Experimental Section
General
The 1H- and 13C-NMR spectra were recorded on a Varian Gemini 2000 spectrometer operating at 200 and 50 MHz, respectively. Chemical shifts were recorded as δ values in ppm referenced to the solvent. Coupling constants (J) are given in Hertz. Infrared (IR) spectra were recorded in cm-1 on a Perkin Elmer 1760x FT-IR spectrometer. Mass spectra were obtained on a Finnigan Polaris GCQ mass spectrometer and accurate masses (HRMS) were obtained on a Bruker Micro TOF in ESI positive mode. Melting points (m.p.) were determined on a SMP3 melting point apparatus and are reported in oC uncorrected. Column chromatography was performed on Scharlau silica gel 60 (70-230 mesh).
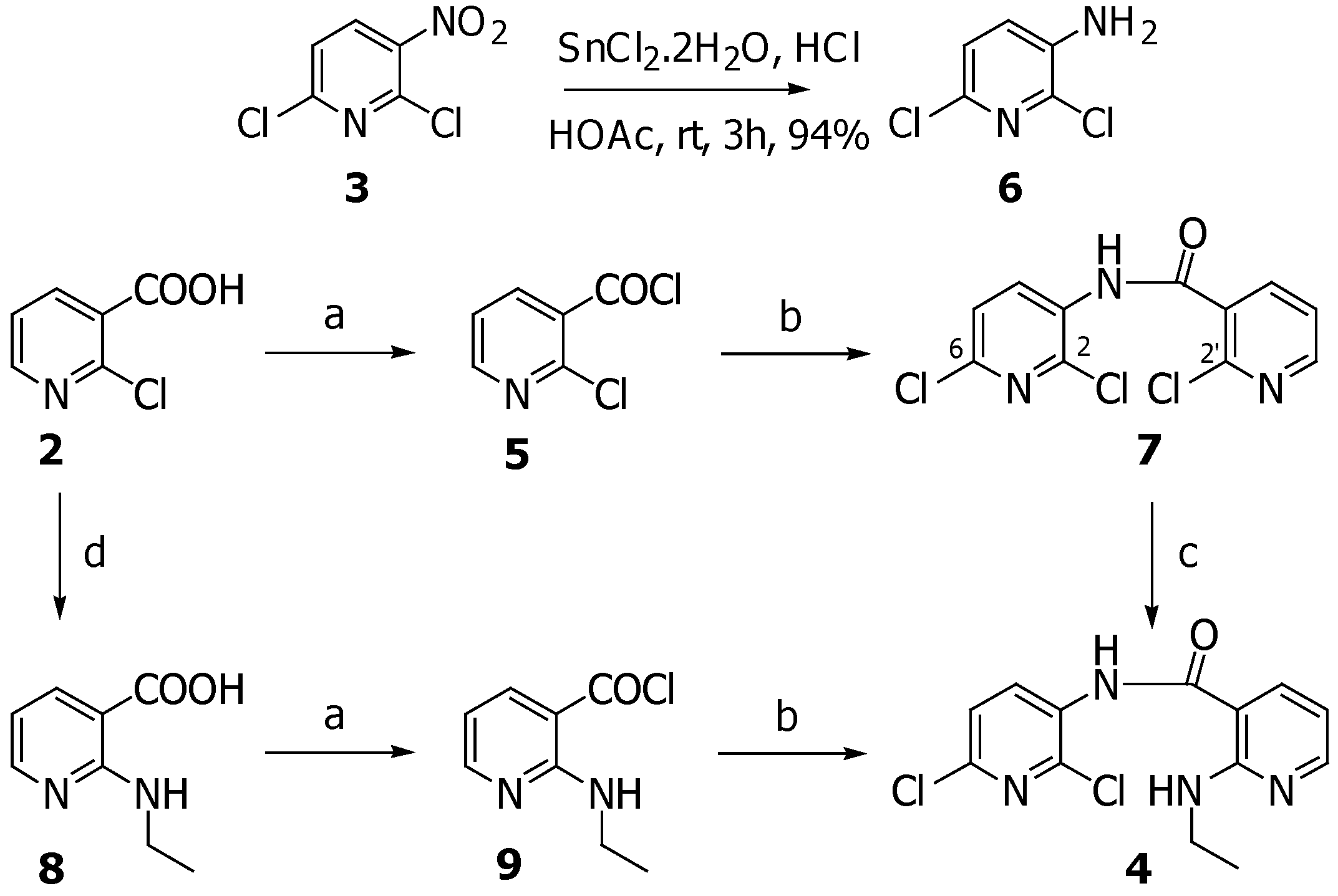
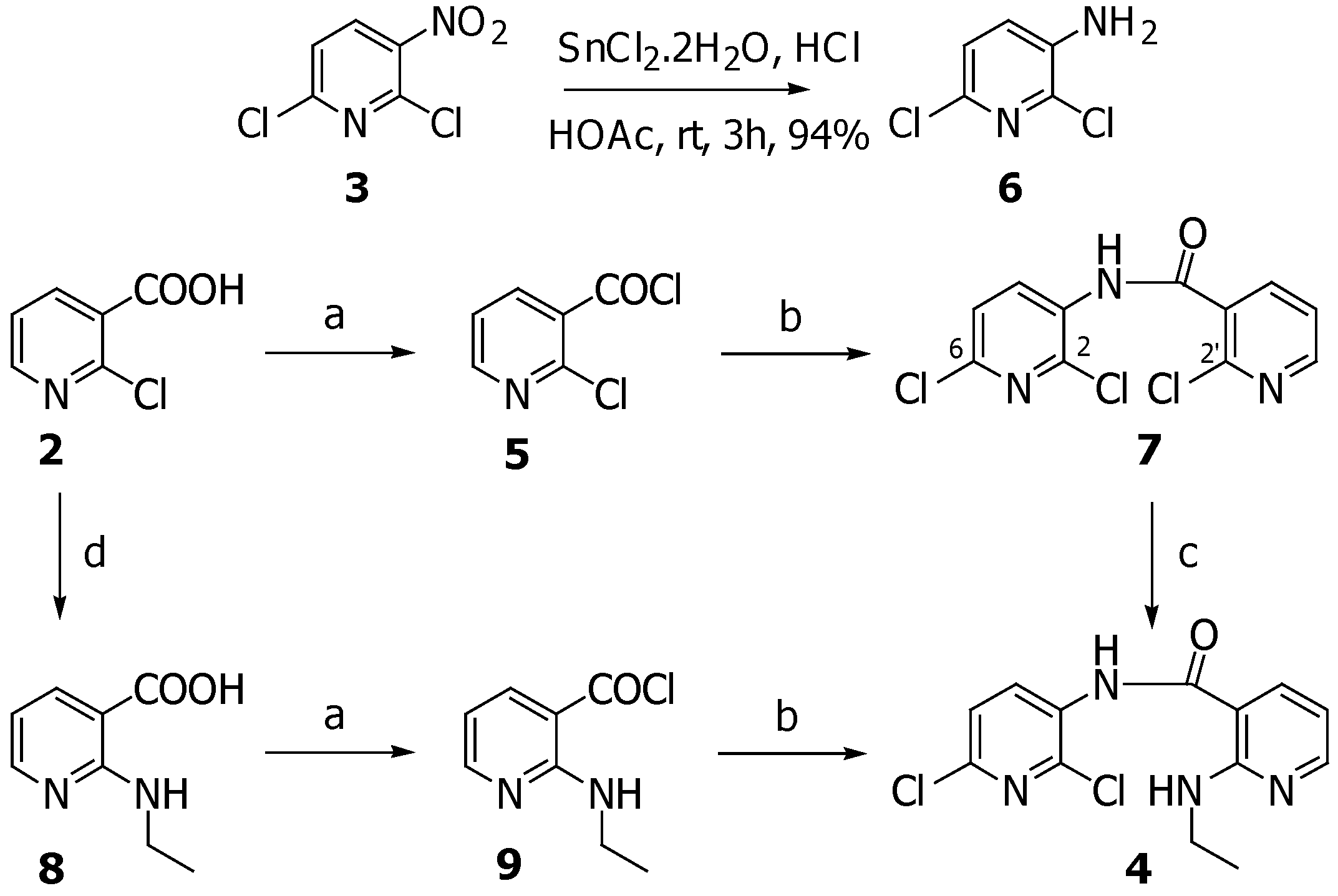
3-Amino-2,6-dichloropyridine (6)
A solution of SnCl2·2H2O (970 mg, 4.3 mmol) in concentrated HCl (0.8 mL) was added dropwise to a stirred solution of 2,6-dichloro-3-nitropyridine (250 mg, 1.3 mmol) in acetic acid (2.6 mL) and the mixture was stirred for 3 hours at room temperature. Then the mixture was cooled in ice bath, and water was added. After stirring for an additional half an hour, the mixture was made basic (pH 12) with aqueous 50% sodium hydroxide. The reaction mixture was extracted with CH2Cl2 and the combined organic layers were washed with water, dried (Na2SO4) and concentrated under reduced pressure to give 6 (200 mg, 94.8%) as white solid which could be used directly in the next step. Recrystallization from hexane/CH2Cl2 afforded needles, m.p. 121-122 oC; FTIR (KBr) νmax: 3460, 3338, 1619, 1559, 1454, 1311, 1145, 720 cm-1; 1H-NMR (CDCl3) δ: 7.06 (d, J=1.47, 2H); 13C-NMR (CDCl3) δ: 138.8, 137.3, 134.6, 125.1, 123.6; MS (EI), m/z (relative intensity): 162 (M+, 100), 126 (20), 99 (10), 64 (30); HRMS (ESI-TOF) calcd. for C5H5Cl2N2 [M+H]+ 162.9824; found: 162.9824.
2-(Ethylamino)-3-pyridine carboxylic acid (8)
