Formal Synthesis of the ACE Inhibitor Benazepril·HCl via an Asymmetric Aza-Michael Reaction

1
Laboratory of Organometallic Chemistry, East China University of Science & Technology, Shanghai, 200237, P.R. China
2
Department of Pharmaceutical and Biochemical Engineering, Sichuan University, Sichuan, 610065, P.R. China
3
Department of Chemistry, National Chung-Hsing University, Taichung, 402, Taiwan
4
Department of Biotechnology and Bioinformatics, Asia University, Wufeng, Taichung, 413, Taiwan
5
Wisdom Pharmaceutical Co. Ltd., Haimen, Jiangsu, 226100, P.R. China
*
Author to whom correspondence should be addressed.
Molecules 2006, 11(8), 641-648; https://doi.org/10.3390/11080641
Submission received: 26 June 2006
/
Revised: 20 July 2006
/
Accepted: 23 August 2006
/
Published: 23 August 2006
Abstract
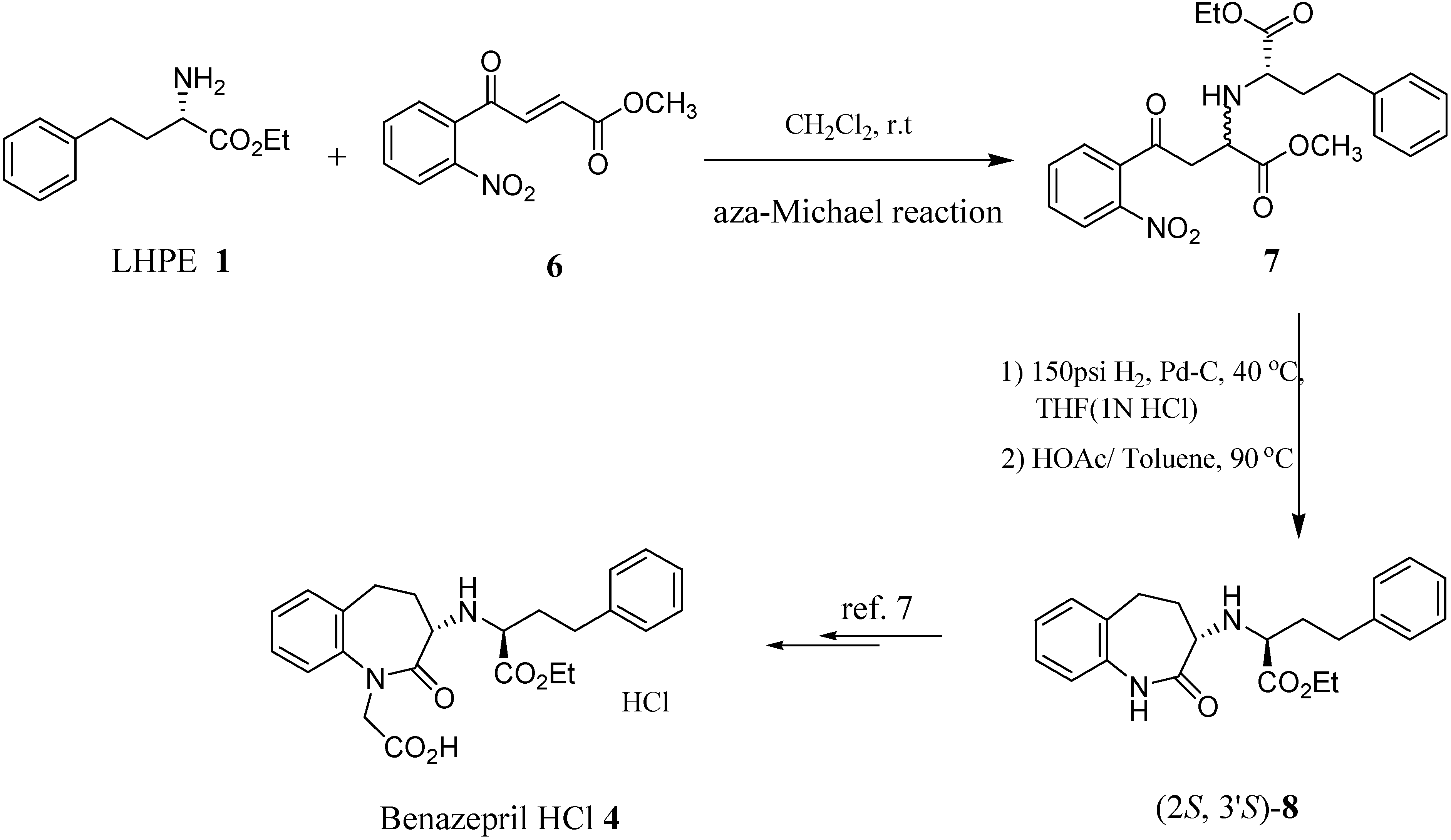
:A formal enantioselective synthesis of benazepril·HCl (4), an anti-hypertensive drug, is reported. Our synthesis employed an asymmetric aza-Michael addition of L-homophenylalanine ethyl ester (LHPE, 1) to 4-(2-nitrophenyl)-4-oxo-but-2-enoic acid methyl ester (6) as the key step to prepare (2S,3’S)-2-(2-oxo-2,3,4,5-tetrahydro-1H-benzo[b]azepin-3-ylamino)-4-phenylbutyric acid ethyl ester (8), which is the key intermediate leading to benazepril·HCl (4).
