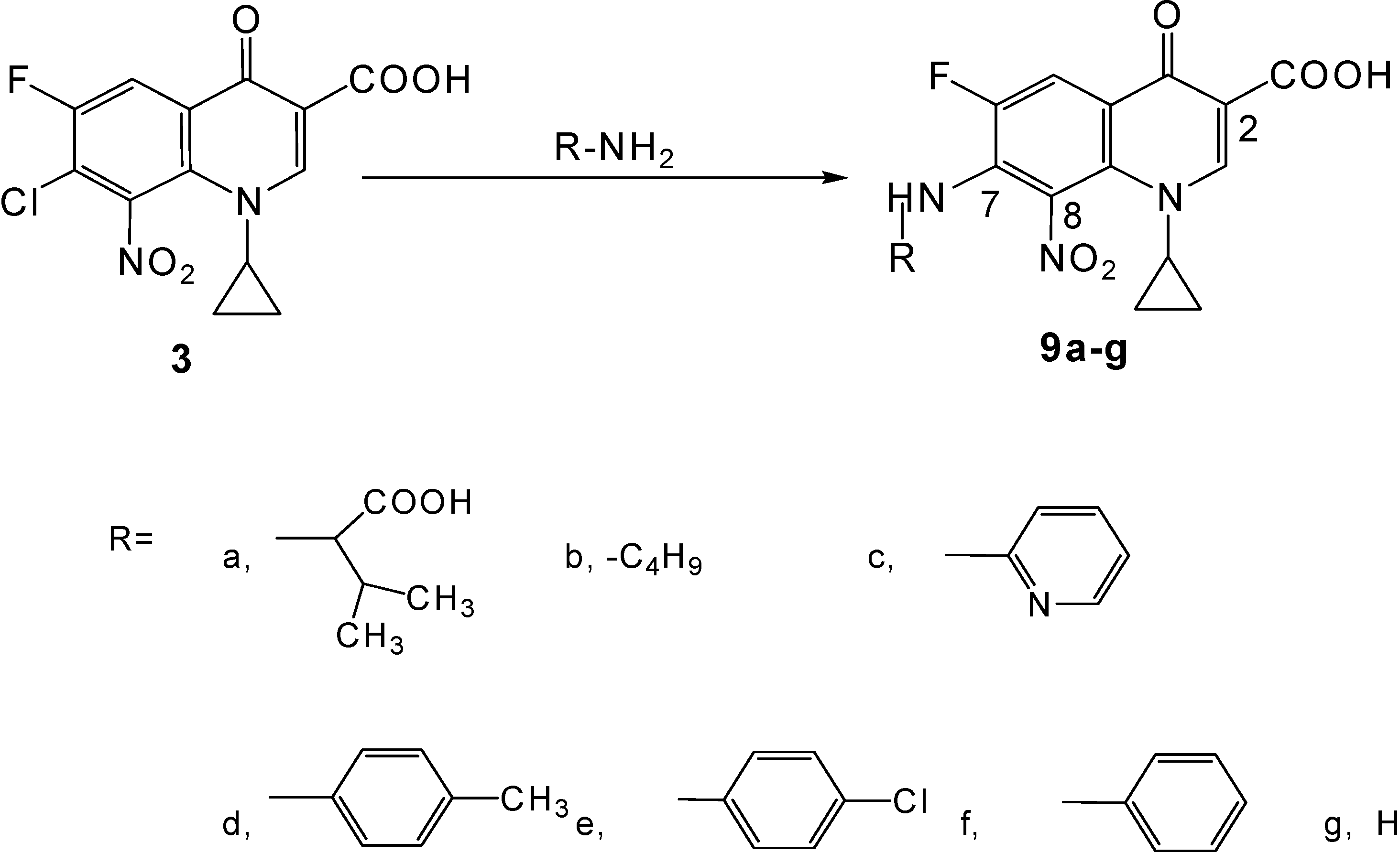
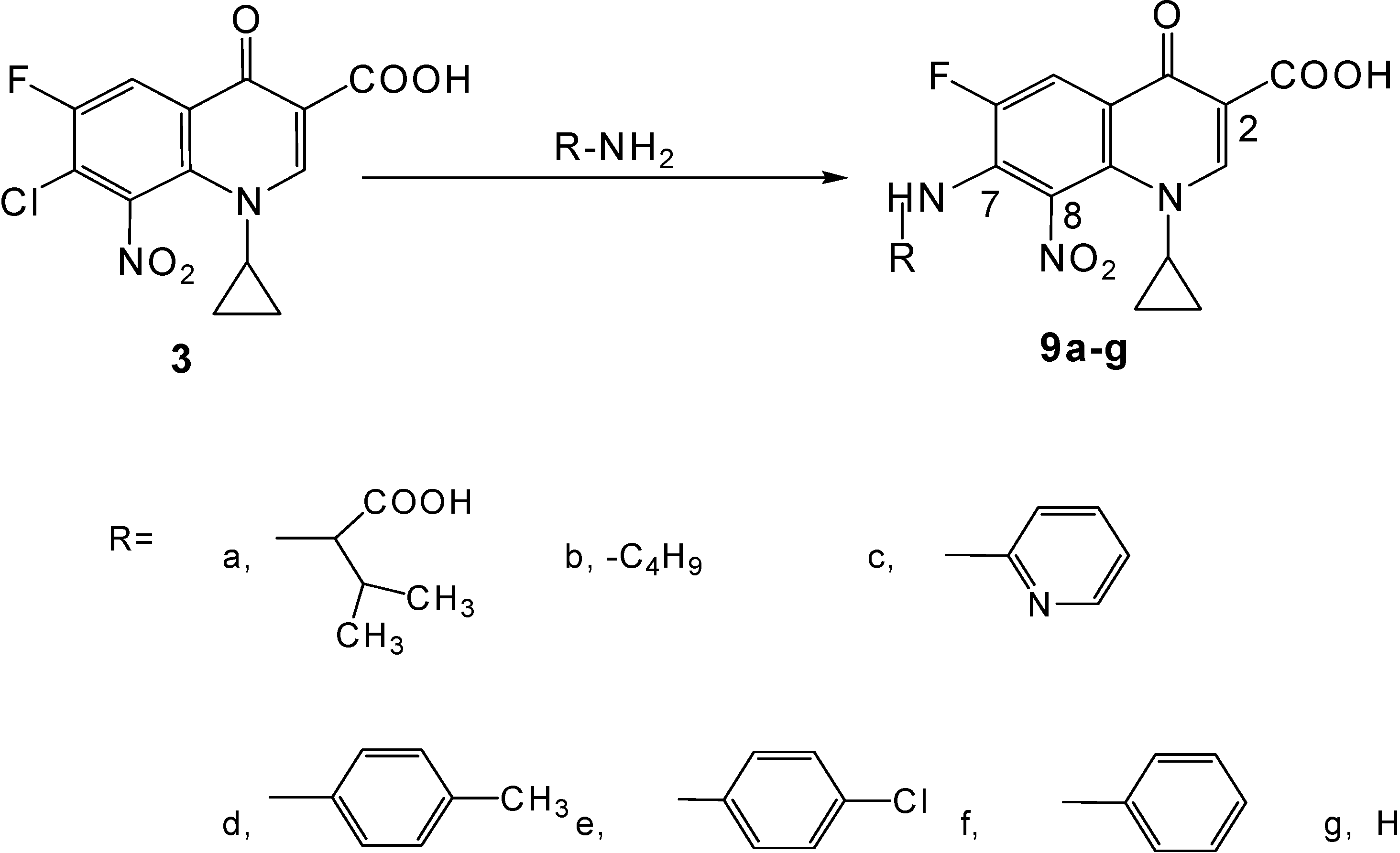
Synthesis of novel title compounds 9a-g (Scheme 2)
General procedure
A stirred mixture of the substituted primary amine (9 mmol), 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3, 1.0 g, 3 mmol) and sodium hydrogen carbonate (1.5 g, 18 mmol) in 50 % aqueous ethanol (140 mL) was heated at 75-80 °C for 144-156 h under reflux conditions. More sodium hydrogen carbonate was added (0.25 g, 3 mmol) and the mixture was heated for another 144-156 h under reflux conditions. Work-up of the resulting reaction mixture was carried out as described for 9a below, producing side products in all attempts.