A stirred mixture of 2-chloronicotinic acid (2) (300 mg, 1.9 mmol), ethylamine (1.3 mL, 19.1 mmol) was heated at 120 oC in a sealed vessel for 4 hours. After cooling down the ethylamine was removed and the residue was purified by silica gel column chromatography (30% methanol/ethyl acetate) to give 8 (315 mg, 99.7%) as a white solid, m.p. 178-180 oC; FTIR (KBr) νmax: 3474, 3228, 1636, 1557, 1341, 1239; 1H-NMR (DMSO-d6) δ: 8.20 (dd, J=4.8, 1.8, 1H), 8.04 (dd, J=7.7, 1.8 Hz, 1H), 6.50 (dd, J=7.6, 5.1, 1H), 4.81 (br s, 1H), 3.43 (q, J=6.9, 2H), 1.15 (t, J=6.9, 3H); 13C-NMR (DMSO-d6) δ: 169.3, 158.4, 153.0, 140.1, 110.8, 106.9, 35.1, 15.0; MS (EI), m/z (relative intensity): 167 (M++1, 20), 151 (60), 133 (100), 122 (30), 93 (50), 78 (96); HRMS (ESI-TOF) calcd. for C8H11N2O2 [M+H]+ 167.0815; found: 167.0812.
N-(2,6-Dichloro-3-pyridinyl)-2-ethylamino-3-pyridinecarboxamide (4)
A stirred solution of 8 (315 mg, 1.9 mmol), in benzene (10 mL) was treated with oxalyl chloride (0.35 ml, 4.1 mmol) followed by a catalytic amount of DMF (2 drops) and the mixture was stirred at room temperature for 1 hour. Then the solvent was removed under reduced pressure to afford acid chloride 9 as yellow solid. This acid chloride was redissolved in 1,4-dioxane (10 mL) and added dropwise to a solution of 6 (200 mg, 1.2 mmol) in 1,4-dioxane (3 mL), cyclohexane (2 mL) and pyridine (0.2 mL, 2.4 mmol). After stirring at room temperature for 16 hours, the resultant precipitate was filtered. The solid was redissolved in CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was washed with water, dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (15% EtOAc/hexane) to yield 4 (308 mg, 80.7%) as a pale yellow solid, m.p. 119-120 oC; FTIR (KBr) νmax: 3431, 3351, 1660, 1572, 1504, 1301, 1235, 1122, 770; 1H-NMR (CDCl3) δ: 8.77 (d, J=7.3, 1H), 8.33 (d, J=4.4, 1H), 8.20 (s, 1H), 7.97 (br t, 1H), 7.74 (d, J=7.3, 1H), 7.34 (d, J=8.1, 1H), 6.60 (dd, J=7.3, 4.4, 1H), 3.61-3.48 (m, 2H), 1.29 (t, J=7.0, 3H); 13C-NMR (CDCl3) δ: 166.4, 158.1, 153.4, 143.5, 138.7, 135.3, 131.2, 130.9, 123.6, 110.5, 108.0, 35.8, 14.7; MS (EI), m/z (relative intensity): 310 (M+, 20), 275 (25), 149 (35), 131 (100), 119 (20); HRMS (ESI-TOF) calcd. for C13H13Cl2N4O [M+H]+ 311.0461; found: 311.0464.