Introduction
Angiotensin converting enzyme (ACE) inhibitors [1] constitute a major class of anti-hypertensive drugs that has been widely studied and developed over the past few decades. Among the familiar ACE inhibitors that have come to the market or to the clinic, most contain an unnatural chiral amino ethyl ester, L-homophenylalanine ethyl ester (LHPE, 1), in their framework, such as enalapril (2) [2], lisinopril (3) [3], and benazepril·HCl (4) [4] (Figure 1).
Figure 1.
LHPE and related ACE inhibitors.
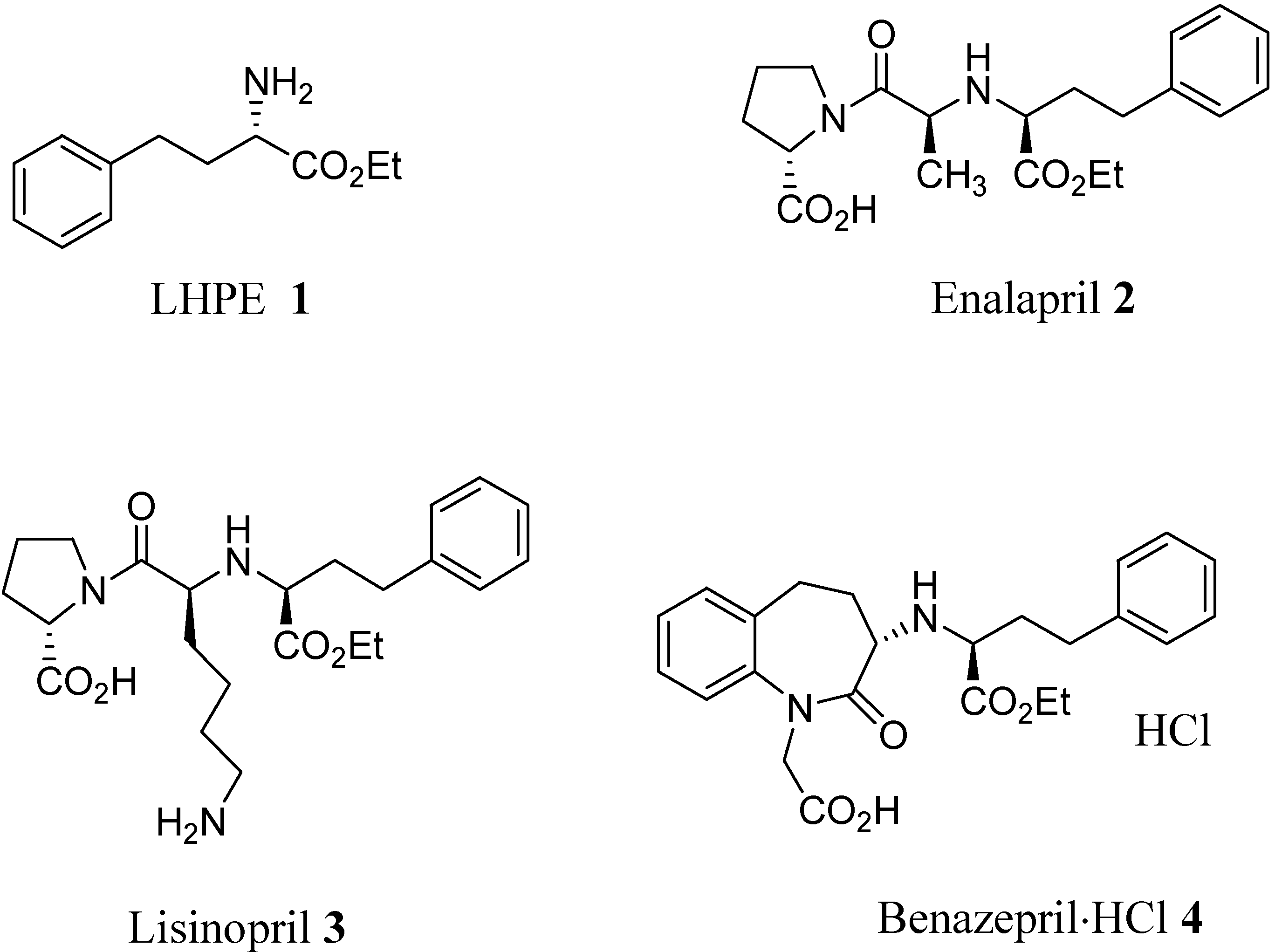
Among them, benazepril·HCl (4) is the most potent one, having less side-effects and better stability due to its unique skeleton, different from that of the others. Compound 4 has been a quite popular subject over the years in both academic and industrial sectors, but only a few papers have been published concerning its synthesis. Both Watthey [4] and Boyer [5] used α-tetralone as starting material to prepare benazepril·HCl (4), and both their synthesis went through rather tedious resolution processes. Herein, we describe a convergent pathway in which compound 8, a key chiral intermediate to benazepril·HCl (4), was prepared through an asymmetric aza-Michael reaction.
