Introduction
Quinones, notably naphthoquinones and anthraquinones, are among the most widely distributed natural products. The majority of them exist as coloured phenolic compounds, useful as dyes and pigments. Isolation of these substances from their natural sources normally requires the use of organic solvents before the required compound can be obtained in a reasonably pure state, either to first extract them from the raw material and then to partition them between various solvent phases or as eluents for chromatographic separations. These organic solvents are usually not only costly but also a potential toxic burden to the environment, even if treated in the proper way.
With the advent of electrocoagulation, it has been known for some time that this process is capable of fractionating a number of organic substances in a rather efficient manner by electrochemically coagulating some of them, while leaving other components in solution. For example, the technique can be used to remove coloured matter from drinking water [
1], textile wastewater [
2,
3] and from crude aqueous plant extracts [
4,
5,
6]. It can coagulate starch and protein, while leaving sugars and polyols intact [
7]. Electrocoagulation is one of the most effective methods for removing tannins from a liquid medium [
8,
9,
10], while for other phenolic substances, their structure, including the number and the location of the hydroxyl groups, seems to dictate whether they will be coagulated or not [
7,
9]. Conversely, many glycosides [
4,
5,
9a] and alkaloids [
11] are unaffected by electrocoagulation and consequently these substances conveniently remain in the electrolysed solutions, relatively free from other impurities which are coagulated out.