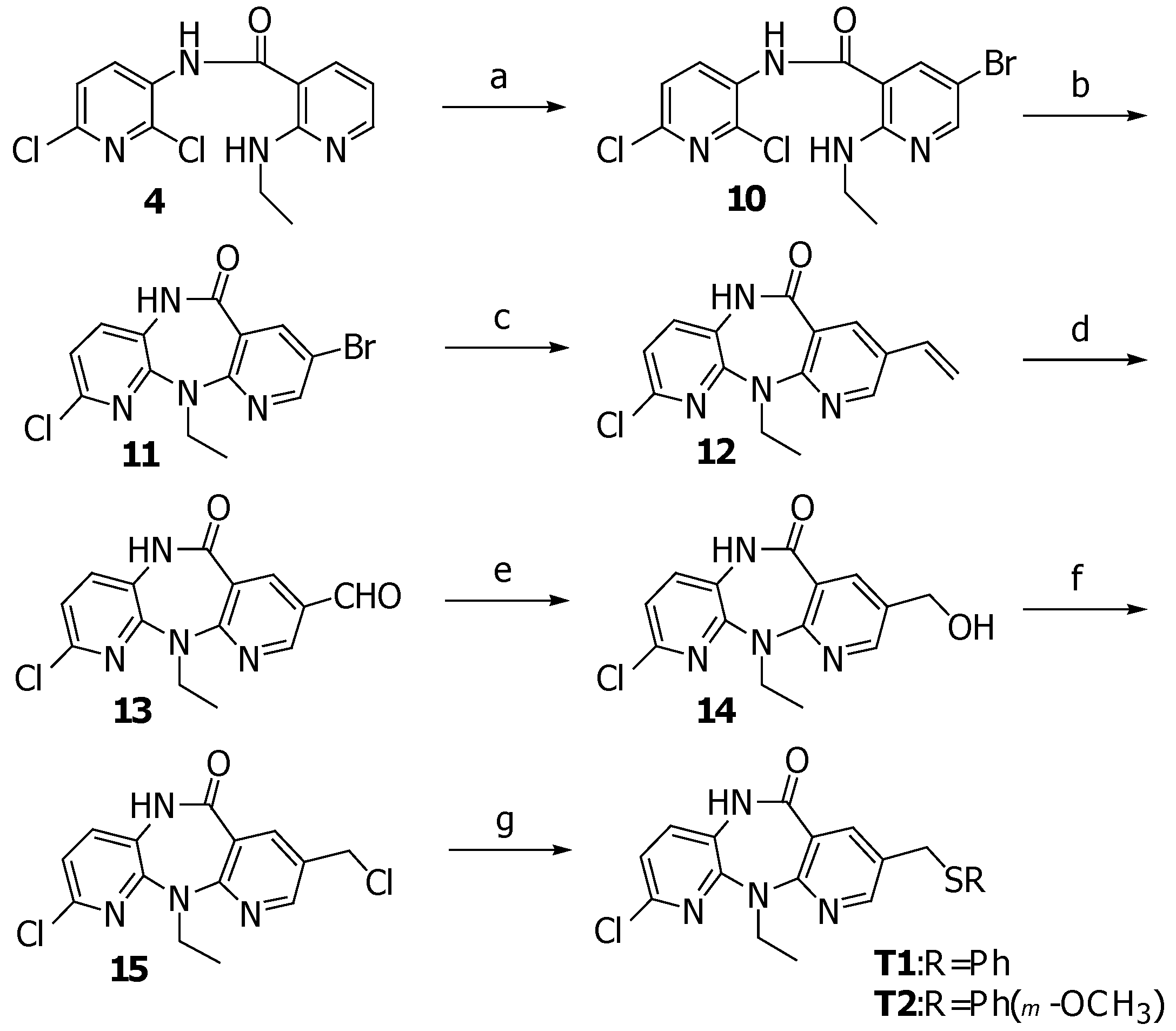
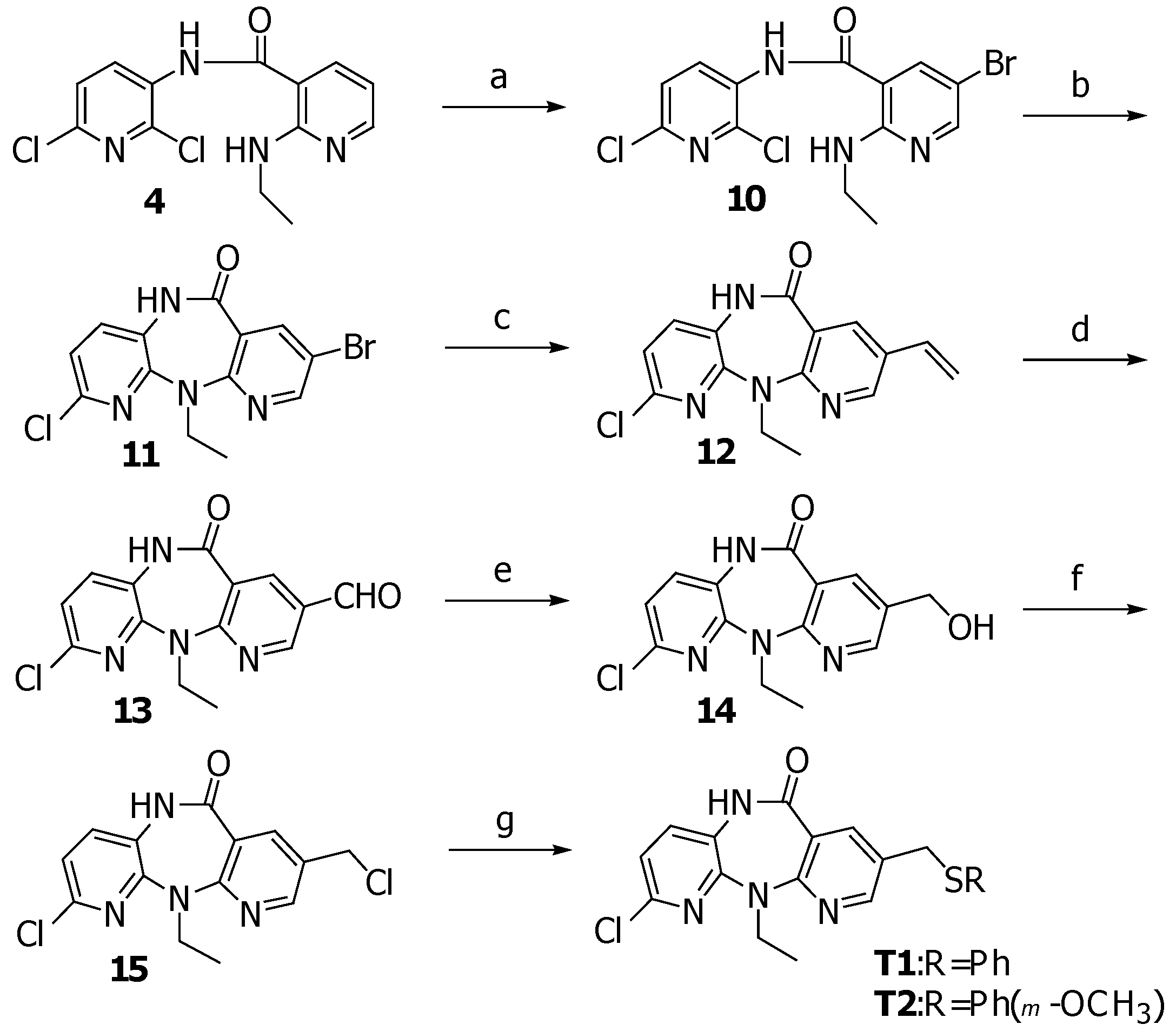
N-(2,6-Dichloro-3-pyridinyl)-5-bromo-2-ethylamino-3-pyridinecarboxamide (10)
A solution of Br2 (0.06 mL, 1.17 mmol) in acetic acid (1 mL) was added dropwise to a stirred solution of 4 (355 mg, 1.14 mmol), and potassium acetate (134 mg, 1.36 mmol) in acetic acid (15 mL). After 15 minutes, to the reaction mixture was added water and the precipitate was collected by suction filtration and washed with water for several times to provide 10 (435 mg, 98.0%) as a yellow solid, m.p. 193-194 oC; FTIR (KBr), νmax: 3439, 3336, 1673, 1575, 1506, 1302, 1248, 787, 528; 1H-NMR (CDCl3) δ: 8.69 (d, J=8.8, 1H), 8.33 (d, J=2.2, 1H), 8.10 (br s, 1H), 7.89 (br t, 1H), 7.79 (d, J=2.2, 1H), 7.30 (d, J=8.1, 1H), 3.58-3.44 (m, 2H), 1.27 (t, J=7.3, 3H); 13C-NMR (CDCl3) δ: 165.4, 156.5, 154.2, 139.1, 137.4, 131.7, 130.7, 123.8, 109.6, 103.9, 36.1, 14.6; MS (EI), m/z (relative intensity): 388 (M+, 60), 373 (15), 353 (20), 227 (70), 209 (60); HRMS (ESI-TOF) calcd. for C13H12BrCl2N4O [M+H]+ 388.9566; found: 388.9579.
8-Bromo-2-chloro-5,11-dihydro-11-ethyl-6H-dipyrido[3,2-b:2’,3’-e][1,4]diazepin-6-one (11)
A solution of 10 (440 mg, 1.13 mmol) in pyridine (10 mL) was heated at 90oC under a nitrogen atmosphere. Then the solution was added 0.6 M sodium bis(trimethylsilyl)amide in toluene (6.3 mL, 3.7 mmol) and the mixture was stirred at 90 oC for 15 minutes. After cooling down, the mixture was poured into ice water and stirred for additional 2 hours. The precipitate was filtered and washed with water to afford 11 (370 mg, 92.8%) as a yellow solid, m.p. 263-264 oC; FTIR (KBr) νmax: 3195, 3074, 2972, 1665, 