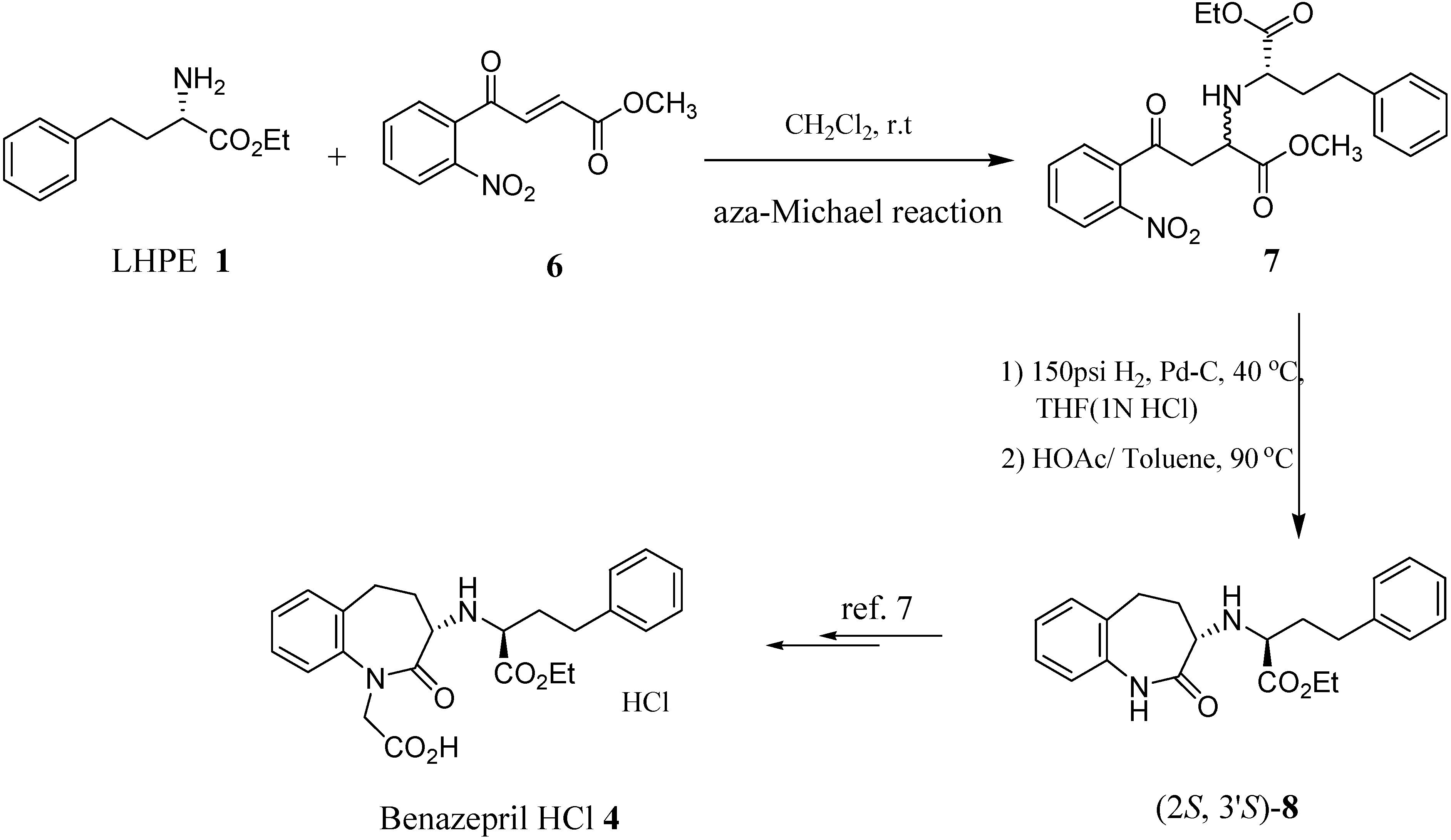
The asymmetric aza-Michael addition reaction of chiral amines to α,β-unsaturated carbonyl compounds is a very useful method to prepare optical active amino acids [6] because of its mild reaction conditions and good chemical yields. In our strategy, commercially available LHPE 1 was employed as nucleophilic reagent toward 4-(2-nitrophenyl)-4-oxo-but-2-enoic acid methyl ester (6) to set up the second chiral center via an asymmetric 1,4-addition reaction. The nitro group of the coupled ethyl ester 7 was then reduced by Pd/C catalyzed hydrogenation, and the resulting amino group reacted in situ intramolecularly with the ester group to give the cyclized (2S,3’S)-caprolactam 8 (Scheme 1). From the compound 8, (2S,3'S)-2-(1-carboxymethyl-2-oxo-2,3,4,5-tetrahydro-1H-benzo[b]azepin-3-ylamino)-4-phenylbutyric acid ethyl ester hydrochloride (benazepril·HCl, 4) could be then prepared efficiently [7].
