Experimental
General
Unless otherwise noted, chemicals were purchased from commercial suppliers and used without further purification. All solvents were distilled prior to use. Flash column chromatography was performed on silica gel 60 from Merck (40-63 μm). Melting points were determined using a Kofler-type Leica Galen III micro hot stage microscope and are uncorrected. NMR spectra were recorded from CDCl3 solutions on a Bruker AC 200 (200 MHz) and chemical shifts are reported in ppm using TMS as internal standard. Combustion analyses were carried out in the Microanalytic Laboratory, University of Vienna.
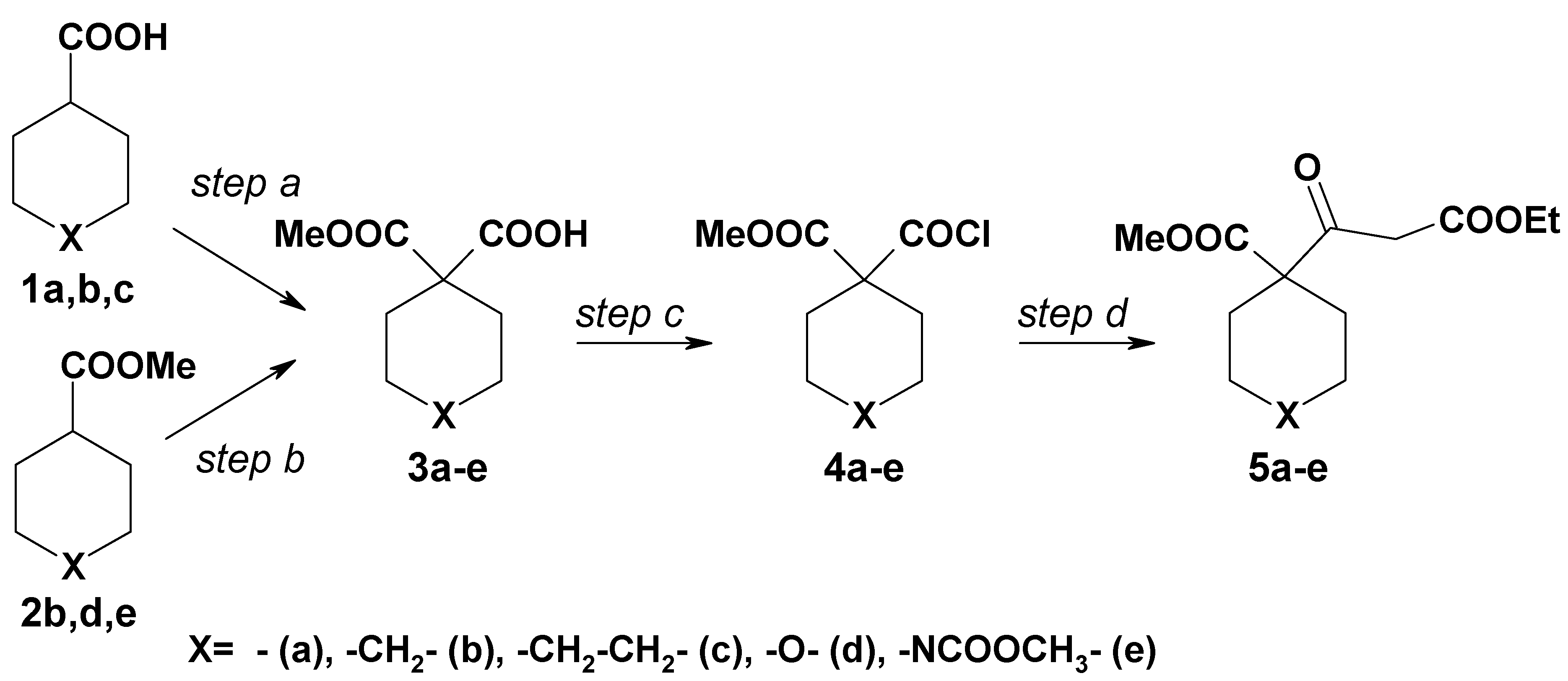
General procedure for the preparation of 1,1-dicarboxylic acid mono-methylesters introducing the ester functionality
Diisopropylamine (2-2.5 equiv.) in dry THF was treated with n-BuLi (2.5M in hexane, 2-2.5 equiv.) under nitrogen and stirred for 30 min at -40 °C. Then carboxylic acid 1 (1 equiv.) was added and the mixture was heated to 50-60 °C (reaction time depending on the acid). The mixture was cooled to -75±5 °C, ethyl chloroformate (1 equiv.) was added, and the reaction was stirred for 30 min at this temperature. The solution was poured into the same amount of deionized water. The aqueous phase was washed three times with diethyl ether. Then the pH of the aqueous layer was adjusted to 2-3 with 2N HCl followed by triple extraction with diethyl ether. The combined organic layers were washed once with brine, dried over Na2SO4, and concentrated. According to NMR the products were pure and were used in the next step without further purification.
