Pyrinodemin A, a cytotoxic
bis-pyridine alkaloid with a unique
cis-cyclopent[
c]isoxazolidine moiety, has been isolated from a marine sponge
Amphimedon sp., and its relative stereostructure was proposed as
1 (Δ
16’,17’) on the basis of spectral data [
1]. The unique structure of pyrinodemin A has prompted synthetic chemists to its total synthesis of
1 as well as syntheses of the double bond isomers
2 (Δ
15’,16’) and
3 (Δ
14’,15’) [
2,
3,
4] followed by different proposals of the structural revision of pyrinodemin A to be
2 [
2] or
3 [
3,
4].
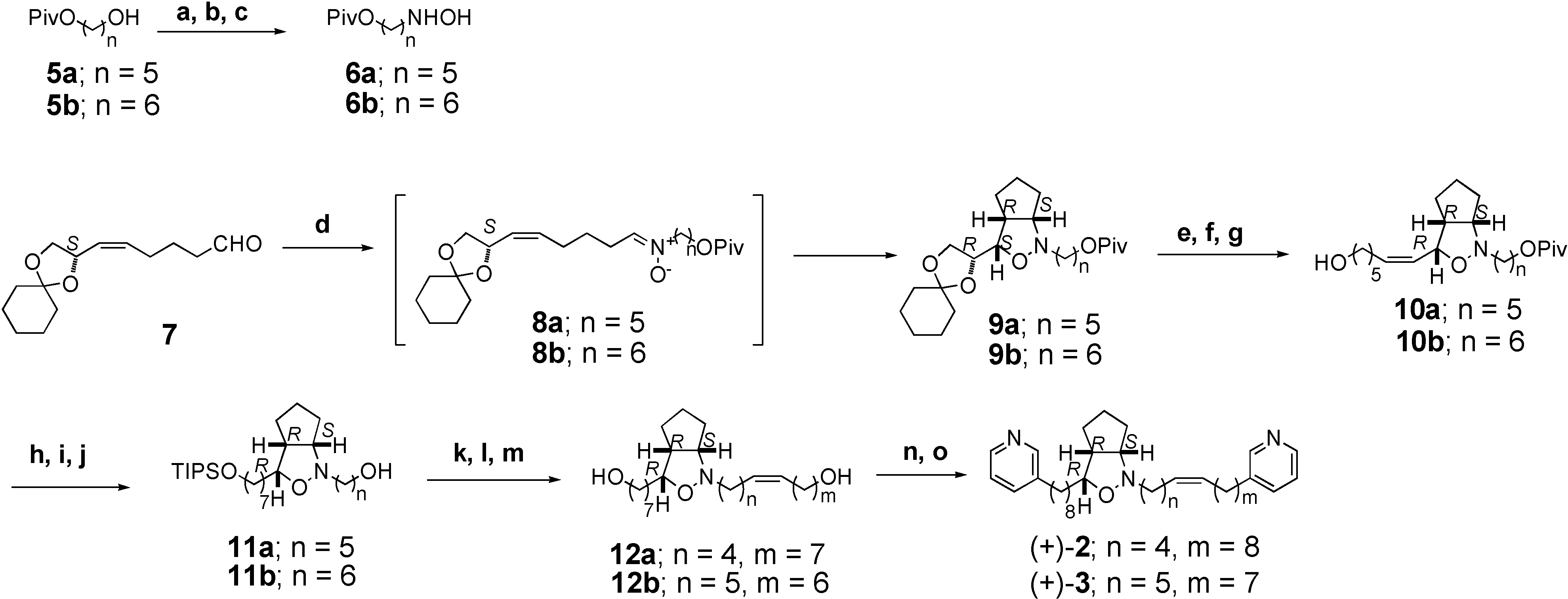
In order to examine the correct structure of pyrinodemin A, we have synthesized (+)-2 and (+)-3, the double bond isomers of 1, as an optically active form, and compared HPLC profiles of the synthetic compounds and pyrinodemin A. In addition, oxidative degradation experiments were performed for a remaining small amount of pyrinodemin A to determine the position of a double bond. In this paper, we describe asymmetric synthesis of (+)-2 and (+)-3, and indication of the structure of pyrinodemin A to be (±)-2.