a) (±)–7-(1-Carboxy-2-methylpropylamino)-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydro-quinoline -3-carboxylic acid (9a)
A solution of dl-valine (1.0 g, 9 mmol) and sodium hydrogen carbonate (NaHCO3) (1.5 g, 18 mmol) in aqueous ethanol (140 mL, 1:1 v/v) was added to (1.0 g, 3 mmol) of 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3). The resulting stirred mixture was heated at 60-65 °C. The mixture slowly developed a light yellow color that changed into bright yellow, then into clear orange solution. The progress of the reaction was monitored by TLC (system (1)), and was completed within 100-120 h. The orange solution was extracted with chloroform (CHCl3, 2 x 50 mL). The aqueous layer was cooled, adjusted with 3.5N HCl addition to get a solution of pH 6-7 and another extraction with CHCl3 (50 mL) was carried out. Further acidification of the leftover of the aqueous layer was performed with 3.5N HCl to pH = 1-2, whereby the title compound was precipitated as yellowish solid which was filtered, washed with cold water (2 x 10 mL) and dried. Re-crystallization of the formed product was carried out from a mixture of chloroform and ethanol (1:1). Yield ≈ 1.1 g (90 %). 1H-NMR (DMSO-d6): δ 0.89, 0.95 (2d, J = 6.8 Hz, 6.0 H, (CH3)2-CH), 0.92 (m, 4H, H2-2′/H2-3′), 2.21 (m, 1H, CHMe2), 3.68 (m, 1H, H-1′), 4.50 (br d, J = 7.2 Hz, CH-CO2H), 7.21 (d, J = 8.0 Hz, 1H, N-H), 8.07 (d, 3JH-F = 13.5 Hz, 1H, H-5), 8.77 (s, 1H, H-2), 13.39 (br s, 1H, CH-CO2H), 14.55 (br s, 1H, C3-CO2H); 13C-NMR (DMSO-d6): δ 10.1 (C-2≈/C-3≈), 18.0, 18.5 ((CH3)2), 31.6 (CHMe2), 40.9 (C-1≈), 63.2 (d, JC-F = 11.3 Hz, CH-NH), 109.8 (C-3), 115.2 (d, 2JC-F = 22.7 Hz, C-5), 117.8 (d, 3JC-F = 7.1 Hz, C-4a), 129.3 (d, 3JC-F = 5.4 Hz, C-8), 135.8 (C-8a), 138.1 (d, 2JC-F = 14.9 Hz, C-7), 150.4 (d, 1JC-F = 248 Hz, C-6), 152.3 (C-2), 165.3 (C3-CO2H), 172.7 (CH-CO2H), 175.6 (d, 4JC-F = 2.5 Hz, C-4); IR (KBr): ν 3308, 3070, 2971, 1733, 1700, 1629, 1549, 1518, 1469, 1321, 1257, 1216, 1144, 1033 cm-1.; Anal. Calcd. for C18H18FN3O7 (407.35): C, 53.07; H, 4.45; N, 10.32. Found: C, 52.83; H, 4.69; N, 10.68; mp = 217–222 °C (decomposition); Rf value in system 1 = 0.44 and in system 2 = 0.175.
b) 1-Cyclopropyl-6-fluoro-8-nitro-4-oxo-7-(butyl-amino)-1,4-dihydro-quinoline-3-carboxylic acid (9b).
A stirred mixture of n-butyl amine (0.66 g, 0.90 mL, 9 mmol), 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3, 1.0 g, 3 mmol) and sodium hydrogen carbonate (1.5 g, 18 mmol) in 50 % aqueous ethanol (140 mL) was heated at 40-45 °C for 136 h under reflux conditions. Work-up of the resulting reaction mixture carried out by extracting the orange solution formed with chloroform (CHCl3) (2 x 80 mL). The aqueous layer pH was adjusted with 3.5N HCl addition to get a solution of pH 6-7 and another extraction with CHCl3 was performed. These two chloroform extracts were collected, dried and the product collected was re-crystallized from ethanol and acetone (1:1) in addition to the early two chloroform extracts (at pH 8-9) producing the title compound 9b as orange crystals. Yield ≈ 0.93 g (85 %). 1H-NMR (CDCl3): δ 0.88, 0.94 (2m, 4H, H2-2′/H2-3′), 1.12-1.19 (t, J = 6.6 Hz, 3H, CH3-), 1.39-1.53 (m, 2H, CH3-CH2-), 1.65-1.83 (m, 2H, -CH2-CH2CH2-), 3.0 (m, 2H, -CH2-NH), 3.68 (m, 1H, H-1′), 7.02 (br s, 1H, NH), 8.10 (d, 3JH-F = 13.5 Hz, 1H, H-5), 8.84 (s, 1H, H-2), 14.55 (s, 1H, CO2H); IR (KBr): ν 3389, 3081, 2960, 2929, 2866, 2638, 2517, 2369, 1627, 1593, 1502, 1462, 1373, 1351, 1318, 1271, 1236, 1189, 1113, 1032 cm-1.; Anal. Calcd. for C17H18FN3O5 (363.34): C, 56.20; H, 4.99; N, 11.56. Found: C, 56.32; H, 5.51; N, 11.65; mp = 185–190 °C (decomposition); Rf value in system 1 = 0.84 and in system 2 = 0.90.
c) 1-Cyclopropyl-6-fluoro-8-nitro-4-oxo-7-(pyridin-2-yl amino)-1,4-dihydro-quinoline-3-carboxylic acid (9c).