Our group has recently demonstrated that compounds of interest may be recovered from the coagulated materials by following a novel specific procedure [
7,
9,
12]. With this technique now available, and with the intention to contributing to “Green Chemistry”, we hypothesized that like glycosides and alkaloids, the application of electrocoagulation to this important class of natural products, viz. quinones, might lead to a simple and environmentally friendly alternative method for their isolation, which is, to the best of our knowledge, an operation that has not been reported elsewhere. The results of the consequent investigation are reported in this paper, in which authentic samples of some typical quinones were electrocoagulated and recovered experimentally, and then attempts were made to isolate some of them electrochemically from their respective natural sources.
Results and Discussion
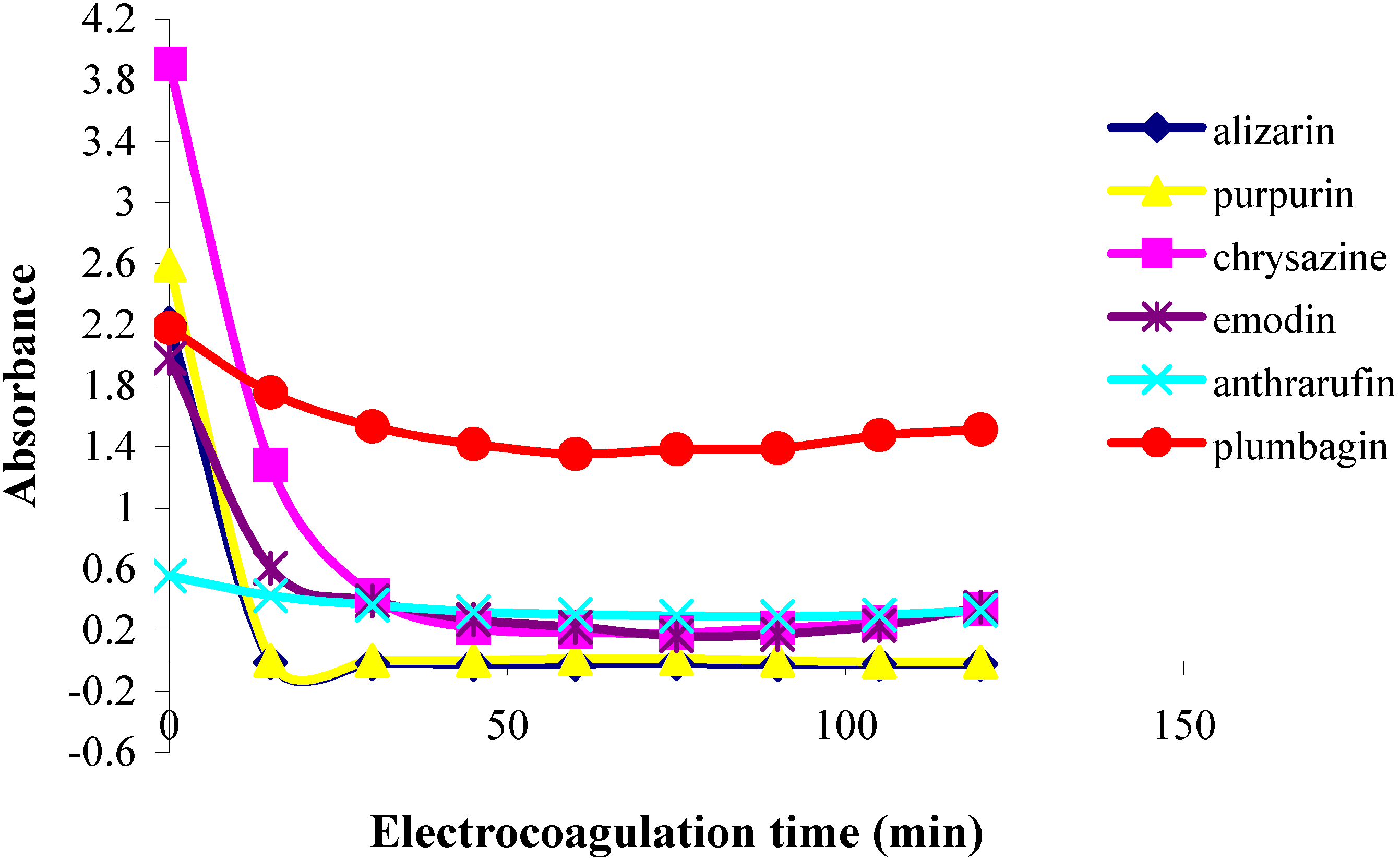
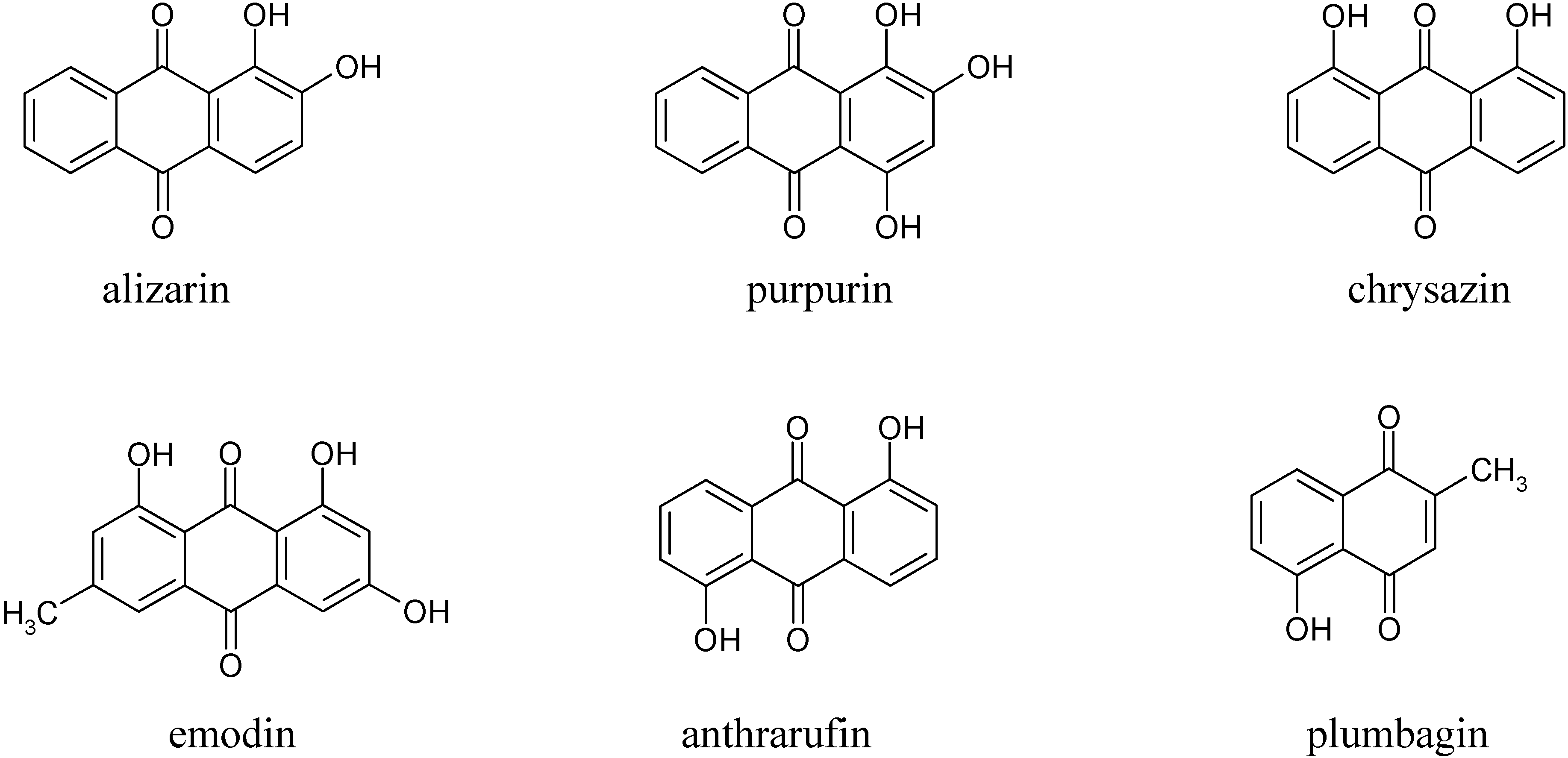
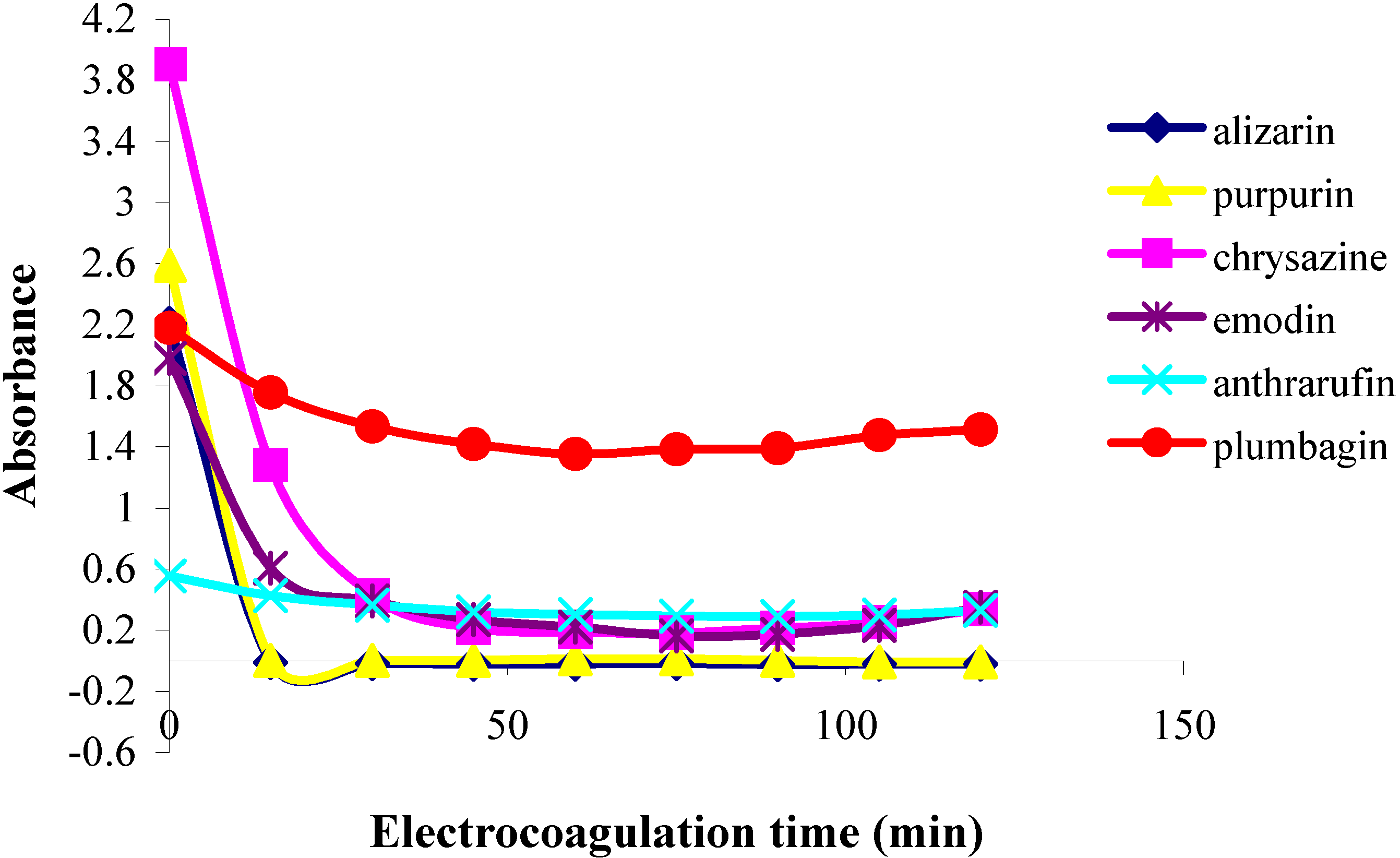
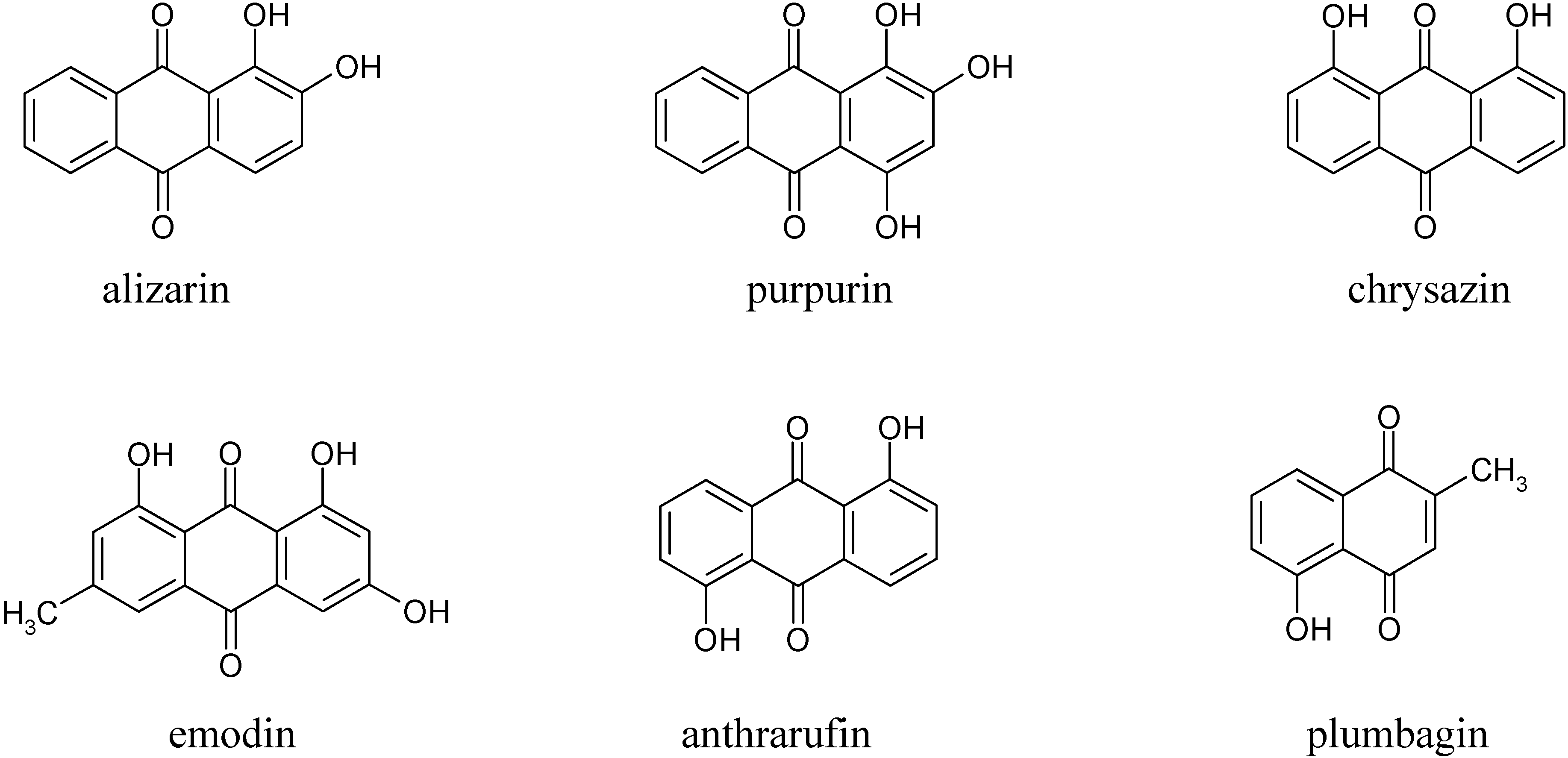
In the first part of this investigation, a number of pure samples of representative quinones, viz. alizarin, purpurin, chrysazin, emodin, anthrarufin and plumbagin (
Figure 1) were separately electrocoagulated using direct current and aluminium plates as electrodes in an aqueous alcoholic solution containing sodium chloride as the supporting electrolyte. The absorbance of each electrolysed sample solution was measured at an appropriate wavelength at regular intervals during electrolysis and electrocoagulation curves were obtained for each quinone as a plot of percent residual weight of the sample or absorbance versus electrocoagulation time, as shown in
Figure 2 and
Figure 3.