![Molecules 10 00312 i001]()
The Δ
15’,16’ double bond isomer (+)-
2 was synthesized as follows (
Scheme 1). The synthesis of hydroxylamine
6a commenced with known pivaloate
5a [
5]. Oxidation of alcohol
5a with 2-iodobenzoic acid (IBX) [
6] in DMSO and THF afforded its aldehyde. Treatment of the aldehyde with NH
2OH·HCl and NaOAc in MeOH provided oxime which was reduced with NaBH
3CN in MeOH to afford hydroxylamine
6a [
7,
8]. Condensation of
6a and optically active aldehyde
7 [
8] in CHCl
3 containing Na
2SO
4 at r.t. gave the nitrone
8a, which was followed by heating to afford
cis-cyclopent[
c]isoxazolidine [
9]
9a in 58% yield.
Reagents and conditions: (a) IBX, DMSO, THF (69%); (b) H2NOH·HCl, AcONa, MeOH (96%); (c) NaBH3CN, MeOH, pH 3, 0 °C; (d) Na2SO4, 6, CHCl3, r.t.~reflux (58% for 2 steps); (e) 3N HCl, dioxane (80%); (f) NaIO4, MeCN, H2O, 0 °C; (g) Br-[Ph3+(CH2)5CH2OH], n-BuLi, THF, 0 °C (51% for 2 steps); (h) TIPSCl, imidazole, CH2Cl2 (75%); (i) H2, Pd-C, MeOH (93%); (j) DIBAL, CH2Cl2, -78 °C (75%); (k) IBX, DMSO (80%); (l) Br-[Ph3P+(CH2)7CH2OH], n-BuLi, THF, 0 °C (81%); (m) 46% HF, MeCN (55%); (n) CBr4, Ph3P (80%); (o) 3-methylpyridine, LDA, DMPU, -40 °C (64%)
Treatment of
9a with 3N HCl in dioxane gave diol, which was converted into its aldehyde by treatment with NaIO
4 and then into alcohol
10a by Wittig reaction [
10]. Protection of alcohol
10a as its TIPS ether followed by reduction with Pd-C gave its saturated TIPS ether, which was converted into alcohol
11a with DIBAL. IBX oxidation of
11a followed by Wittig reaction [
10] afforded its unsaturated alcohol, which was subjected to deprotection with HF to give diol
12a in 55 %. Treatment of diol
12a with CBr
4 and PPh
3 provided its dibromide, which was coupled with 3-methypyridine using LDA and DMPU [
11] in THF to furnish optically active compound (+)-
2. This is the first synthesis of optical active form of
2, although its racemic form ((±)-
2) has been synthesized [
2,
3,
4]. The Δ