A stirred mixture of 2-aminopyridine (3.46 g, 9 mmol), 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3, 1.0 g, 3 mmol) and sodium hydrogen carbonate (1.5 g, 18 mmol) in 50 % aqueous ethanol (140 mL) was heated at 75-80 °C for 156-162 h under reflux conditions. Work-up of the resulting reaction mixture was carried out as described for 9a above, producing 9c as yellow solid. Yield ≈ 0.65 g (57 %). 1H-NMR (DMSO-d6): δ 0.89, 0.91 (2m, 4H, H2-2′/H2-3′), 3.58 (m, 1H, H-1′), 6.81 (dd, J = 6.61 Hz, 6.56 Hz, 1H, H-5′′), 6.91 (d, J = 7.8 Hz, 1H, H-3′′), 7.56 (d, 3JH-F = 11.73 Hz, 1H, H-5), 7.83-7.89 (m, 3H, superimposed H-3′′ and H-6′′ and N-H), 8.45 (s, 1H, H-2), 16.06 (br s, 1H, C(3)-CO2H); 13C-NMR (DMSO-d6): δ 10.4 (C-2≈/C-3≈), 40.7 (C-1≈), 106.9 (d, 3JC-F = 7.0 Hz, C-4a), 107.5 (C-3), 108.7 (d, 2JC-F = 20.6 Hz, C-5), 112.6 (C-3≈′), 113.7 (C-5≈′), 133.0 (d, 3JC-F = 7.4 Hz, C-8), 136.4 (C-8a), 136.9 (C-4≈′), 144.3 (C-6≈′), 148.6 (C-2), 154.6 (NH-C-2≈′), 155.6 (d, 1JC-F = 247 Hz, C-6), 160.4 (d, 2JC-F = 20.4 Hz, C-7), 166.7 (C(3)-CO2H), 174.7 (d, 4JC-F = 3.6 Hz, C-4); IR (KBr): ν3372, 3194, 2691, 1706, 1669, 1627, 1541, 1445, 1404, 1316, 1227, 1111, 1054 cm-1.; Anal. Calcd. for C18H13FN4O5 (384.32): C, 56.25; H, 3.41; N, 14.58. Found: C, 56.07; H, 3.29; N, 14.95; mp = 234–237 °C (decomposition); Rf value in system 1 = 0.075 and in system 2 = 0.013.
d) 1-Cyclopropyl-6-fluoro-8-nitro-4-oxo-7-p-tolylamino-1,4-dihydro-quinoline-3-carboxylic acid (9d)
A stirred mixture of p-toluidine (0.97 g, 9 mmol), 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3, 1.0 g, 3 mmol) and sodium hydrogen carbonate (0.75 g, 9 mmol) in 50 % aqueous ethanol (140 mL) was heated at 70-75 °C for about 120 h. Then extra p-toluidine (0.97 g, 9 mmol) and sodium hydrogen carbonate (0.25 g, 3 mmol) were added and the mixture was heated for 65 h under reflux conditions. The resulting reaction mixture was a suspension; suction filtration was carried out to get the precipitate, which was not pure. Re-crystallization of the crude product from chloroform (20 mL) was carried out to yield the pure 9d as orange solid. Yield ≈ 1.13 g (92 %). 1H-NMR (DMSO-d6): δ 0.99 (m, 4H, H2-2′/H2-3′), 2.22 (s, 3H, CH3), 3.70 (m, 1H, H-1′), 6.93 (d, J = 8.0 Hz, 2H, H-2′′/H-6′′), 7.05 (d, J = 8.2 Hz, 2H, H-3′′/H-5′′), 8.13 (d, 3JH-F = 11.91 Hz, 1H, H-5), 8.80 (s, 1H, H-2), 8.95 (br s, 1H, NH-Ar), 14.48 (br s, 1H, COOH); 13C-NMR (DMSO-d6): δ 10.54 (C-2≈/C-3≈), 20.9 (CH3), 40.4 (C-1≈), 109.4 (C-3), 114.7 (d, 2JC-F = 21.83 Hz, C-5), 119.2 (C-4≈′), 120.0 (C-2≈′, C-6≈′), 120.6 (d, 3JC-F = 7.8 Hz, C-4a), 129.7 (C-3≈′, C-5≈′), 133.0 (C-8a), 133.5 (d, 2JC-F = 15.4 Hz, C-7), 134.3 (C-8), 139.4 (C-1≈′), 152.5 (C-2), 152.8 (d, 1JC-F = 253 Hz, C-6), 165.4 (C(3)-CO2H), 175.8 (C-4); IR (KBr): ν 3449, 3360, 3063, 2921, 1730, 1620, 1531, 1450, 1362, 1318, 1223, 1102, 1048, 1020 cm-1; Anal. Calcd. for C20H16FN3O5 (397.36): C, 60.45; H, 4.06; N, 10.57. Found: C, 60.71; H, 3.76; N, 10.65; mp = 240–247 °C (decomposition); Rf value in system 1 = 0.78 and in system 2 = 0.39.