1575, 1456, 1381, 1226, 687, 630; 1H-NMR (CDCl3) δ: 8.53 (br s, 1H), 8.49 (d, J=2.2, 1H), 8.24 (d, J=2.2, 1H), 7.27 (d, J=8.1, 1H), 7.06 (d, J=8.1, 1H), 4.81 (q, J=7.3, 2H), 1.25 (t, J=7.3, 3H); 13C-NMR (DMSO-d6) δ: 165.4, 156.8, 151.8, 150.5, 143.0, 142.6, 133.3, 126.3, 122.2, 120.4, 113.6, 41.5, 13.4; MS (EI), m/z (relative intensity): 352 (M+, 80), 337 (42), 324 (75), 309 (20); HRMS (ESI-TOF) calcd. for C13H11BrClN4O [M+H]+ 352.9799; found: 352.9803.
2-Chloro-5,11-dihydro-11-ethyl-8-vinyl-6H-dipyrido[3,2-b:2’,3’-e][1,4]diazepin-6-one (12)
A solution of 11 (500 mg, 1.4 mmol) in DMF (7 mL) was treated with tetrakis(triphenylphosphine)palladium(0) (70 mg, 0.06 mmol) followed by vinyl tributyltin (1 mL, 3.42 mmol) and heated at 100 oC under nitrogen atmosphere for half an hour. After cooling down, the reaction mixture was poured into water and extracted with ethyl acetate. The combined organic layers were washed with 15% aqueous ammonia, brine and water, then dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (30% EtOAc/hexane) to provide 12 (318 mg, 74.6%) as a yellow solid, m.p. 201-202 oC; FTIR (KBr) νmax: 3195, 2965, 1665, 1585, 1455, 1383, 1239, 688; 1H-NMR (CDCl3) δ: 9.37 (s, 1H), 8.46 (d, J=2.2, 1H), 8.20 (d, J=2.2, 1H), 7.35 (d, J=8.1, 1H), 7.04 (d, J=8.1, 1H), 6.66 (dd, J=17.6, 11.0, 1H), 5.78 (d, J=17.6, 1H), 5.35 (d, J=11.0, 1H), 4.22 (q, J=7.3, 2H), 1.27 (t, J=7.3, 3H); 13C-NMR (CDCl3) δ: 168.8, 157.9, 151.9, 150.1, 145.1, 137.7, 131.9, 131.8, 128.8, 125.0, 119.7, 119.6, 115.8, 42.1, 13.7; MS (EI), m/z (relative intensity): 300 (M+, 100), 285 (35), 272 (92), 257 (35); HRMS (ESI-TOF) calcd. for C15H14ClN4O [M+H]+ 301.0851; found: 301.0855.