Scheme 1.
Results and Discussion
First, we used commercially 40% aqueous glyoxylic acid and o-nitroacetophenone (5) as starting materials to prepare 4-(2-nitrophenyl)-4-oxo-but-2-enoic acid methyl ester (6) via an aldol condensation reaction [8] (Scheme 2). A large coupling constant (15.6 Hz) for the olefinic protons, which appeared at δ 7.3 and 6.4, suggested a trans geometry for the double bond.
Scheme 2.
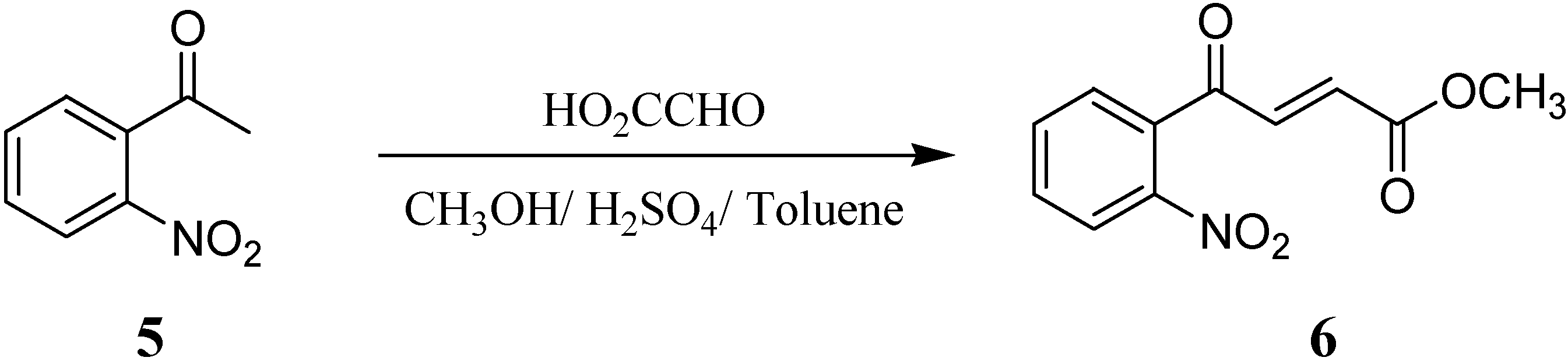
With an optically active amine as the chiral nucleophile, the aza-Michael addition to α,β-unsaturated carbonyl compounds should proceed in an asymmetric fashion, and thus one diastereomer should be generated predominately. However, equal amount of the two diastereomers would be produced through the equilibrium of a reversible addition-elimination process. To resolve this problem, we have tested the following two ways: 1) selecting suitable aza-Michael reaction conditions to achieve dynamic resolution, in which solvent would be the main factor. The differences in solubility or solvent effect between the two diastereoisomers can be employed to improve the diastereoselectivity through the equilibrium of the reversible aza-Michael reaction [6]. In addition, the reaction stoichiometry, temperature, reaction time, and acidity may also exert certain influences on the diastereoselectivity. 2) introducing metallic counterions to affect the reversibility of the aza-Michael reaction through metallic chelation, or formation of amino-metal complex to modify the nucleophilicity of the amino group [9]. The results are discussed in detail below:

{kind=link}
{kind=link}
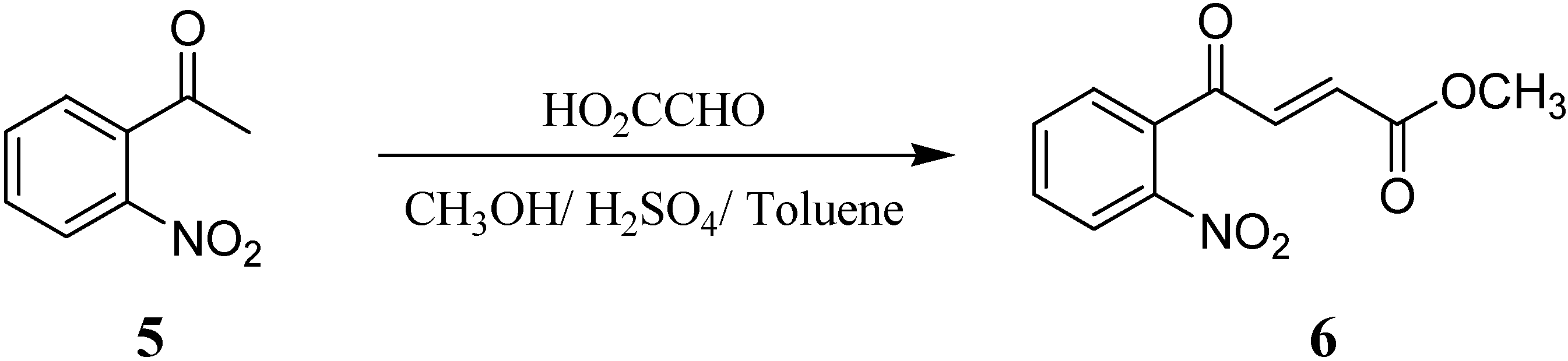{kind=link}
| Entrya | Solvent | Reaction time (hr) | Diastereomeric ratio of 7 (S, S) : (R, S)b | Conversion (%)c,d |
| 1 | Dichloromethane | 17 | 4.20 : 1 | 96.0 |
| 2 | Acetonitrile | 26 | 3.75 : 1 | 99.0 |
| 3 | Ethanol | 18 | 2.15 : 1 | 98.0 |
| 4 | Isopropanol | 16 | 1.91 : 1 | 98.8 |
| 5 | Xylene | 16 | 1.88 : 1 | 99.3 |
| 6 | Pyridine | 33 | 1.87 : 1 | 98.2 |
| 7 | Ether | 25 | 1.86 : 1 | 98.2 |
| 8 | Benzene | 16 | 1.83 : 1 | 98.6 |
| 9 | Acetonitrile/H2O | 35 | 1.81 : 1 | 99.2 |
| 10 | Isobutanol | 33 | 1.80 : 1 | 96.9 |
| 11 | tert-Butanol | 18 | 1.69 : 1 | 96.7 |
| 12 | DME | 25 | 1.68 : 1 | 97.5 |
| 13 | Ethyl acetate | 18 | 1.67 : 1 | 95.2 |
| 14 | Toluene | 16 | 1.60 : 1 | 99.1 |
| 15 | THF | 16 | 1.53 : 1 | 98.6 |