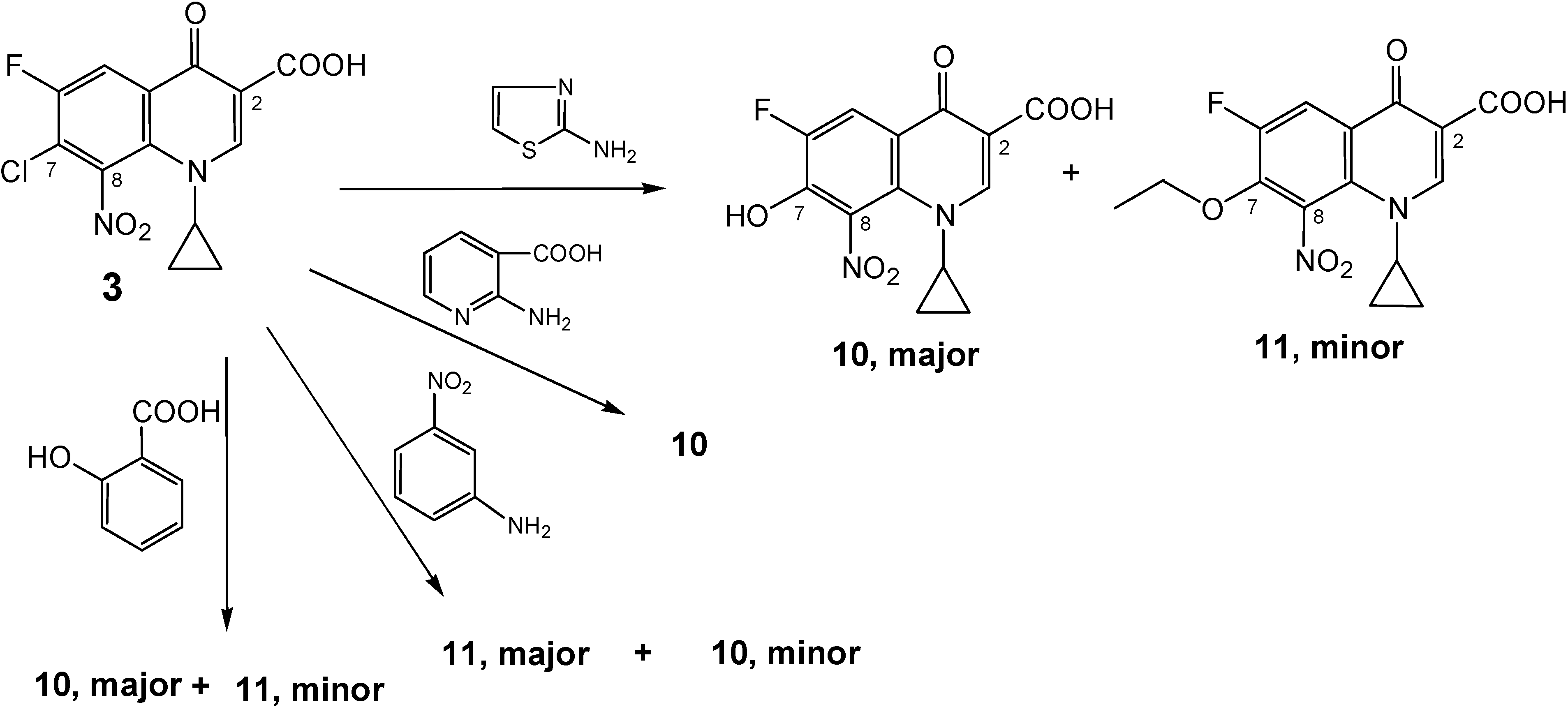
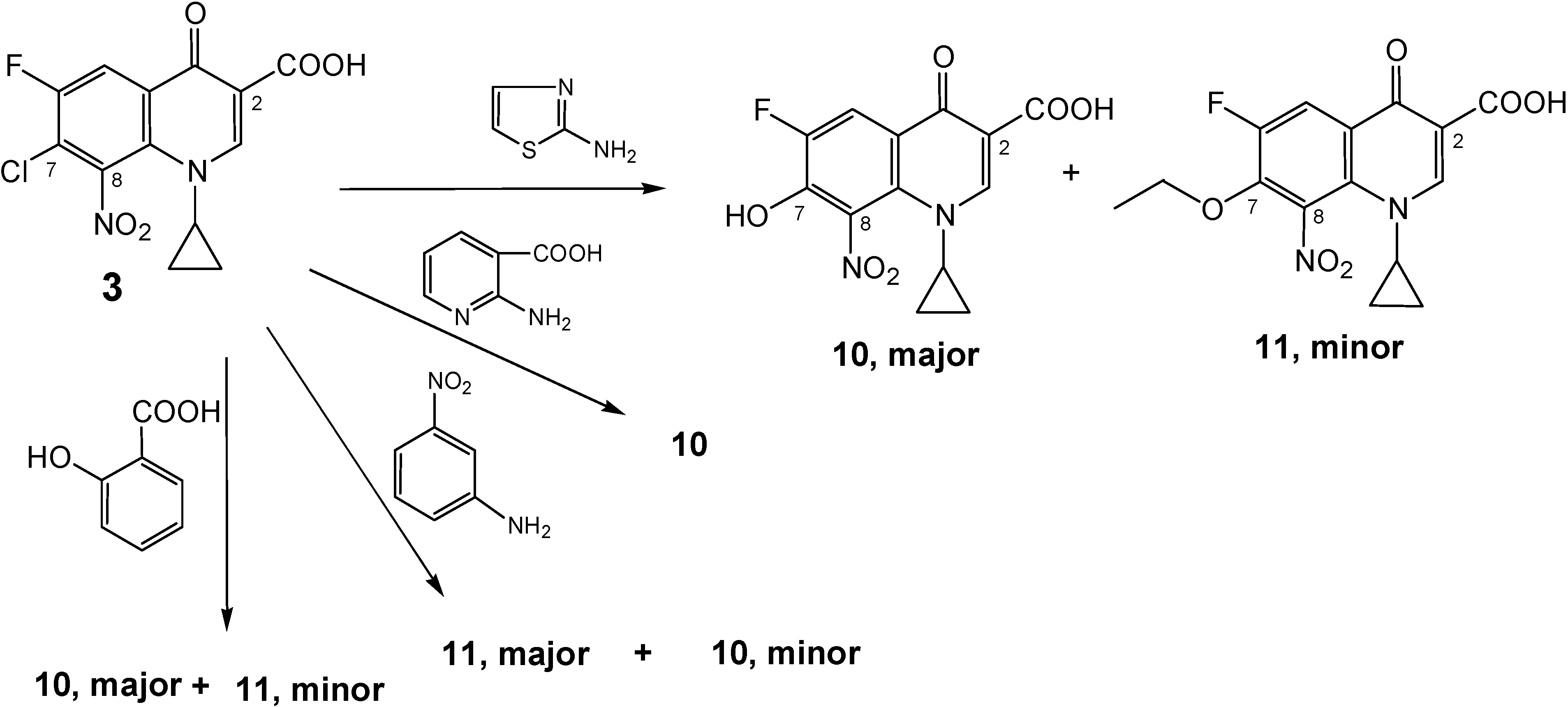
e)1-Cyclopropyl-6-fluoro-8-nitro-4-oxo-7-(4-chloro-phenylamino)-1,4-dihydro-quinoline-3-carboxylic acid (9e)
A stirred mixture of 4-chloroaniline (1.15 g, 9 mmol), 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3, 1.0 g, 3 mmol) and sodium hydrogen carbonate (1.5 g, 18 mmol) in 50 % aqueous ethanol (140 mL) was heated at 70-75 °C for 230-240 h under reflux conditions. Work-up of the resulting reaction mixture was carried out as described for 9a above, producing the title compound as pale brown solid. Crystallization of the crude mixture gave two spots on TLC; one was faint and the other was condensed. NMR analysis showed more than one compound but the peaks of 9e were distinct and could be recognized. First attempt: The 7-hydroxy product 10 was major but not the product. Second attempt: Minor with the 7-ethoxy side product 11. 1H-NMR (DMSO-d6/CO(CD3)2), minor: δ 1.01 (m, 4H, H2-2′/H2-3′), 3.87 (m, 1H, H-1′), 7.05 (d, J = 8.7 Hz, 2H, H-2′′/H-6′′), 7.31 (d, J = 8.7 Hz, 2H, H-3′′/H-5′′), 8.22 (d, 3JH-F = 11.4 Hz, 1H, H-5), 8.84 (s, 1H, H-2), 9.10 (br s, 1H, NH-Ar), 14.25 (br s, 1H, COOH); IR (KBr): ν 3376, 3093, 2918, 2849, 2362, 1621, 1585, 1542, 1472, 1366, 1285, 1175, 1090, 1015 cm-1.; mp = 175–180 °C (decomposition); Rf value in system 1 = 0.81 and in system 2 = 0.875.
f) 1-Cyclopropyl-6-fluoro-8-nitro-4-oxo-7-phenylamino-1,4-dihydro-quinoline-3-carboxylic acid (9f).