Cyclopentane-1,1-dicarboxylic acid methylester (3a)
Yield: 12.46cg (83%) as a yellow oil from diisopropylamine (30.7 mL, 219 mmol), dry THF (200 mL), n-BuLi (92.5 mL, 219 mmol), cyclopentanecarboxylic acid 1a (10.0 g, 87.6 mmol in 30 mL dry THF) and ethyl chloroformate (6.7 mL, 87.6 mmol in 15 mL dry THF) with a deprotonation time of 3 hours at 50-60 °C. 1H-NMR δ: 1.36-2.35 (m, 8H, -(CH2)4-), 3.72 (s, 3H, -OCH3), 11.36 (s, 1H, -COOH); 13C-NMR δ: 25.4 (t, C3/C4), 34.6 (t, C2/C5), 52.7 (q, -OCH3), 60.3 (s, C1), 172.8 (s, -COOMe), 178.8 (s, -COOH)
Cyclohexane-1,1-dicarboxylic acid methylester (3b)
Yield: 13.35 g (92%) as yellow crystals [
6] from diisopropylamine (21.9 mL, 156 mmol), dry THF (150 mL),
n-BuLi (64.7 mL, 156 mmol), cyclohexanecarboxylic acid
1b (10.0 g, 78 mmol in 30 mL dry THF) and ethyl chloroformate (6 mL, 78 mmol in 15 mL dry THF) with a deprotonation time of 2 hours at 50-60 °C. Mp: 74-77 °C;
1H-NMR δ: 1.39–2.06 (m, 10H, -(CH
2)
5-), 3.75 (s, 3H, -OCH
3), 10.17 (s, 1H, -COOH);
13C-NMR δ: 22.6 (t, C3/C5), 24.9 (t, C4), 31.2 (t, C2/C6), 52.5 (q, -OCH
3), 55.0 (s, C1), 171.9 (s, -COOMe), 178.1 (s, ‑COOH)
Cycloheptane-1,1-dicarboxylic acid methylester (3c)
Yield: 11.67g (83%) as a yellow oil from diisopropylamine (29.6 mL, 210.9 mmol), dry THF (200 mL), n-BuLi (89.1 mL, 210.9 mmol), cycloheptanecarboxylic acid 1c (10.0 g, 70.3 mmol in 30 mL dry THF) and ethyl chloroformate (5.4 mL, 70.3 mmol in 15 mL dry THF) with a deprotonation time of 4 hours at 50-60 °C. 1H-NMR δ: 1.36-2.19 (m, 12H, -(CH2)6-), 3.66 (s, 3H, -OCH3), 9.97 (s, 1H, -COOH); 13C-NMR δ: 23.8 (t, C4/C5), 29.8 (t, C3/C6), 33.6 (t, C2/C7), 52.5 (q, -OCH3), 57.7 (s, C1), 172.9 (s, -COOMe), 178.9 (s, -COOH)
General procedure for the preparation of 1,1-dicarboxylic acid monomethylesters by introducing the acid functionality
n-BuLi (2.5M in hexane, 1.5-2 equiv.) was added to a stirred solution of diisopropylamine (1.5-2 equiv.) in dry THF (N2, -10 to -15 °C). After 15 min this mixture was treated dropwise (during 30 min) with a solution of methyl ester 2 (1 equiv.), followed by passing a fast and dry stream of CO2 through the mixture for 15 min. The solution was then poured into the same amount of deionized water. The aqueous phase was separated and washed three times with diethyl ether. Then the pH of the aqueous layer was adjusted to 2-3 with 2N HCl and it was extracted three times with diethyl ether. The organic layer was washed once with brine, dried over Na2SO4 and evaporated. According to NMR the products were pure and were used in the next step without purification.