a. All reactions were carried out under room temperature. And the ratio of unsaturated ester 6 to LHPE 1 was 1.00:1.05b. Diastereomeric ratios were taken directly from 1H-NMR by comparison of the characteristic peaks of the two diastereomers at δ3.74 for (S,S)-form and δ3.71 for (R,S)-form.c. The conversions were determined from the characteristic 1H-NMR peak of the unsaturated ester 6 at δ3.80.d. The isolated yields of ester 7 were around 90% for most of the reactions listed in Table 1.
According to our results (Table 1), higher diastereomeric ratios of the compound 7 were observed in polar aprotic solvents such as dichloromethane (4.20:1) and acetonitrile (3.73:1), while all of the other solvents gave poor diastereomeric ratios (2.15:1~1.53:1) for this asymmetric addition process.
The stoichiometric ratio of unsaturated ester 6 to LHPE 1 could be adjusted from 2.00 to 0.50, but some excess LHPE 1 accelerated the reaction rate, so the best ratio of ester 6 to LHPE 1 was 1.00:1.10 (Table 2, entry 5), which lead to the highest observed diastereomeric selectivity of 4.50:1. In addition, the suitable reaction concentrations of unsaturated ester 6 were 1%~20% (w/w), as lower concentrations resulted in lower conversion yields and higher concentrations resulted in lower diastereomeric ratios (Table 2, entries 21~22).
The optimum reaction temperature range was 20~40°C. Lower temperatures prolonged the reaction times, which might lower the diastereomeric ratios, while higher reaction temperatures would enhance the reversibility of the aza-Michael reaction, which likewise might also lower the diastereomeric ratio (Table 2, entries 7~9). Consequently, we did most reactions at a temperature of 20°C.
Reaction times of 15~35 hours are suitable for this asymmetric aza-Michael reaction (Table 2, entries 3, 10~16), because shorter times would result in lower conversion and prolonged times would lower the diastereomeric ratios. This may due to the fact that the aza-Michael reaction is a kinetically controlled process at the beginning and then becomes a thermodynamic equilibrium. Noteworthy, although a high diastereselectivity was achieved in CH2Cl2 in short time, decreasing diastereomeric ratios of product 7 and significant amounts of the starting reactants were observed after a long time. This phenomenon may be accounted for by the occurrence of the retro-Michael process [10]. We also found that alkaline additives such as NEt3 could accelerated the aza-Michael reaction but could not improve the diastereoselectivity (Table 2, entries 23~25).