From the results above it can be seen that the quinones under study may be roughly divided into three categories. The two most rapidly and completely coagulated quinones were alizarin and purpurin, being completely coagulated within 15 minutes under the experimental conditions used. It can also be noted that these two compounds have adjacent phenolic hydroxyl groups (
Figure 1). This catechol-like structure thus seems to be particularly prone to coagulation by a metal ion, probably by complexation, a fact consistent with similar cases encountered and reported earlier [
9].
Figure 1.
Structures of the quinones studied.
Figure 1.
Structures of the quinones studied.
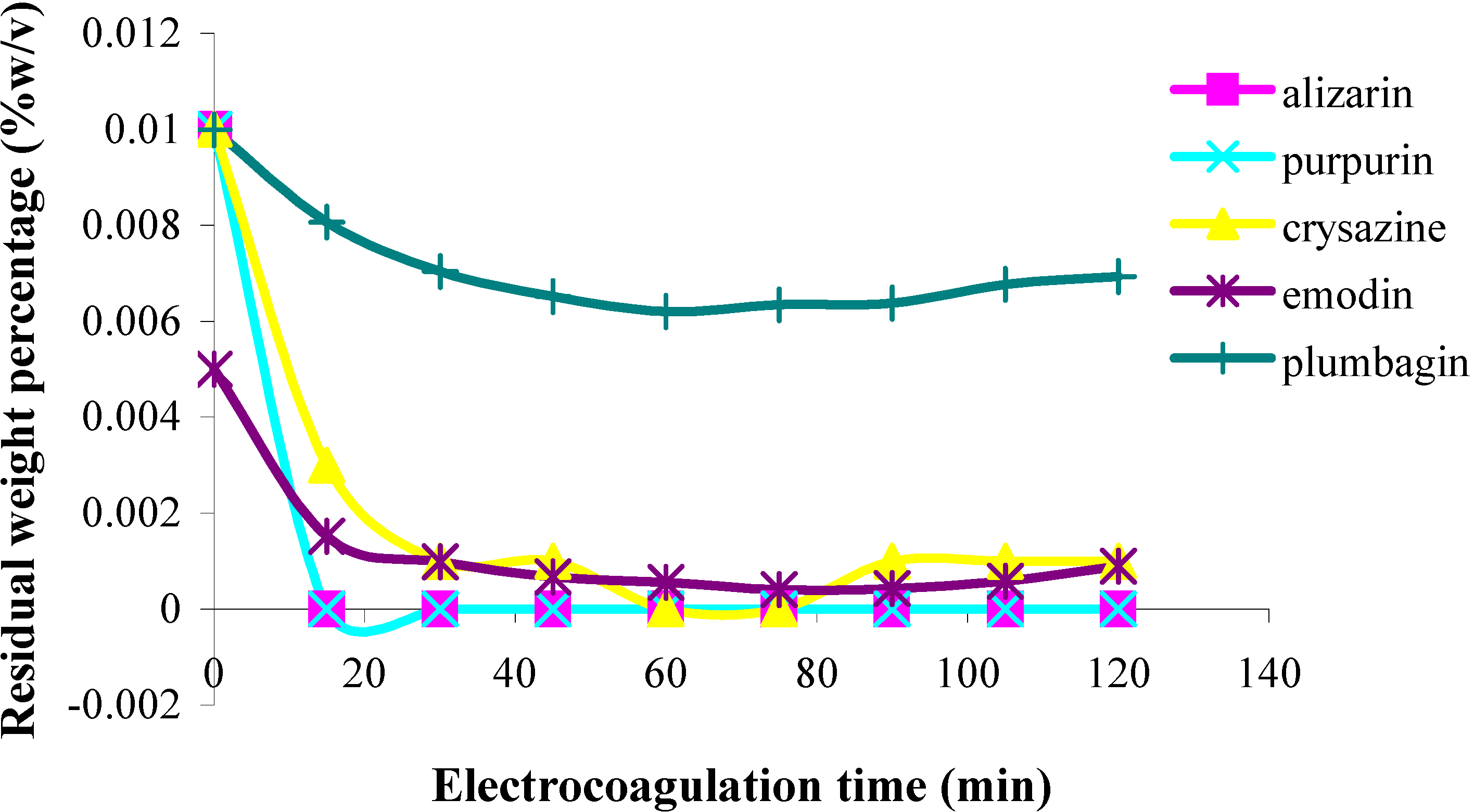
Figure 2.