Cyclohexane-1,1-dicarboxylic acid methylester (3b)
Yield: 4.55 g (68%) as yellow crystals [
6] from diisopropylamine (5.9 mL, 42 mmol), dry THF (50 mL),
n-BuLi (25.2 mL, 42 mmol), and
2b (5.09 g, 36 mmol).
Tetrahydropyran-4,4-dicarboxylic acid methylester (3d)
Yield: 9.5 g (72%) as colorless crystals from diisopropylamine (19.7 mL, 140.8 mmol), dry THF (200 mL), n-BuLi (59.5 mL, 140.8 mmol), and 2d (10.0 g, 70.4 mmol).. Mp: 113-116 °C; 1H-NMR δ: 2.01-2.18 (m, 4H, H3/H5), 3.57-3.76 (m, 4H, H2/H6), 3.72 (s, 3H, -OCH3), 10.25 (s, 1H, -COOH); 13C-NMR δ: 30.8 (t, C3/C5), 52.2 (s, C4), 52.9 (q, -OCH3), 64.6 (t, C2/C6), 170.9 (s, -COOMe), 175.1 (s, -COOH)
1,4,4-Piperidinetricarboxylic acid 1,4-dimethylester (3e)
Yield: 4.22 g (84%) as colorless crystals from diisopropylamine (5.8 mL, 41.1 mmol), dry THF (60 mL), n-BuLi (17.4 mL, 41.1 mmol), and 2e (4.14 g, 20.55 mmol). Mp: 123-126 °C; 1H-NMR δ: 2.02 (t, J=6Hz, 4H, H3/H5), 3.43 (t, J=7Hz, 4H, H2/H6), 3.63 (s, 3H, -OCH3), 3.69 (s, 3H, -NCOOCH3), 10.15 (bs, 1H, -COOH); 13C-NMR δ: 30.3 (t, C3/C5), 40.9 (t, C2/C6), 52.8 (q, -OCH3), 52.9 (q, -OCH3), 53.0 (s, C4), 156.1 (s, -NCOOMe), 170.1 (s, -COOMe), 173.9 (s, -COOH)
1-Chlorocarbonylcylopentane-1-carboxylic acid methyl ester (4a)
Yield: 4.22 g (84%) as a red brown oil from compound 3a (12.46 g, 72.4 mmol) and thionyl chloride (10.5 mL, 144.8 mmol). 1H-NMR δ: 1.36-2.35 (m, 8H, ‑(CH2)4-), 3.72 (s, 3H, -OCH3); 13C-NMR δ: 25.3 (t, C3/C4), 34.9 (t, C2/C5), 53.1 (q, -OCH3), 70.4 (s, C1), 170.6 (s,-COOMe), 173.1 (s, -COCl)
1-Chlorocarbonylcyclohexane-1-carboxylic acid methyl ester (4b)
Yield: 15.9 g (96%) as a red brown oil from compound 3b (14.032 g, 75.35 mmol) and thionyl chloride (10.9 mL, 150.7 mmol). 1H-NMR δ: 1.33–2.15 (m, 10H, (CH2)5), 3.72 (s, 3H, -OCH3); 13C-NMR δ: 22.5 (t, C3/C5), 24.8 (t, C4), 31.8 (t, C2/C6), 52.9 (q, ‑OCH3), 64.7 (s, C1), 169.9 (s, -COOMe), 173.2 (s, -COCl)
1-Chlorocarbonylcycloheptane-1-carboxylic acid methyl ester (4c)
Yield: 11.35 g (99%) as a red brown oil from compound 3c (10.47 g, 52.3 mmol) and thionyl chloride (7.6 mL, 104.6 mmol). 1H-NMR δ: 1.41-2.21 (m, 12H, -(CH2)6-), 3.71 (s, 3H, -OCH3); 13C-NMR δ: 23.5 (t, C4/C5), 29.6 (t, C3/C6), 33.9 (t, C2/C7), 52.9 (q, -OCH3), 67.7 (s, C1), 170.9 (s, ‑COOMe), 173.7 (s, -COCl)
4-Chlorocarbonyltetrahydopyran-4-carboxylic acid methyl ester (4d)