| Entry | Solvent a /addictive | LHPE (equiv.) | Reaction time (hr) | Reaction temperature (°C) | Proportion of diastereomers (S, S) : (R, S) | Conversionb (%) |
| 1 | CH2Cl2 | 0.50 | 53 | 20 | 2.89 : 1 | 86.7 |
| 2 | CH2Cl2 | 0.90 | 53 | 20 | 3.10 : 1 | 92.5 |
| 3 | CH2Cl2 | 1.05 | 16 | 20 | 4.15 : 1 | 96.0 |
| 4 | CH2Cl2 | 1.10 | 16 | 20 | 4.50 : 1 | 96.0 |
| 5 | CH2Cl2 | 1.50 | 16 | 20 | 3.85 : 1 | 98.0 |
| 6 | CH2Cl2 | 2.00 | 16 | 20 | 3.07 : 1 | 97.5 |
| 7 | CH2Cl2 | 1.05 | 35 | 0 | 2.70 : 1 | 90.8 |
| 8 | CH2Cl2 | 1.05 | 16 | 40 | 3.75 : 1 | 96.2 |
| 9 | CH2Cl2 | 1.05 | 16 | 60 | 2.20 : 1 | 98.0 |
| 10 | CH2Cl2 | 1.05 | 10 | 20 | 3.36 : 1 | 90.5 |
| 11 | CH2Cl2 | 1.05 | 26 | 20 | 3.68 : 1 | 95.3 |
| 12 | CH2Cl2 | 1.05 | 35 | 20 | 3.76 : 1 | 96.2 |
| 13 | CH2Cl2 | 1.05 | 40 | 20 | 3.60 : 1 | 96.0 |
| 14 | CH2Cl2 | 1.05 | 53 | 20 | 3.29 : 1 | 94.4 |
| 15 | CH2Cl2 | 1.10 | 120 | 20 | 0.62 : 1 | [b] |
| 16 | CH2Cl2 | 1.10 | 168 | 20 | 0.54 : 1 | [b] |
| 17 | CH2Cl2 | 0.95 | 10 | 20 | 3.82 : 1 | 88.7 |
| 18 | CH2Cl2 | 0.95 | 26 | 20 | 3.87 : 1 | 88.5 |
| 19 | CH2Cl2 | 0.95 | 35 | 20 | 3.79 : 1 | 91.3 |
| 20 | CH2Cl2 | 0.95 | 40 | 20 | 3.56 : 1 | 91.6 |
| 21 | CH2Cl2 | 1.05c | 24 | 20 | 2.56 : 1 | 98.0 |
| 22 | CH2Cl2 | 1.05d | 24 | 20 | - | - |
| 23 | CH2Cl2/Et3N | 1.15 | 12 | 20 | 1.59 : 1 | 99.0 |
| 24 | C2H5OH | 1.15 | 18 | 20 | 2.15 : 1 | 98.3 |
| 25 | C2H5OH/Et3N | 1.15 | 12 | 20 | 2.11 : 1 | 100.0 |
a. The amount of solvent was based on the concentration 1~10% of unsaturated ester 6.b. For diastereomeric ratios, conversions, and isolated yields, please see Table 1. b. The starting materials were separated out.c. The concentration of unsaturated ester 6 was >30%.d. The concentration of unsaturated ester 6 was <0.1% and the addition product was non-detectable over a long reaction time.
On the other hand, we had also studied the effects of introducing metallic counterions in the aza-Michael reaction. We originally wished to cause a “metal chelate effect” in which the metal ion becomes chelated with the heteroatoms of compound 7 to form a rather stable conformation that mayn decrease the reversibility of the aza-Michael reaction. However, the current results indicated that we did not observe any significant change in diastereselectivity with or without the additive metallic compounds. In fact, some transition metal salts even blocked the aza-Michael reaction [11].
After the aza-Michael reaction, a 4:1 mixture of the two diastereomers was obtained. The mixture was reduced by Pd/C catalyzed hydrogenation, and then the resulting amino functionality reacted in situ with the ester group intramolecularly to give the cyclized (2S,3’S)-caprolactam 8 (Scheme 1) and according to the literature [7], benazepril·HCl 4 could be easily obtained from compound 8.
Conclusions
This report has offered a novel strategy for the asymmetric synthesis of the ACE inhibitor benazepril·HCl (4). Through an asymmetric aza-Michael reaction, we kinetically synthesized (2S,3’S)-2-(2-oxo-2,3,4,5-tetrahydro-1H-benzo[b]azepin-3-ylamino)-4-phenylbutyric acid ethyl ester (8), a known key intermediate for the preparation of compound 4. Our results indicated that the diastereoselectivity of this 1,4-addition reaction was greatly influenced by solvent effects, but was not so sensitive to other influencing factors, such as stoichiometry, reaction temperatures, and counterion effects. Thus, this report not only offers an ecomomical synthesis to benazepril·HCl (4), but also offers useful knowledge for the preparation of chiral β-keto-aminoesters in general.
Experimental
General
Starting materials were obtained from commercial suppliers and used without further purification unless otherwise stated. Flash column chromatography was carried out using Merck silica gel 60 (0.063-0.200 mm). Melting points are uncorrected. Optical rotations were measured on a Perkin Elmer 241 polarimeter. Infrared spectra were recorded on a Hitachi 270-30 Infrared Spectrophotometer. NMR spectra were recorded for CDCl3 solutions on a Varian Mercury 400 instrument operating at 400 MHz (for 1H) and 10 MHz (for 13C), respectively. The chemical shifts are reported as δ values in ppm relative to TMS in CDCl3 (δ=0), used as internal standard for 1H-NMR spectra and the center peak of CDCl3 (δ=77.0 ppm) used as the internal standard in the 13C-NMR spectra. FAB-mass were collected on a JMS-700 Double Focusing Mass Spectrometer. Elemental analyses were run on a Foss Heraeus CHN-O-RAPID Elemental Analyzer.