Plots of residual weight percentage vs. electrolysis time for quinones in 85% ethanol solution;
![Molecules 11 00514 i001]()
alizarin,
![Molecules 11 00514 i002]()
purpurin,
![Molecules 11 00514 i003]()
chrysazine,
![Molecules 11 00514 i004]()
emodin, and
![Molecules 11 00514 i005]()
plumbagin.
Figure 2.
Plots of residual weight percentage vs. electrolysis time for quinones in 85% ethanol solution;
![Molecules 11 00514 i001]()
alizarin,
![Molecules 11 00514 i002]()
purpurin,
![Molecules 11 00514 i003]()
chrysazine,
![Molecules 11 00514 i004]()
emodin, and
![Molecules 11 00514 i005]()
plumbagin.
The second category of quinones comprised chrysazin and emodin, which were coagulated somewhat less completely and more slowly than the first two ones. In this case, it can also be seen from their structures that they have no adjacent hydroxyl groups, although they both possess two phenolic groups flanking a quinone carbonyl group (
Figure 1). This may then be the next best functional group arrangement of a quinone for complexing with metal ions released by the electrolytic process, although exactly why this should be so remains to be explained. In any case, however, this is better than the arrangement seen in the third category, in which only
one phenolic hydroxyl group flanks a carbonyl group, as seen in anthrarufin and plumbagin (
Figure 1), which were barely coagulated and give nearly horizontal coagulation curves (
Figure 2 and
Figure 3).
Next, all the coagulated quinones, i.e. alizarin, purpurin, chrysazin, and emodin, were recovered using the previously reported procedure [
7,
9,
12], which essentially consists of dissociating the aluminium complexes of each phenolic quinone with dilute mineral acid (HCl) and separating the thus freed quinones by extraction with a polar solvent (in this case 1-butanol). The integrity of each recovered quinone was confirmed by established chemical and physical methods. Having done this successfully, we then embarked on the isolation of some natural quinones, namely plumbagin, morindone, and erythrolaccin, from the roots of
Plumbago rosea, Morinda angustifolia and from seedlac, respectively. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) is a well-known natural naphthoquinone [
13], while morindone (1,2,5-trihydroxy-6-methylanthraquinone) is a less well-known isomer of emodin, useful as a red dye for cotton [
14]. Erythrolaccin (1,3,5,6-tetrahydroxy-8-methyl-anthraquinone) is a yellow dyestuff present in seedlac obtained from crude lac, a resinous substance secreted by the scale insect
Laccifer lacca [
15a].
Figure 3.
Plots of absorbance vs. electrolysis time for quinones in 85% ethanol solution;
![Molecules 11 00514 i006]()
alizarin,
![Molecules 11 00514 i003]()
purpurin,
![Molecules 11 00514 i001]()
chrysazine,
![Molecules 11 00514 i004]()
emodin,
![Molecules 11 00514 i002]()
anthrarufin, and
![Molecules 11 00514 i007]()
plumbagin.
Figure 3.
Plots of absorbance vs. electrolysis time for quinones in 85% ethanol solution;
![Molecules 11 00514 i006]()
alizarin,
![Molecules 11 00514 i003]()
purpurin,
![Molecules 11 00514 i001]()
chrysazine,
![Molecules 11 00514 i004]()
emodin,
![Molecules 11 00514 i002]()
anthrarufin, and
![Molecules 11 00514 i007]()
plumbagin.
Plumbagin was extracted with 50% aqueous ethanol and the resulting extract was then electrolysed using conditions similar to those used in the experimental electrocoagulation of pure plumbagin. After filtering off the precipitate formed, the remaining electrolysed solution contained nearly pure plumbagin, plus a little of the salt added as supporting electrolyte. This can be eliminated by ion exchange, but we used distillation, whereby we obtained an aqueous alcoholic solution of purified plumbagin, which sublimed out from the distillation flask into the collection flask. Partitioning of this solution with a little dichloromethane afforded pure plumbagin in 0.49 g (0.54 % yield). By applying a reported isolation method involving organic solvent extraction and chromatographic purification [
16], a slightly lower yield (0.47g, 0.51%) was obtained from the same root sample.
In the isolation of morindone, the situation was somewhat different, due its being coagulated by the electrochemical process. Thus, after filtering the electrolysed solution, the coagulum or precipitate formed was saved for recovery of the desired phenolic anthraquinone. When this was done, a moderately pure morindone was obtained (matching IR, but broad melting point).