Yield: 10.02 g (97%) as a red brown oil from compound 3d (9.438 g, 50.2 mmol) and thionyl chloride (7.3 mL, 100.4 mmol). 1H-NMR δ: 2.09-2.20 (m, 4H, H3/H5), 3.62-3.68 (m, 4H, H2/H6), 3.76 (s, 3H, -OCH3); 13C-NMR δ: 31.4 (t, C3/C5), 53.3 (q, ‑OCH3), 61.9 (s, C4), 64.0 (t, C2/C6), 168.8 (s, -COOMe), 172.4 (s, -COCl)
4-Chlorocarbonylpiperidine-1,4-dicarboxylic acid dimethyl ester (4d)
Yield: 4.13 g (91%) as a red brown oil from compound 3d (4.22 g, 17.2 mmol) and thionyl chloride (2.5 mL, 34.4 mmol). As the reaction was not complete after 3 days another 2 equiv. of thionyl chloride were added and stirring was continued for another 3 days. 1H-NMR δ: 2.11-2.25 (m, 4H, H3/H5), 3.50-3.58 (m, 4H, H2/H6), 3.70 (s, 3H, -NCOOCH3), 3.83 (s, 3H, -COOCH3); 13C-NMR δ: 30.9 (t, C3/C5), 40.4 (t, C2/C6), 52.8 (q, -OCH3), 53.4 (q, -OCH3), 62.8 (s, C4), 155.6 (s, -NCOOMe), 168.8 (s, -COOMe), 172.4 (s, -COCl)
1-(2-Ethoxycarbonylacetyl)cyclopentanecarboxylic acid methyl ester (5a)
Yield: 10.00 g (57%) as a yellow oil from diisopropylamine (20.2 mL, 144 mmol), dry THF (150 mL), n-BuLi (60.6 mL, 144 mmol), dry ethyl acetate (7 mL, 72 mmol), and 4a (13.69 g, 72 mmol), followed by hydrolysis with saturated NH4Cl-solution (8 mL). Product purification was achieved by chromatography on silica gel (300 g, 20:1 LP-EtOAc). Elemental analysis: calculated: C 59.49%, H 7.49% found: C 59.67%, H.7.32%; 1H-NMR keto δ: 1.18 (t, J=7.18Hz, 3H, -CH3), 1.48-2.18 (m, 8H, -(CH2)4-), 3.43 (s, 2H,-CH2-), 3.65 (s, 3H, -OCH3), 4.11 (q, J=7.16Hz, 2H, -OCH2-); enol δ: 1.18 (t, J=7Hz, 3H, -CH3), 1.48-2.18 (m, 8H, ‑(CH2)4-), 3.65 (s, 3H, ‑OCH3), 4.11 (q, J=7Hz, 2H, -OCH2-), 5.03 (s, 1H, =CH-), 12.29 (s, 1H, -OH); 13C-NMR keto δ: 13.9 (q, -CH3), 25.5 (t, C3/C4), 32.9 (t, C2/C5), 45.7 (t, C-α), 52.5 (q, -OCH3), 61.2 (t, -OCH2), 66.9 (s, C1), 166.7 (s, -COOMe), 173.2 (s, -COOEt), 198.3 (s, C=O); enol δ: 13.9 (q, -CH3), 25.5 (t, C3/C4), 32.9 (t, C2/C6), 52.5 (q, -OCH3), 61.2 (t, -OCH2-), 66.9 (s, C1), 88,1(d, C-α), 166.7 (s, ‑COOMe), 173.2 (s, -COOEt), 178.1 (s, =C-OH)
1-(2-Ethoxycarbonylacetyl)cyclohexanecarboxylic acid methyl ester (5b)