Preparation of 4-(2-nitrophenyl)-4-oxo-but-2-enoic acid methyl ester (6)
An aqueous solution of glyoxylic acid (72 g, 40%) was concentrated to 2/3 volume under reduced pressure, and then concentrated sulfuric acid (0.8 g), methanol (120 mL), and toluene (90 mL) were added. The mixture was refluxed for 5 hours. The methanol was distilled off and then o-nitro-acetophenone (22.3 g, 135 mmol) in toluene (90 mL) was added. The reaction mixture was heated to 110~130 °C for 36 hours to remove water by azeotropic distillation. After cooling, the toluene phase was collected and washed with saturated sodium bicarbonate (20 mL x 2) and water (20 mL x 2). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The crude product was recrystallized in ether (120 mL) to give 20.67 g of a yellowish solid (20.67 g, 65%); mp 55-56 °C; IR (neat): 1730, 1682, 1529, 1348, 1309 cm-1; 1H-NMR: δ 8.21 (d, J = 8 Hz, 1H, CH), 7.81-7.77(m, 1H,CH), 7.72-7.68(m, 1H, CH), 7.47(d, J = 8 Hz, 1H, CH), 7.31(d, J = 15.6 Hz, 1H, CH), 6.39(d, J = 15.6 Hz, 1H, CH), 3.80(s, 3H, CH3); 13C- NMR: δ 191.7, 165.2, 146.2, 139.4, 135.0, 134.5, 132.2, 131.3, 128.6, 124.5, 52.4; MS (EI): m/z 235 (M+ ), 204, 189, 150, 134 (100%), 113, 104, 76, 59; HRMS (EI): Calcd. for C11H9NO5 M+ 235.0581, Found M+ 235.0477; Anal. Calcd for C11H9NO5: C, 56.17; H, 3.86; N, 5.96; Found: C, 56.16; H, 3.94; N, 6.06.
General procedure for Aza-Michael addition reactions
A 50 mL flask was charged with 4-(2-nitrophenyl)-4-oxo-but-2-enoic acid methyl ester (6, 2.35 g, 10 mmol) and L-homophenylalanine ethyl ester (2.28 g, 11 mmol) in the desired solvent (20 mL) and the mixture was stirred at ambient temperature (or another suitable temperature) for the given reaction times. The reaction mixture was concentrated to give a crude product that was purified by chromatography to afford (2S,2’S)-2-(1-(methoxycarbonyl)-3-(2-nitrophenyl)-3-oxopropylamino)-4-phenylbutyric acid ethyl ester (7) as a mixture of two diastereomers. The diastereoselectivities and yields obtained are shown in Table 1. Mixture of compound 7: 1H-NMR: δ 8.11 (d, 1H, Ph), 7.75-7.50 (m, 3H, Ph), 7.30-7.15 (m, 5H, Ph), 4.25-4.15 (m, 2H, OCH2), 3.90-3.85 (m, 1H, NHCH), 3.74 & 3.71 (s, 3H, OCH3), 3.45-3.15 (m, 3H, NHCHCH2), 2.80-2.65 (m, 2H, CH2), 2.10-1.80 (m, 2H, CH2), 1.35-1.25 (m, 3H, OCH2CH3).
Preparation of (2S,3’S)-2-(2-oxo-2,3,4,5-tetrahydro-1H-benzo[b]azepin-3-ylamino)-4-phenylbutyric acid ethyl ester (8).
To a solution of methyl 4-oxo-4-nitrophenyl-2-aminobutanoate 7 (1.46 g, 1.0 mmol) in THF (20 mL) was added Pd-C (5%, 0.14 g) in a pressure vessel. The reaction mixture was hydrogenated at 40 °C under 150 psi of H2 for 24 hours, then HCl (1 N, 6 mL) was added to the reaction mixture and hydrogenated at 40 °C under 150 psi of H2 for another 16 hours. The reaction mixture was filtered and basified with saturated aqueous sodium bicarbonate (30 mL). The mixture was extracted with ethyl acetate (50 mL x 2). The combined organic layer was dried with MgSO4 and purified by flash column chromatography to obtain the (S,S)-diastereomer (0.69 g). It was then refluxed in 10% acetic acid-toluene solution for 24 hours. The reaction mixture was concentrated to give the crude product (0.55 g). The crude product was purified chromatography to afford the title compound 8 as a pale yellow solid (0.15 g, 40%); [α]D -168.1 (c 1.09, CHCl3); mp 110-111 °C; IR (neat): 3224, 2948, 1734, 1676, 1494, 1160 cm-1; 1H-NMR: δ 7.76 (br, 1H, CONH), 7.29-7.13 (m, 8H, CH), 6.99 (d, J = 8.0 Hz, 1H, CH), 4.12-4.03 (m, 2H, CO2CH2CH3), 3.33-3.26 (m, 2H, CHNH), 2.91-2.84 (m, 1H, CH2), 2.75-2.61 (m, 3H, CH2), 2.55-2.42 (m, 1H, CH2), 2.10-1.88 (m, 3H, CH2), 1.15 (t, J = 7.2 Hz, 3H, CO2CH2CH3); 13C-NMR: δ 175.3, 174.2, 141.3, 136.6, 134.3, 129.6, 128.3, 128.2, 127.5, 125.9, 125.8, 122.0, 60.5, 60.0, 56.6, 37.8, 35.0, 32.1, 28.9, 14.1; MS (FAB): m/z 367 (MH+, 100%), 293, 206, 154, 91, 77, 51; Anal. Calcd. for C22H26N2O3: C, 72.11; H, 7.15; N, 7.64. Found: C, 71.88; H, 7.36; N, 7.63.