A stirred mixture of aniline (0.84 g, 0.82 mL, 9 mmol), 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3, 1.0 g, 3 mmol) and sodium hydrogen carbonate (1.5 g, 18 mmol) in 50 % aqueous ethanol (140 mL) was heated at 40-45 °C for 132-144 h under reflux conditions. Work-up of the resulting reaction mixture was carried out as indicated for 7b to give a dark yellow powder. NMR analysis of the product showed that the 7-ethoxy compound 11 was the major product, but the presence of the title compound was detected. Chromatography failed to separate the product in pure form. Yield ≈ 0.43 g (44 %); 1H-NMR (DMSO-d6), minor: δ 0.92, 1.10 (2m, 4H, H2-2′/H2-3′), 3.65 (m, 1H, H-1′), 6.95-7.27 (m, 5H, Ar-H), 7.85 (br s, 1H, NH), 8.33 (d, 3JH-F = 13.0 Hz, 1H, H-5), 8.62 (s, 1H, H-2), 14.30 (s, 1H, CO2H); IR (KBr): ν 3430, 3083, 2924, 2851, 1702, 1604, 1539, 1464, 1390, 1340, 1265, 1191, 1096, 1039 cm-1; Anal. Calcd. for C19H14 FN3O5 (383.33): C, 59.53; H, 3.68; N, 10.96. Found: C, 59.40; H, 3.61; N, 10.90; mp = 250-256 °C (decomposition); Rf value in system 1 = 0.70 and in system 2 = 0.85;
g) 7-Amino-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (9g).
Method A: A mixture of 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (
3, 1.3 g, 4 mmol), ammonium hydroxide (28 %, 8 mL) and pyridine (8 mL) was stirred for 17 hours in a round bottom flask at R.T. Excess ammonia (4 mmol) was added every 2-3 h until the starting material spot disappeared on TLC. The reaction mixture was evaporated to dryness to give an orange residue. The orange residue was triturated with water-acetic acid (10:1, 44 mL), filtered, washed with water, and then re-crystallized to give 7-amino-1-cyclopropyl-6-flouro-8-nitro-1,4-dihydro-4-oxo-3-quinoline carboxylic acid
9g as orange crystals (
Scheme 2). Yield ≈ 0.92 g (75 %);
1H-NMR (CDCl
3): δ 1.17, 1.29 (2m, 4H, H
2-2′/H
2-3′), 3.78 (m, 1H, H-1′), 6.7 (br s, 2H, NH
2, exchangeable), 8.47 (d,
3JH-F = 7.8 Hz, 1H, H-5), 8.94 (s, 1H, H-2), 13.71 (s, 1H, CO
2H);
13C- NMR (CDCl3):
δ 11.3 (C-2′/C-3′), 38.8(C-1′), 109.9 (C-3), 115.0 (d,
2JC-F = 23 Hz, C-5), 127.6 (d,
3JC-F = 6.7 Hz, C-4a), 131.0 (C-8), 134.3 (d,
2JC-F = 23.2 Hz, C-7), 140.0 (C-8a), 151.8 (C-2), 155.1 (d,
1JC-F = 255 Hz, C-6), 164.7 (
CO
2H), 175.8 (d,
4JC-F = 1.8 Hz, C-4); IR (KBr):
ν 3435, 3076, 2923, 1718, 1604, 1539, 1436, 1385, 1330, 1261, 1193, 1125, 1051 cm
-1; Anal. Calcd. for C
13H
10FN
3O
5 (307.23): C, 50.82; H, 3.28; N, 13.68. Found: C, 50.76; H, 3.33; N, 13.49; mp = 264-267 °C (decomposition);
Rf value in system 1 = 0.725 and in system 2 = 0.875.
Method B: This compound was also prepared by acid hydrolysis of ethyl 7-Aminoamino-1-cyclopropyl-6-flouro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylate [
15].
1H-NMR (DMSO-d
6): δ 1.06, 1.18 (2m, 4H, H
2-2′/H
2-3′), 3.74 (m, 1H, H-1′), 6.8 (br s, 2H, NH
2, exchangeable), 8.51 (d,
3JH-F = 8.4 Hz, 1H, H-5), 8.82 (s, 1H, H-2), 13.81 (s, 1H, CO
2H).
Side products:
a) 1-Cyclopropyl-6-fluoro-7-hydroxy-8-nitro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (10).