In our attempt to isolate erythrolaccin from seedlac, an aqueous alcoholic solution of this substance was treated under similar electrocoagulation conditions and using the same subsequent recovery operations as described above for the isolation of morindone. The recovered colouring matter obtained, however, was not identical to that which was isolated from seedlac by an established procedure for erythrolaccin isolation [
15a], thus suggesting that the recovered pigment, though potentially useful in itself, might be an artefact. This might occur due to the fact that the coagulation time required in this case was rather long (1-2 hours), which according to our experience, can be somewhat detrimental in general to the coagulated compounds. Also, it has been mentioned that in a prolonged electrolysing operation of this type, certain compounds present in the solutions may undergo reactions (e.g. oxidation or reduction) before coagulating [
10,
17]. As to the reason why erythrolaccin was only slowly electrocoagulated when it possesses adjacent phenolic groups like alizarin or purpurin, this is not clear, although it was mentioned some time ago that erythrolaccin does not all occur in a free state in lac or seedlac but is partially bound with the resinous substance [
15b]. On the other hand, however, it was noted that the electrolysed seedlac was very pale in colour. Normally, this material is decolourised using various forms of carbon but the results are usually far from satisfactory. We have found that electrocoagulation is far more efficient than carbon treatment in decolourising seedlac by coagulating out the yellow erythrolaccin. However, in our hand the yield of decolourised seedlac was still low, due to the fact that a good part of the resinous substances themselves tended to co-coagulate with the colouring matter.
Experimental Section
General
The tested compounds were used as received. Alizarin (1,2-dihydroxyanthraquinone), anthrarufin (1,5-dihydroxyanthraquinone) and purpurin (1,2,4-trihydroxyanthraquinone) were of standard grade and were purchased from Fluka Chemica AG (Buchs, Switzerland). Chrysazin (1,8-dihydroxy-anthraquinone, 90-95%) and emodin (1,3,8-trihydroxy-6-methylanthraquinone, 95%) were purchased from ACROS Organics (New Jersey, USA). Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) was of standard reagent grade and was purchased from Sigma (USA). Authentic morindone was a donated product, courtesy of Dr. Suree Phutrakul, Chemistry Department, Chiang Mai University. Sodium chloride (99.9%, AR grade) was purchased from Ajax Chemical Co. (Sydney, Australia). 1-Butanol (AR grade) was purchased from Fisher Scientific UK Limited (UK). Hydrochloric acid (37%) was purchased from Carlo Erba Reagent Co. (Ronando, MI, Italy), and 95% ethanol was of a commercial grade and purchased from a local brewery. Aluminium plate was purchased from a local store. Direct current was sustained by a GW Instek DC power supply. Absorbance was measured on a Genesys 10 spectrophotometer. 1H- and 13C-NMR spectra were recorded on a Bruker AVANCE 300 spectrometer. GC-MS and FT-IR were performed on Agilent 6890(GC)/HP 5975(MS) and Perkin Elmer 1725X instruments, respectively. The roots of Plumbago rosea were obtained from the botanical garden of the Faculty of Agricultural Production, Maejo University, Chiang Mai, Thailand. The roots of Morinda angustifolia were from Mae Hong Son Province, courtesy of Dr. Suree Phutrakul of the Chemistry Department, Chiang Mai University, Thailand. The seedlac was a local product obtained courtesy of Dr. Pichai Pirakitikulr of the Faculty of Pharmacy, Payap University, Chiang Mai, Thailand.
Preparation of sample solutions for electrocoagulation
0.01% (w/v) sample solutions of each of the four quinone compounds, viz. alizarin, chrysazin, purpurin, and plumbagin were prepared in aqueous (85% v/v) ethanol. In the case of anthrarufin, its concentration in this solvent system was somewhat less than 0.01%, due to incomplete dissolution, and for emodin its concentration in the same solvent was reduced to 0.005% due to poor solubility.
Electrocoagulation experiments
A pair of 15 X 4 cm aluminium plates spaced 3 cm apart was immersed 5.5 cm into each sample solution (250 mL) contained in a 400-mL beaker. The solution was agitated throughout the experiment with a magnetic stirrer. Sodium chloride (0.5 g or 0.2% w/v) was added as supporting electrolyte. Direct current (0.3 A, 24-31 V) was passed through the solution
via the two electrodes. At 15-minute intervals over a 2-hour period of electrolysis, a 4-mL aliquot of the solution was withdrawn, centrifuged, and the absorbance of the supernatant solution was measured at an appropriate wavelength corresponding to the absorption maximum for each quinone solution as follows: alizarin 435 nm, anthrarufin 420 nm, chrysazin 430 nm, emodin 440 nm, purpurin 485 nm and plumbagin, 420 nm. The data thus obtained for each compound were used to construct the electrocoagulation curves as presented in
Figure 2 and
Figure 3.