Yield: 14.2 g (80%) as a yellow oil from diisopropylamine (20.2 mL, 143.8 mmol), dry THF (150 mL), n-BuLi (60.6 mL, 143.8 mmol), dry ethyl acetate (7 mL, 71.9 mmol), and 4b (15.88 g, 71.9 mmol), followed by hydrolysis using saturated NH4Cl-solution (8 mL). Product purification was achieved by chromatography on silica gel (300 g, 10:1 LP-EtOAc). Elemental analysis: calculated: C 60.92%, H 7.80% found: C 61.12%, H.7.62%; 1H-NMR keto δ: 1.23 (t, J=7Hz, 3H, -CH3), 1.17-2.12 (m, 10H, -(CH2)5-), 3.45 (s, 2H, -CH2-), 3.71 (s, 3H, -OCH3), 4.14 (q, J=7Hz, 2H, -OCH2-); enol δ: 1.23 (t, J=7Hz, 3H, -CH3), 1.17-2.12 (m, 10H, ‑(CH2)5), 3.71 (s, 3H, ‑OCH3), 4.14 (q, J=7Hz, 2H, -OCH2-), 5.09 (s, 1H, =CH-), 12.38 (s, 1H, -OH); 13C-NMR keto δ: 13.9 (q, ‑CH3), 22.6 (t, C3/C5), 25.1 (t, C4), 30.4 (t, C2/C6), 44.9 (t, C-α), 52.5 (q, -OCH3), 61.3 (t, -OCH2), 61.4 (s, C1), 166.8 (s, -COOMe), 171.9 (s, -COOEt), 199.9 (s, C=O); enol δ: 13.9 (q, -CH3), 22.6 (t, C3/C5), 25.1 (t,C4), 30.4 (t,C2/C6), 52.5 (q,-OCH3), 61.3 (t, -OCH2-), 61.4 (s, C1), 88.5 (d, C-α), 166.8 (s, -COOMe), 171.9 (s, -COOEt), 176.2 (s, =C-OH)
1-(2-Ethoxycarbonylacetyl)cycloheptanecarboxylic acid methyl ester (5c)
Yield: 10.02 g (72%) as a yellow oil from diisopropylamine (14.4 mL, 103 mmol), dry THF (100 mL), n-BuLi (43.5 mL, 103 mmol), dry ethyl acetate (5 mL, 51.5 mmol), and 4c (11.26 g, 51.5 mmol) followed by hydrolysis using saturated NH4Cl-solution (7 mL). Product purification was achieved by chromatography on silica gel (300g, 10:1 LP:EtOAc). Elemental analysis: calculated: C 62.20%, H 8.20% found: C 62.45%, H.7.93%; 1H-NMR keto δ: 1.29 (t, J=7Hz, 3H, -CH3), 1.39-2.12 (m, 12H, -(CH2)6-), 3.39 (s, 2H,-CH2-), 3.66 (s, 3H, -OCH3), 4.12 (q, J=7Hz, 2H, -OCH2-); enol δ: 1.29 (t, J=7Hz, 3H, -CH3), 1.39-2.12 (m, 12H ‑(CH2)6), 3.66 (s, 3H, -OCH3), 4.12 (q, J=7.11Hz, 2H, -OCH2-), 5.03 (s, 1H, =CH-), 12.33 (s, 1H, -OH); 13C-NMR keto δ: 13.9 (q, -CH3), 23.5 (t, C4/C5), 29.9 (t, C3/C6)), 32.1 (t, C2/C7), 45.1 (t, C-α), 52.4 (q, -OCH3), 61.2 (t, -OCH2-), 63.8 (s, C1), 166.7 (s, -COOMe), 173.3 (s, -COOEt), 199.4 (s, C=O); enol δ: 13.9 (q, -CH3), 23.5 (t, C4/C5), 29.9 (t, C3/C6), 32.1 (t, C2/C7), 52.4 (q,-OCH3), 61.2 (t, -OCH2-), 63.8 (s, C1), 87.9 (d, C-α), 166.7 (s, -COOMe), 173.3 (s, -COOEt), 179.6 (s, =C-OH)
4-(2-Ethoxycarbonylacetyl)tetrahydropyran-4-carboxylic acid methyl ester (5d)