2-Chloro-5,11-dihydro-11-ethyl-8-formyl-6H-dipyrido[3,2-b:2’,3’-e][1,4]diazepin -6-one (13)
A cooled (–78 oC) solution of 12 (343 mg, 1.14 mmol) in 1:1 CH2Cl2-MeOH (20 mL) was treated with O3 for 30 minutes. After the completion of reaction, the solution was purged with O2 for 5 minutes. Then the reaction mixture was stirred with triphenylphosphine (598 mg, 2.28 mmol) for additional 1 hour at room temperature. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (30%EtOAc/hexane) to yield 13 (295 mg, 85.4%) as a yellow solid, m.p. 216-217 oC; FTIR (KBr) νmax: 3203, 2939, 1702, 1674, 1596, 1456, 1354, 1231, 975, 692; 1H-NMR (CDCl3) δ: 10.02 (s, 1H), 9.53 (s, 1H), 8.60 (d, J=2.2, 1H), 8.64 (d, J=2.2, 1H), 7.41 (d, J=8.1, 1H), 7.13 (d, J=8.1, 1H), 4.34 (q, J=7.0, 2H), 1.32 (t, J=7.0, 3H); 13C-NMR (CDCl3) δ: 188.6, 168.1, 162.1, 153.8, 149.9, 145.3, 142.3, 132.3, 126.9, 125.0, 120.8, 118.8, 43.0, 13.7; MS (EI), m/z (relative intensity): 302 (M+, 80), 287 (30), 274 (100), 260 (30), 245 (35); HRMS (ESI-TOF) calcd. for C14H12ClN4O2 [M+H]+ 303.0643; found: 303.0645.
2-Chloro-5,11-dihydro-11-ethyl-8-hydroxymethyl-6H-dipyrido[3,2-b:2’,3’-e][1,4]diazepin-6-one (14)
To a solution of 13 (228 mg, 0.75 mmol) in THF (12 mL) were added water (0.1 mL) and sodium borohydride (28.5 mg, 0.75 mmol). The mixture was stirred for half an hour, then diluted with water. THF was removed under reduced pressure and the precipitate was filtered and washed with water to yield 14 (214 mg, 93%) as a white solid, m.p. 197-198 oC; FTIR (KBr) νmax: 3319, 3191, 2959, 1666, 1590, 1456, 1390, 1232, 1041; 1H-NMR (acetone-d6) δ: 9.50 (br s, 1H), 8.46 (d, J=2.2, 1H), 8.11 (d, J=2.2, 1H), 7.60 (d, J=8.1, 1H), 7.18 (d, J=8.1, 1H), 4.65 (d, J=5.13, 2H), 4.56 (t, J=5.86, 1H), 4.15 (q, J=7.3, 2H), 1.21 (t, J=7.3, 3H); 13C-NMR (acetone-d6) δ: 167.8, 158.7, 152.7, 151.0, 144.5, 140.3, 134.1, 133.4, 127.3, 121.2, 120.5, 61.5, 42.4, 14.0; MS (EI), m/z (relative intensity): 304 (M+, 69), 289 (39), 276 (100), 261 (29), 247 (20), 164 (31); HRMS (ESI-TOF) calcd. for C14H14ClN4O2 [M+H]+ 305.0800; found: 305.0805.
2-Chloro-5,11-dihydro-11-ethyl-8-chloromethyl-6H-dipyrido[3,2-b:2’,3’-e][1,4]diazepin-6-one (15)
A suspension of 14 (177 mg, 0.58 mmol) in CH2Cl2 (100 mL) was treated with thionyl chloride (0.3 mL) followed by triethylamine (1 mL). The reaction mixture was stirred at room temperature for 1 hour and a clear solution was obtained. Then, saturated aqueous NaHCO3 was added and the mixture was extracted with CH2Cl2. The organic layer was washed with water, then dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20%EtOAc/hexane) to give 15 (163 mg, 87%) as a pale yellow solid, m.p. 226-227 oC; FTIR (KBr) νmax: 3195, 2969, 1671, 1588, 1455, 1382, 1247, 699; 1H-NMR (CDCl3) δ: 9.15 (br s, 1H), 8.47 (d, J=2.9, 1H), 8.19 (d, J=2.9, 1H), 7.34 (d, J=8.1, 1H), 7.06 (d, J=8.1, 1H), 4.58 (s, 2H), 4.23 (q, J=7.3, 2H), 1.27 (t, J=7.3, 3H); 13C-NMR (CDCl3) δ: 168.3, 158.7, 151.7, 151.5, 145.2, 141.4, 131.9, 128.3, 125.0, 119.9, 119.6, 42.3, 42.2, 13.7; MS (EI), m/z (relative intensity): 322 (M+, 54), 307 (31), 294 (62), 287 (37), 259 (100), 244 (28), 231 (21); HRMS (ESI-TOF) calcd. for C14H13Cl2N4O [M+H]+ 323.0461; found: 323.0470.