References
- Kori, M.; Itoh, K.; Inada, Y.; Katoh, T.; Sumino, Y.; Nishikawa, K.; Sugihara, H. Chem. Pharm. Bull. 1994, 42, 580–585. [CrossRef]Kubota, H.; Nunami, K.; Yamagishi, M.; Nishimoto, S.; Hayashi, K. Chem. Pharm. Bull. 1991, 39, 1374–1377. [CrossRef]
- Patchett, A. A.; Harris, E; Tristram, E. W.; Wyvratt, M. J.; Wu, M. T.; Taub, D.; Peterson, E. R.; Ikeler, T. J.; Broeke, J. T.; Payne, L. G.; Ondeyka, D. L.; Thorsett, E. D.; Greenlee, W. J.; Lohr, N. S.; Hoffsommer, R. D.; Joshua, H.; Ruyle, W. V.; Rothrock, J. W.; Aster, S. D.; Maycock, A. L.; Robinson, F. M.; Hirschmann, R.; Sweet, C. S.; Ulm, E. H.; Gross, D. M.; Vassil, T. C.; Stone, C. A. Nature 1980, 288, 280–283. [CrossRef]
- Barton, J. N.; Piwinski, J. J.; Skiles, J. W.; Regan, J. R.; Menard, P. R.; Desai, R.; Golec, F. S.; Reilly, L. W.; Goetzen, T.; Ueng, S. N.; Warus, J. D.; Schwab, A.; Samuels, A. I.; Neiss, E. S.; Suh, J. T. J. Med. Chem. 1990, 33, 1600–1606. [CrossRef]Iwasaki, G.; Kimura, R.; Numao, N.; Kondo, K. Chem. Pharm. Bull. 1989, 37, 280–283. [CrossRef]Blacklock, T. J.; Shuman, R. F.; Butcher, J. W.; Shearin, Jr. W. E.; Budavari, J.; Grenda, V. J. J. Org. Chem. 1988, 53, 836–844. [CrossRef]Iwasaki, G.; Kimura, R.; Numao, N.; Kondo, K. Chem. Lett. 1988, 1691–1694. [CrossRef]
- Watthey, J. W. H.; Stanton, J. L.; Desai, M.; Babiarz, J. E.; Finn, B. M. J. Med. Chem. 1985, 28, 1511–1516. [CrossRef]
- Boyer, S. K.; Pfund, R. A.; Portmann, R. E.; Sedelmeier, G. H.; Wetter, H. F. Helv. Chim. Acta 1988, 71, 337–343.
- Urbach, H.; Henning, R. Tetrahedron Letters 1984, 25, 1143–1146. [CrossRef]Yamada, M; Nagashima, J.; Takahashi, S. Tetrahedron Lett. 1998, 39, 9019–9022. [CrossRef]Knollmüller, M.; Ferencic, M.; Gärtner, P.; Girreser, U.; Klinge, M.; Gaischin, L.; Mereiter, K.; Noe, C. R. Monstsh. Chem. 1999, 130, 769–782.
- Chang, C.-Y.; Yang, T.-K. Tetrahedron: Asymmetr. 2003, 14, 2239–2245. [CrossRef]
- Yanagida, Y.; Matsumoto, S.; Takahashi, S. U.S. Patent 5264611, 1993.
- Hawkins, J.M.; Gregory, C.F. J. Org. Chem. 1986, 52, 2820–2822. [CrossRef]
- Tan, K.; Alvarez, R.; Nour, M.; Cave, C.; Chiaroni, A.; Riche, C.; d'Angelo, J. Tetrahedron Lett. 2001, 42, 5021–5023. [CrossRef]
- Kobayashi, S.; Kakumoto, K.; Sugiura, M. Org. Lett. 2002, 4, 1319–1322. [CrossRef]
- Sample availability: Samples of compounds 1 and 4 are available on request from the authors.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Yu, L.-T.; Huang, J.-L.; Chang, C.-Y.; Yang, T.-K. Formal Synthesis of the ACE Inhibitor Benazepril·HCl via an Asymmetric Aza-Michael Reaction. Molecules 2006, 11, 641-648. https://doi.org/10.3390/11080641
AMA Style
Yu L-T, Huang J-L, Chang C-Y, Yang T-K. Formal Synthesis of the ACE Inhibitor Benazepril·HCl via an Asymmetric Aza-Michael Reaction. Molecules. 2006; 11(8):641-648. https://doi.org/10.3390/11080641
Chicago/Turabian StyleYu, Luo-Ting, Ji-Ling Huang, Ching-Yao Chang, and Teng-Kuei Yang. 2006. "Formal Synthesis of the ACE Inhibitor Benazepril·HCl via an Asymmetric Aza-Michael Reaction" Molecules 11, no. 8: 641-648. https://doi.org/10.3390/11080641