Compound recovery experiments
A sample solution (200 mL) of each of the four well-coagulated quinone compounds, viz. alizarin, chrysazin, purpurin and emodin, was placed in a 250-mL beaker. Two aluminium plates (15 X 4 cm) were used as electrodes. These were placed 3 cm apart and dipped 6.5 cm into the magnetically-stirred solution. Sodium chloride (0.4 g) was added as supporting electrolyte. Direct current (0.3 A, 22-24 V) was then passed through the solution via the two electrodes for an appropriate time as follows: 40 min for alizarin, 75 min for chrysazin and emodin, and 15 min for purpurin. The resulting mixture was filtered through a Buchner funnel. The precipitate was collected and stirred in a sufficient volume of 10% hydrochloric acid solution to completely dissolve it. The acidic solution obtained was extracted with 1‑butanol (50 mL), the alcoholic solution was evaporated to dryness and the residual solid treated again with a small amount of 10% HCl, then filtered, washed with water and dried to afford the recovered compound. TLC, IR, m.p., and mixed m.p. were used to confirm the integrity of each recovered quinone (except in the case of emodin, whose m.p. could not be determined due to insufficient sample). The percentages of recovery obtained were 55, 28, 56 and 64 % for alizarin, purpurin, chrysaszin, and emodin, respectively.
Isolation of plumbagin
Air-dried roots of
Plumbago rosea (91.7 g) were immersed in 50% ethanol for 24 hours and then the mixture was refluxed for 2 hours. Filtration gave a dark solution, which was concentrated to a volume of 1 L, transferred to a beaker, sodium chloride (0.2%) was added as supporting electrolyte and the solution was electrocoagulated with a pair of 10 x 30 cm aluminium electrodes spaced 3 cm apart and dipped into the magnetically-stirred mixture. After 2 hours of electrolysis with direct current (2.0 A, 12 V), the mixture was filtered and the filtrate re-electrolysed under the same conditions for an additional 30 minutes, refiltered, and distilled on a rotary evaporator. The yellow distillate was extracted with a small volume of dichloromethane and the dichloromethane layer was concentrated to afford orange needles (0.49 g, 0.54%), m.p. 74-75
oC (lit. [
13] 78-79
oC), with spectroscopic data (IR, NMR, MS) identical with those of authentic plumbagin.
Isolation of morindone
Dry roots of
Morinda angustifolia (29 g) were extracted with methanol (500 mL) in a Soxhlet extractor for 8 hrs. The resulting solution, after diluting with a little water to an 85% alcohol concentration, was transferred to a beaker and electrocoagulated with a pair of 9 x 20 cm aluminium electrodes, spaced 1.5 cm apart and dipped 5.5 cm into the magnetically-stirred solution which contained 0.1% sodium chloride as supporting electrolyte. After 30 minutes of electrolysis with direct current (1.7 A, 30V) the mixture was filtered and the precipitate collected was dissolved in 10% HCl and the acidic solution extracted with 1-butanol. After evaporating off the alcohol, the extract yielded a coloured solid (2.72 g), a portion of which was recrystallised from acidified methanol to give a solid product (m.p. 274-280
oC) with an IR spectrum identical to that of authentic morindone (m.p. [
14] 280-284
oC).
Example of attempted isolation of erythrolaccin
A 1% solution of seedlac in 85% ethanol (250 mL) contained in a 400-mL beaker was electrocoagulated with a pair of 15 x 4 cm aluminium electrodes, spaced 3 cm apart and dipped 5.5 cm into the magnetically-stirred solution containing 0.2% sodium chloride as supporting electrolyte. After 1 hour of electrolysis with direct current (0.3 A, 22-27 V), the mixture was filtered and the filtrate was evaporated. The resinous brown residue was dissolved in absolute ethanol and the remaining undissolved inorganic matter was then filtered off from the solution which, after evaporating the solvent, gave a pale-coloured resinous lac (0.62 g). The precipitate obtained from the electrocoagulation above was dissolved in 10% hydrochloric acid solution and the resulting acidic solution was extracted with 1-butanol. The alcoholic solution was evaporated and the solid residue was collected, treated with a little 10% HCl, filtered, and washed free of acid with water. The IR spectrum of the resulting dried solid (0.37 g) was compared with that of a yellow pigment obtained from seedlac by the known method of erythrolaccin isolation [15a] and found to be non-identical.

{kind=link}
{kind=link}
{kind=link}
 alizarin,
alizarin,  purpurin,
purpurin,  chrysazine,
chrysazine,  emodin, and
emodin, and  plumbagin.
plumbagin.

 alizarin,
alizarin,  plumbagin.
plumbagin.