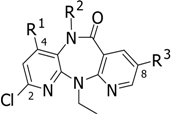
2-Chloro-5,11-dihydro-11-ethyl-8-(phenylthio)-methyl-6H-dipyrido[3,2-b:2’,3’-e][1,4]diazepin-6-one (T1)
A solution of thiophenol (0.1 ml, 0.97 mmol) in DMF (2 mL) was treated with 60% sodium hydride (65 mg, 1.6 mmol) under a N2 atmosphere. After 10 minutes, a solution of 15 (100 mg, 0.31 mmol) in DMF (3 mL) was added and the mixture was stirred at room temperature for 1 hour. The reaction was quenched by addition of water and the mixture was extracted with EtOAc. The organic layer was washed with water, then dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20%EtOAc/hexane) to give T1 (85.5 mg, 70%) as a yellow solid, m.p. 188.5-189.5 oC; FTIR (KBr) νmax: 3182, 2965, 1665, 1585, 1455, 1383, 1239, 739, 688; 1H-NMR (CDCl3) δ: 9.33 (s, 1H), 8.29 (d, J=2.2, 1H), 8.09 (d, J=2.2, 1H), 7.34-7.20 (m, 6H), 7.04 (d, J=8.1, 1H), 4.18 (q, J=7.3, 2H), 4.05 (s, 2H), 1.23 (t, J=7.3, 3H); 13C-NMR (CDCl3) δ: 168.6, 157.8, 151.8, 145.1, 141.2, 134.9, 131.8, 130.6(2C), 129.1(2C), 128.6, 127.0(2C), 125.1, 119.7, 119.6, 42.0, 35.6, 13.6; MS (EI), m/z (relative intensity): 396 (M+, 7), 287 (100), 259 (52), 231 (14); HRMS (ESI-TOF) calcd. for C20H18ClN4OS [M+H]+ 397.0884; found: 397.0894
2-Chloro-5,11-dihydro-11-ethyl-8-(3-methoxyphenylthio)-methyl-6H-dipyrido[3,2-b:2’,3’-e][1,4] diazepin-6-one (T2)
A solution of 3-methoxythiophenol (0.14 ml, 1.14 mmol) in DMF (2 mL) was treated with 60% sodium hydride (76 mg, 1.88 mmol) under a N2 atmosphere. After 10 minutes, a solution of 15 (122 mg, 0.38 mmol) in DMF (3 mL) was added and the mixture was stirred at room temperature for 1 hour. The reaction was quenched by addition of water and the mixture was extracted with EtOAc. The organic layer was washed with water, then dried (Na2SO4) and concentrated under reduced pressure. The residue was recrystallized from hexane/EtOAc to afford T2 (140 mg, 87%) as yellow crystals, m.p. 188-189oC; FTIR (KBr) νmax: 3181, 2965, 1662, 1589, 1458, 1385, 1229, 1044, 767, 693; 1H- NMR (CDCl3) δ: 9.56 (br s, 1H), 8.30 (d, J=2.2, 1H), 8.08 (d, J=2.2, 1H), 7.33 (d, J=8.1, 1H), 7.18 (t, J=8.1, 1H), 7.04 (d, J=8.1, 1H), 6.90-6.72 (m, 3H), 4.06 (s, 2H), 3.73 (s, 3H), 1.23 (t, J=7.3, 3H); 13C- NMR (CDCl3) δ: 168.8, 159.9, 157.9, 151.8(2C), 145.1, 141.2, 136.2, 131.8, 129.9, 128.6, 125.2, 122.6, 119.7, 119.6, 115.8, 112.9, 55.3, 42.1, 35.4, 13.6; MS (EI), m/z (relative intensity): 426 (M+, 6), 287 (100), 259 (57), 231 (15); HRMS (ESI-TOF) calcd. for C21H20ClN4O2S [M+H]+ 427.0990; found: 427.0988.
Biological methods: Materials
DA5 (HIV-1 subtype E) cells were obtained from HIV-1 infected pregnant women. These viruses were X4 strain with the syncytium-inducing (SI) formation property, causing morphological changes (cytopathic effect) in infected cells. White blood cells: peripheral blood mononuclear cells were obtained from blood donors and used for HIV virus isolation. H9 T-lymphoblastoid cell line obtained from Medical Research Centre, UK, was used to prepare the viral stock. C8166 T-lymphoblastoid cell line was used to test the activities of bioactive compounds in all experiments. Bioactive compounds were dissolved in 70-95% DMSO. Three anti-retroviral drugs (reverse transcriptase inhibitors) manufactured by the Government Pharmaceutical Organization (GPO), including ANTIVIR (100 mg AZT), GPO-vir (containing 200 mg nevirapine, 150 mg lamivudine, and 30 mg stavudine), and NERAVIR (200 mg nevirapine), were used as controls in all experiments. All experiments were performed in duplicate.
Toxicity of bioactive compounds, anti-retroviral drugs, and DMSO against white blood cell culture.
The solutions of the bioactive compounds and anti-retroviral drugs (diluted to 10, 1, and 0.1 µg/mL in the culture medium), as well as DMSO (diluted to 1%, 0.1%, and 0.01%), were dispensed into tissue culture plates in duplicates. C8166 cells were then added into each well and examined everyday for 7 days. Moreover, the cells were counted, stained with 1% tryphan blue in order to examine for cell viability under the microscope on day 0, day 4, and day 7, and compared with drug-free control cells.
Virustatic and virucidal tests
The viruses were used at 100 TCID50 (50% tissue cell infectivity dose) level, which was determined by diluting the viruses in quadruplicates, and the potency of the viruses was calculated using Karber equation. Additionally, the following controls were used in all experiments: control viruses diluted to 100, 10, and 1 TCID50 in order to verify that the viruses used in all experiments were at 100 TCID50 level. C8166 control cells were used for cell examination throughout the experiments. Bioactive control was used to investigate the effect of the compounds on the cells. DMSO control was used to evaluate the effect of DMSO on virus proliferation in the cells. Anti-retroviral drug controls (AZT, GPO-vir, and Nevirapine) were used to investigate the effect of the drugs on the cells.