Yield: 9.39g (82%) as a yellow oil from diisopropylamine (12.56 mL, 89.6 mmol), dry THF (100 mL), n-BuLi (37.9 mL, 89.6 mmol), dry ethyl acetate (4.4 mL, 44.8 mmol), and 4d (9.26 g, 44.8 mmol) followed by hydrolysis using saturated NH4Cl-solution (6 mL). Product purification was achieved by chromatography on silica gel (300g, 20:1 LP:EtOAc). Elemental analysis: calculated: C 55.81%, H 7.02% found: C 55.65%, H.6.74%; 1H-NMR keto δ: 1.18 (t, J=7Hz, 3H, -CH3), 1.81-2.14 (m, 4H, H3/H5), 3.41-3.75 (m, H2/H6), 3.44 (s, 2H, -CH2-), 3.69 (s, 3H, -OCH3), 4.11 (q, J=7Hz, 2H, -OCH2-); enol δ: 1.18 (t, J=7Hz, 3H, -CH3), 1.81-2.14 (m, 4H, H3/H5), 3.41-3.75 (m, H2/H6), 3.69 (s, 3H, -OCH3), 4.11 (q, J=7Hz, 2H, -OCH2-), 5.04 (s, 1H, =CH), 12.35 (s, 1H, ‑OH); 13C-NMR keto δ: 13.9 (q, -CH3), 29.9 (t, C3/C5), 44.5 (t, C-α), 52.8 (q, -OCH3), 58.8 (s, C4), 61.4 (t, -OCH2-), 64.4 (t, C2/C6), 166.4 (s, -COOMe), 170.9 (s, -COOEt), 198.5 (s, C=O); enol δ: 13.9 (q, -CH3), 29.9 (t, C3/C5), 52.8 (q, -OCH3), 58.8 (s, C4), 61.4 (t, -OCH2-), 64.4 (t, C2/C6), 88.9 (d, C-α), 166.4 (s, -COOMe), 170.9 (s, -COOEt), 176.5 (s, =C-OH)
4-(2-Ethoxycarbonylacetyl)piperidine-1,4-dicarboxylic acid dimethyl ester (5e)
Yield: 4.54 g (93%) as a yellow oil from diisopropylamine (4.34 mL, 31 mmol), dry THF (150 mL), n-BuLi (13.1 mL, 31 mmol), dry ethyl acetate (1.5 mL, 15.5 mmol), and 4e (4.08 g, 15.5 mmol) followed by hydrolysis using saturated NH4Cl-solution (4 mL). Product purification was achieved by chromatography on silica gel (150 g, 7:1 LP:EtOAc). Elemental analysis: calculated: C 53.33%, H 6.71%, N 4.44% found: C 53.61%, H.6.78%, N 4.33%; 1H-NMR keto δ: 1.27 (t, J=7Hz, 3H, -CH3), 1.78-2.27 (m, 4H, H3/H5), 3.15-3.35 (m, H2/H6), 3.53 (s, 2H, ‑CH2), 3.68 (s, 3H, -OCH3), 3.79 (s, 3H, -NCOOCH3), 4.19 (q, J=7Hz, 2H, -OCH2-); enol δ: 1.27 (t, J=7Hz, 3H, -CH3), 1.78-2.27 (m, 4H, H3/H5), 3.15-3.35 (m, H2/H6), 3.68 (s, 3H, -OCH3), 3.79 (s, 3H, -NCOOCH3), 4.19 (q, J=7Hz, 2H, -OCH2-), 5.12 (s, 1H, =CH-), 12.47 (s, 1H, -OH); 13C-NMR keto δ: 13.9 (q, -CH3), 29.3 (t, C3/C5), 40.6 (t, C2/C6), 44.7 (t, C-α), 52.5 (q,-OCH3), 52.8 (q, ‑NCOOCH3), 59.5 (s, C4), 61.3 (t, -OCH2-), 155.5 (s, -NCOOMe), 166.3 (s, -COOMe), 170.7 (s, ‑COOEt), 198.5 (s, C=O); enol δ: 13.9 (q, -CH3), 29.3 (t, C3/C5), 40.6 (t, C2/C6), 52.5 (q, -OCH3), 52.8 (q, -NCOOCH3), 59.5 (s, C4), 61.3 (t, -OCH2-), 88.9 (d, C-α), 155.5 (s, -NCOOMe), 166.3 (s, ‑COOMe), 170.7 (s, -COOEt), 176.2 (s, =C-OH).


{kind=link}