14’,15’ double bond isomer (+)-
3 was prepared from pivaloate
5b by almost same procedure as described for synthesis of (+)-
2 (
Scheme 1).
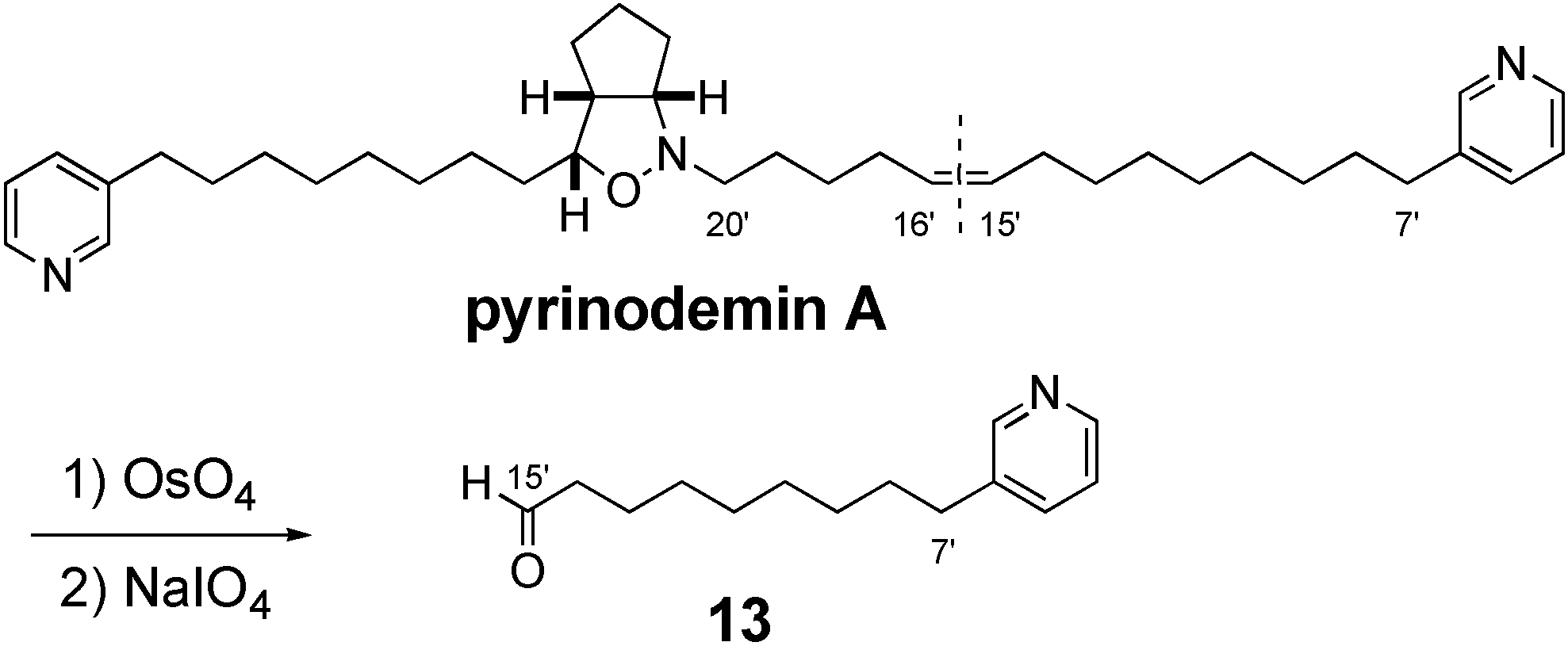
The position of a double bond and the stereochemistry of pyrinodemin A were examined as follows. Compounds (±)-
1 [
2], (±)-
2 [
2], and (+)-
3 were subjected to C
18 HPLC [Wako sil-II 5C18 RS, Wako Ind., Ltd., 4.6 x 250 mm; flow rate 1.0 mL/min: eluent; MeOH/H
2O (91:9); UV detection at 263 nm] and found to be separated (
1, t
R 21.6 mim;
2, t
R 17.0 min;
3, t
R 15.8 min), while the retention time (t
R 17.0 min) of pyrinodemin A was identical with that of
2 under the same condition, indicating that the position of a double bond of pyrinodemin A corresponded to that (Δ
15’,16’) of
2. To elucidate the stereochemistry of pyrinodemin A, compound (±)-
2 was subjected to chiral HPLC [CHIRALCELL OD-H, Daicel Co., Ltd., 4.6 x 250 mm; flow rate 1.0 mL/min: eluent: hexanes/
i-PrOH (95:5); UV detection at 263 nm] and found to be separated (t
R 44 and 47 min), while the retention time of (+)-
2 was 47 min (
Figure 1). On the other hand, pyrinodemin A gave the two peaks corresponding to those of (±)-
2 in a ratio of 1:1 under the same conditions, indicating that pyrinodemin A is a 1:1 racemic mixture of
2. Furthermore, pyrinodemin A was treated with OsO
4 and then NaIO
4 to give degradation products, one of which showed an ESIMS fragment ion peak at
m/z 242 (M+Na)+, corresponding to an aldehyde (
13) of C-7’~C-15’ segment connected to a pyridine ring (
Scheme 2). From the results described above, it was indicated that the olefin position of pyrinodemin A was C-15’ and C-16’ (
2), as proposed by Snider’s group [
2], and that pyrinodemin A was a 1:1 racemic mixture of
2.
Figure 1.
Chiral HPLC profiles of (a) synthetic compounds (±)-2, (b) (+)-2, and (c) pyrinodemin A
Figure 1.
Chiral HPLC profiles of (a) synthetic compounds (±)-2, (b) (+)-2, and (c) pyrinodemin A

{kind=link}
{kind=link}
{kind=link}