Virustatic test
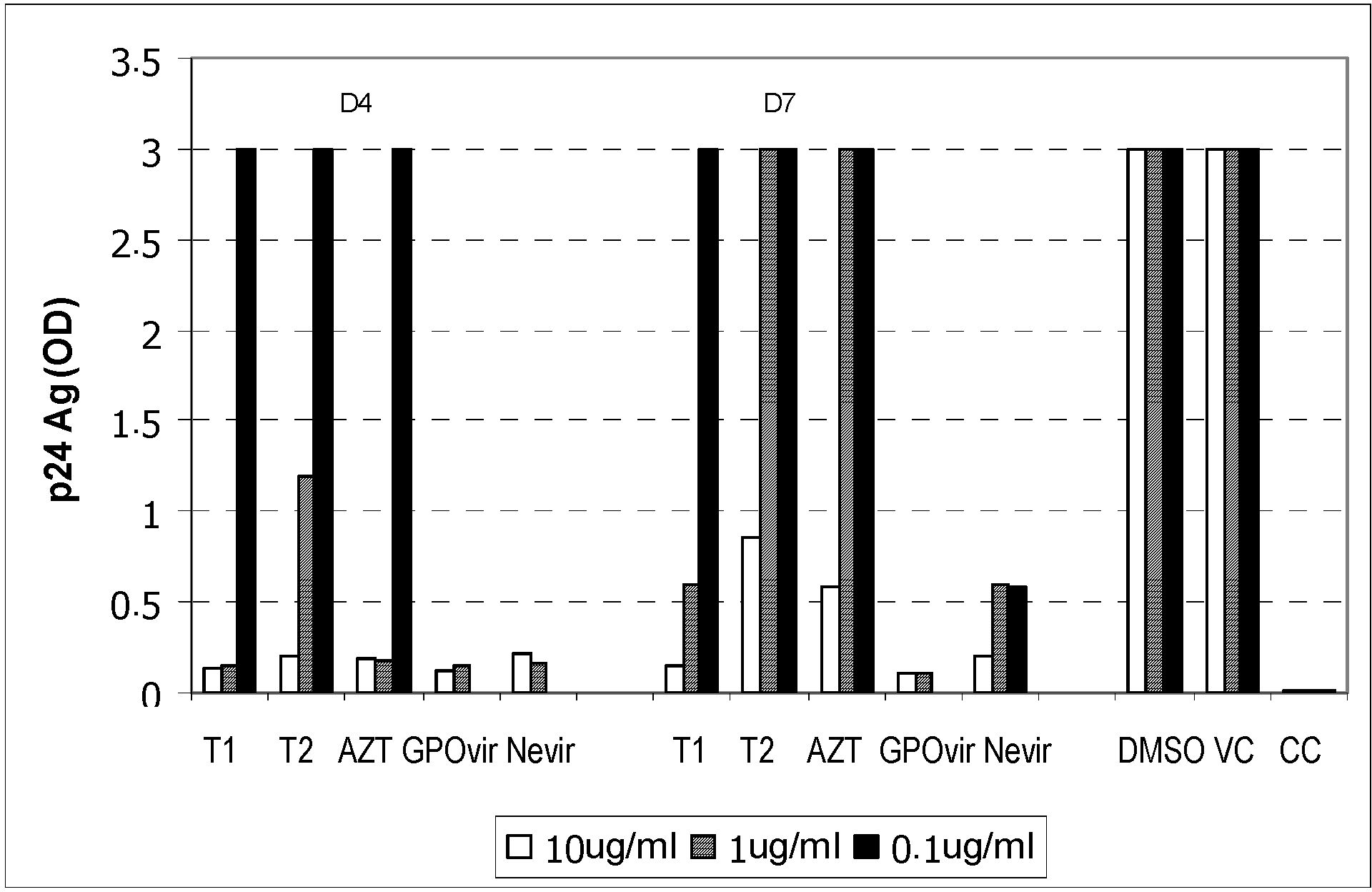
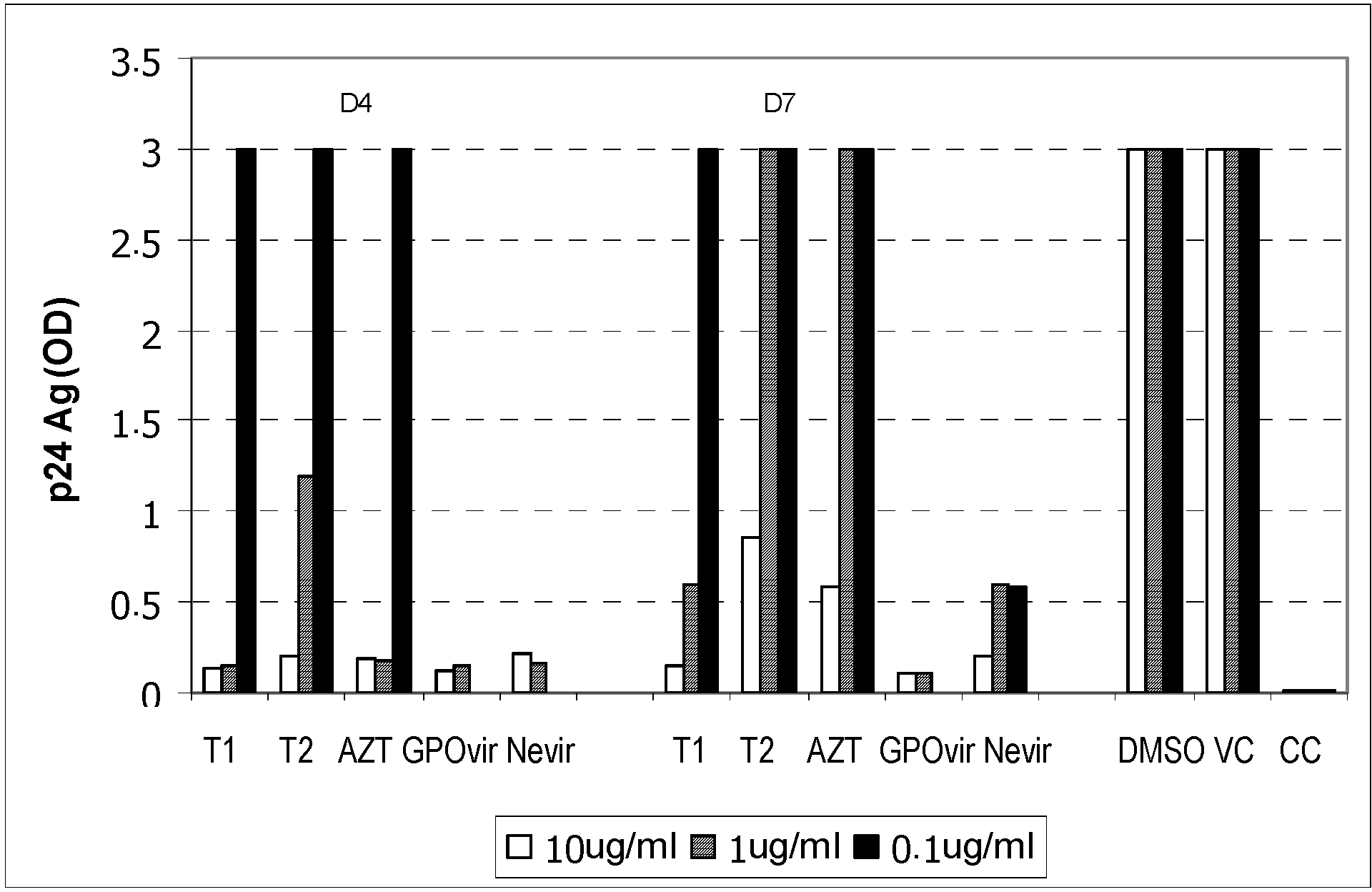
C8166 cells were incubated with the solutions of bioactive compounds for 30 minutes. The viruses were then added into each well at 100 TCID50 level, and the samples were further incubated for 3 hours. After that, the cells were washed 4 times, and maintained for 7 days. The culture medium was changed on day 3, and the cells were examined for cytopathic effect on days 4 and 7. Additionally, the level of the p24 antigen in the culture medium was determined on day 7 using commercially-available ELISA kits in order to determine the EC50, which was the concentration of each bioactive compound that inhibited the viruses by 50%.