A stirred mixture of 2-aminonicotinic acid (1.25 g, 9 mmol), 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3, 1.0 g, 3 mmol) and sodium hydrogen carbonate (1.5 g, 18 mmol) in 50 % aqueous ethanol (140 mL) was heated at 70-75 °C under reflux conditions. TLC of the resulted solution at different intervals showed that the reaction proceeded very slowly, producing two faint yellow spots. After 5 weeks, more NaHCO3 (0.25 g, 3 mmol) and 2-amino-nicotinic acid (0.6 g, 4.5 mmol) were added and the reaction mixture was heated at 80 °C under reflux conditions for another 3 weeks. Extraction with CHCl3 was carried out three times and a fourth extraction was also carried out after decreasing pH to 7 using 3.5 N HCL.HCl. The chloroform extracts were collected and, the side product 11 was re-crystallized from chloroform and ethanol (3:1) as yellow crystals. The pH of the aqueous solution was decreased to about 2 and the resulting precipitate was collected by suction filtration, washed with water and dried. Recrystallization from ethanol (30 mL) produced 10 as an off-white powder. Yield ≈ 0.75 g (75 %); 1H-NMR (DMSO-d6): δ 0.97, 1.07 (2m, 4H, H2-2′/H2-3′), 3.66 (m, 1H, H-1′), 4.95 (br s, 1H, OH, disappeared in acetone), 8.10 (d, 3JH-F = 10.57 Hz, 1H, H-5), 8.66 (s, 1H, H-2), 14.39 (br s, 1H, CO2H in acetone only, did not appear in DMSO); 13C-NMR (DMSO-d6): δ 10.8, 10.9 (C-2′/C-3′), 39.59 (C-1′), 108.38 (C-3), 113.0 (d,2JC-F = 19.4 Hz, C-5), 116.99 (d, 3JC-F = 5.2 Hz, C-4a), 132.36 (C-8), 133.05 (C-8a), 148.75 (d, 2JC-F = 20.8 Hz, C-7), 150.92 (d, 1JC-F = 247 Hz, C-6), 151.58 (C-2), 165.57 (CO2H), 175.92 (d, 4JC-F = 2.7 Hz, C-4); IR (KBr): ν 3431, 3076, 2362, 1717, 1619, 1538, 1470, 1339, 1292, 1253, 1220, 1113, 1032 cm-1.; Anal. Calcd. for C13H9FN2O6 (308.22): C, 50.66; H, 2.94; N, 9.09. Found: C, 49.98; H, 2.93; N, 9.01; mp = 240-249 °C (decomposition); Rf value in system 1 = 0.413 and in system 2 = 0.15.
b) 1-Cyclopropyl-7-ethoxy-6-fluoro-8-nitro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (11).
The same reaction mentioned above for production of 10, also produced as an additional side product ethoxy cipro acid (1-cyclopropyl-7-ethoxy-6-fluoro-8-nitro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid, 11). Work up and re-crystallization were as mentioned above. This product was isolated from chloroform extracts at pH above 7. Yield ≈ 0.25 g (25 %); 1H-NMR (DMSO-d6): δ 1.02, 1.15 (2m, 4H, H2-2′/H2-3′), 1.28 (t,J = 7.0 Hz, 3H, CH3), 3.69 (m, 1H, H-1′), 4.46 (q,J = 6.95 Hz, 2H, OCH2CH3), 8.31 (d, 3JH-F = 11.47 Hz, 1H, H-5), 8.78 (s, 1H, H-2), 14.05 (br s, 1H, CO2H); 13C-NMR (CDCl3): δ 10.95 (C-2′/C-3′), 15.66 (CH3), 39.45 (C-1′), 72.96 (d, J = 7.5 Hz, OCH2CH3), 108.89 (C-3), 115.66 (d,2JC-F = 20.8 Hz, C-5), 122.74 (d, 3JC-F = 6.53 Hz, C-4a), 131.61 (C-8), 136.42 (C-8a), 145.78 (d, 2JC-F = 16.68 Hz, C-7), 152.11 (d, 1JC-F = 247.13 Hz, C-6), 152.72 (C-2), 165.06 (CO2H), 175.89 (C-4); IR (KBr): ν 3440, 3054, 2921, 2852, 2363, 1722, 1614, 1544, 1456, 1363, 1286, 1102, 1014 cm-1.; Anal. Calcd. for C15H13FN2O6 (336.27): C, 53.58; H, 3.90; N, 8.33. Found: C, 53.65; H, 4.31; N, 7.97; mp = 203–206 °C (decomposition); Rf value in system 1 = 0.825 and in system 2 = 0.925.


{kind=link}
{kind=link}
{kind=link}
{kind=link}