Virucidal test
The viruses at 100 TCID50 level were incubated with the solutions of bioactive compounds for 1 hour. C8166 cells were then added to each well, and the samples were further incubated for 1 hour. After that, the cells were washed 4 times, and maintained for 7 days. The culture medium was changed on day 3, and the cells were examined for cytopathic effect on days 4 and 7. Additionally, the level of the p24 antigen in the culture medium was also determined on day 7 using commercially-available ELISA kits.

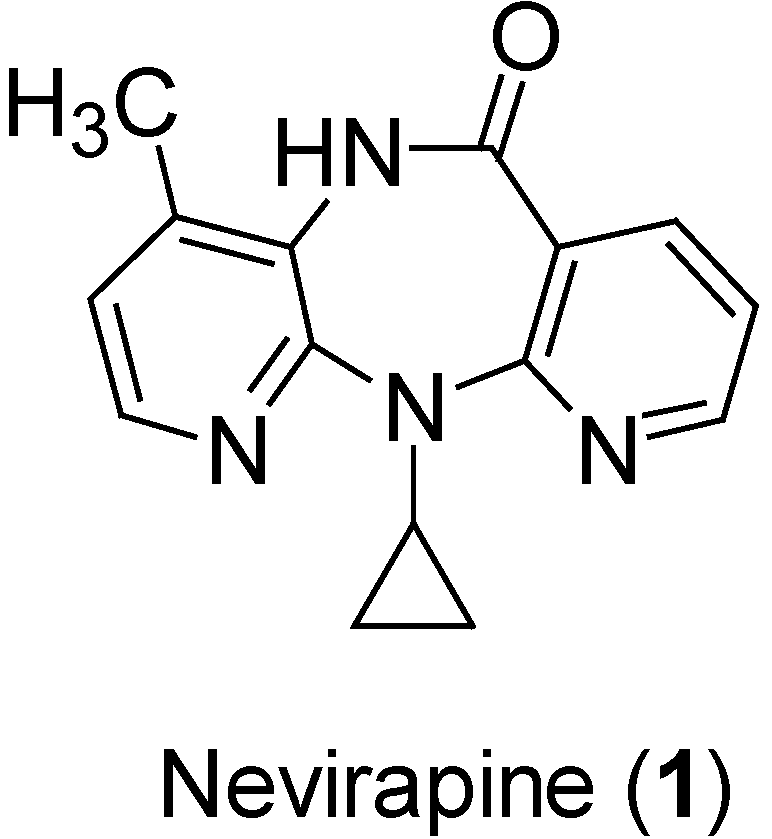

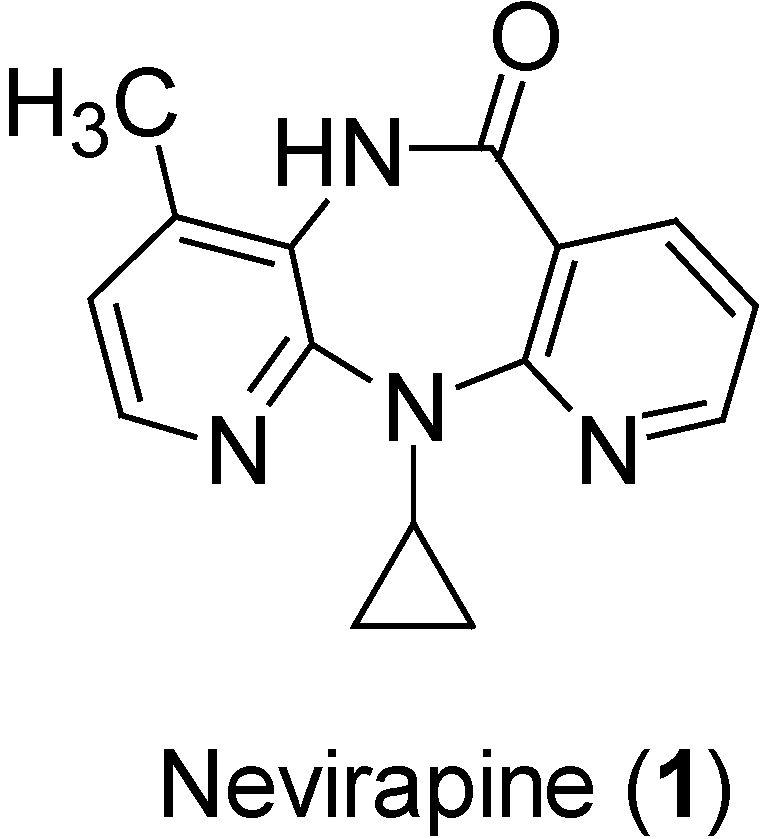{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}